Resumos
Acredita-se que o controle glicêmico e a duração do diabetes sejam os fatores de risco mais importantes para o desenvolvimento das microangiopatias diabéticas, contudo, as velocidades de progressão da nefropatia, da retinoaptia e da polineuropatia variam consideravelmente entre os pacientes. Além da presença de fatores de risco, como a hipertensão arterial, a dislipidemia e o fumo, existem evidências sugerindo que uma predisposição genética desempenha um papel na susceptibilidade para as complicações microvasculares. Com base na patogênese dessas complicações crônicas do diabetes, polimorfismos de vários genes candidatos que atuam em diferentes vias desse processo têm sido investigados, como os genes relacionados aos mecanismos dos danos induzidos pela hiperglicemia (os produtos finais de glicação avançada, o aumento na formação de espécies reativas de oxigênio e a atividade aumentada da via da aldose-redutase), os genes relacionados ao sistema renina-angiotensina; os genes que codificam a síntese das citoquinas, dos fatores de crescimento e dos seus receptores e dos transportadores de glicose entre muitos outros. Este artigo discute alguns estudos que corroboram com a importância da predisposição genética no desenvolvimento da microangiopatia diabética.
Diabetes do tipo 1; Hiperglicemia; Genética; Predisposição; Microangiopatia
Glycemic control and diabetes duration are believed to be the most important risk factors for the development of diabetic microangiopathy; however, the rate of progression of nephropathy, retinopathy and polyneuropathy varies considerably among patients. Besides the presence of risk factors such as hypertension, dyslipidaemia and smoking, there is evidence suggesting that genetic predisposition plays a role in the susceptibility to microvascular complications. Based on underlying pathogenesis, polymorphisms of several candidate genes belonging to multiple pathways have been investigated, like the genes related to mechanisms of hyperglycaemia-induced damage (such as advanced glycation end-products and reactive oxygen species increased formation, augmented activity of the aldose reductase pathway); genes related to the renin-angiotensin system; genes coding for cytokines, growth factors and its receptors, glucose transporter; among many others. This article reviews some studies that corroborate the importance of the genetic background in the development of diabetic microangiopathy.
Type 1 diabetes; Microvascular complications; Hyperglycemia; Genetic; Predisposition
REVISÃO
A predisposição genética para o desenvolvimento da microangiopatia no DM1
Genetic susceptibility to microangiopathy development in Type 1 diabetes mellitus
Maria Lúcia Corrêa-Giannella; Suzana Maria Vieira
Laboratório de Endocrinologia Celular e Molecular (LIM-25) da Faculdade de Medicina da Universidade de São Paulo (USP), SP, Brasil
Endereço para correspondência Endereço para correspondência: Maria Lúcia Corrêa-Giannella Av. Dr. Arnaldo, 455, sala 4305 01246-903 São Paulo, SP E-mail: malugia@lim25.fm.usp.br
RESUMO
Acredita-se que o controle glicêmico e a duração do diabetes sejam os fatores de risco mais importantes para o desenvolvimento das microangiopatias diabéticas, contudo, as velocidades de progressão da nefropatia, da retinoaptia e da polineuropatia variam consideravelmente entre os pacientes. Além da presença de fatores de risco, como a hipertensão arterial, a dislipidemia e o fumo, existem evidências sugerindo que uma predisposição genética desempenha um papel na susceptibilidade para as complicações microvasculares. Com base na patogênese dessas complicações crônicas do diabetes, polimorfismos de vários genes candidatos que atuam em diferentes vias desse processo têm sido investigados, como os genes relacionados aos mecanismos dos danos induzidos pela hiperglicemia (os produtos finais de glicação avançada, o aumento na formação de espécies reativas de oxigênio e a atividade aumentada da via da aldose-redutase), os genes relacionados ao sistema renina-angiotensina; os genes que codificam a síntese das citoquinas, dos fatores de crescimento e dos seus receptores e dos transportadores de glicose entre muitos outros. Este artigo discute alguns estudos que corroboram com a importância da predisposição genética no desenvolvimento da microangiopatia diabética.
Descritores: Diabetes do tipo 1; Hiperglicemia; Genética; Predisposição; Microangiopatia
ABSTRACT
Glycemic control and diabetes duration are believed to be the most important risk factors for the development of diabetic microangiopathy; however, the rate of progression of nephropathy, retinopathy and polyneuropathy varies considerably among patients. Besides the presence of risk factors such as hypertension, dyslipidaemia and smoking, there is evidence suggesting that genetic predisposition plays a role in the susceptibility to microvascular complications. Based on underlying pathogenesis, polymorphisms of several candidate genes belonging to multiple pathways have been investigated, like the genes related to mechanisms of hyperglycaemia-induced damage (such as advanced glycation end-products and reactive oxygen species increased formation, augmented activity of the aldose reductase pathway); genes related to the renin-angiotensin system; genes coding for cytokines, growth factors and its receptors, glucose transporter; among many others. This article reviews some studies that corroborate the importance of the genetic background in the development of diabetic microangiopathy.
Keywords: Type 1 diabetes; Microvascular complications; Hyperglycemia; Genetic; Predisposition
INTRODUÇÃO
A PREVENÇÃO DAS COMPLICAÇÕES crônicas é uma questão-chave no manejo do diabetes melito (DM) por causa da morbimortalidade a elas associadas. O Diabetes Control and Complications Trial (DCCT) (1) demonstrou que o controle intensivo da glicemia reduz a incidência de complicações microvasculares em diabéticos melito tipo 1 (DM1), reforçando a hiperglicemia como o fator de risco mais importante para o aparecimento dessas complicações. Sabe-se, entretanto, que outros fatores de risco modificáveis, como a hipertensão arterial sistêmica (HAS), a dislipidemia e o tabagismo, participam do desenvolvimento da microangiopatia diabética.
Ainda que o papel da hiperglicemia crônica, modulado por determinantes, como idade de início e duração do DM, e dos demais fatores de risco estejam bem estabelecidos, o aparecimento das complicações crônicas em um indivíduo é determinado por sua susceptibilidade genética à lesão induzida pelas anormalidades metabólicas desencadeadas pela hiperglicemia crônica. A importância da predisposição genética é ilustrada por dados recém-publicados em uma coorte de 6.707 famílias com DM1 que demonstrou que a presença de complicação em um irmão aumenta o risco para aquela complicação no paciente, com uma razão de risco de 9,9 para retinopatia, 6,2 para a nefropatia e 2,2 para a neuropatia. Além disso, o risco para nefropatia aumenta significativamente em pacientes que têm um dos pais com diabéticos melito tipo 2 (DM2) (2).
Tendo em vista as dificuldades de aderência ao tratamento intensivo, a identificação de marcadores genéticos para o desenvolvimento das complicações pode ajudar a definir que pacientes necessitam de um controle glicêmico mais (ou menos) rigoroso (3), além de permitir melhor compreensão da patogênese das complicações, o que pode possibilitar o desenvolvimento de novas terapias direcionadas para prevenção e tratamento.
EPIDEMIOLOGIA E FATORES DE RISCO PARA O DESENVOLVIMENTO DA MICROANGIOPATIA
Nefropatia
Várias evidências apontam para a participação de fatores genéticos no risco de desenvolvimento de nefropatia diabética (ND), como a agregação familiar de nefropatia (4,5), a concordância da gravidade da lesão glo-merular em indivíduos de uma mesma família (6) e a maior predisposição para desenvolvimento de ND em pacientes diabéticos cujos pais apresentam hipertensão ou doença cardiovascular, o que sugere que genes envolvidos no controle da pressão arterial (PA) e no risco cardiovascular também desempenhem algum papel na susceptibilidade à ND (7,8).
A própria história natural da ND corrobora a importância da susceptibilidade genética, pois se a hiperglicemia fosse o único fator de risco, a prevalência da ND aumentaria ao longo do tempo até que a maioria dos pacientes diabéticos desenvolvesse essa complicação (9). Sabe-se, no entanto, que o pico de incidência de ND ocorre entre 15 e 20 anos após o início do DM1 e diminui após esse período, com a incidência cumulativa de acometimento renal nos estudos mais antigos variando de 25% a 40% após 25 anos de diabetes (10). Assim, determinado subgrupo de pacientes parece destinado a desenvolver a ND, enquanto a maioria dos pacientes não a desenvolve, ainda que apresente controle glicêmico ruim.
Mesmo reconhecendo a participação de fatores genéticos na predisposição ao desenvolvimento da ND, as intervenções para minimizar o impacto dos fatores de risco modificáveis são fundamentais. No DCCT, a terapia intensiva reduziu o risco de desenvolvimento de microalbuminúria em 34% e, na coorte de prevenção secundária, a terapia intensiva reduziu o risco de microalbuminúria em 43% e o risco de albuminúria em 56% (11). Além disso, a incidência cumulativa de ND após 20 anos de doença em estudos mais recentes está menor do que a previamente relatada, variando de 3,6% a 13,7% em algumas populações avaliadas, o que provavelmente reflete o melhor controle glicêmico e pressórico dos pacientes nas últimas décadas (10).
Já foi demonstrado em um estudo prospectivo que avaliou a taxa de perda da função renal ao longo do tempo que apenas um terço dos pacientes com ND experimenta perda rápida da função renal, enquanto os outros dois terços apresentam evolução lenta ou mesmo função renal inalterada, apesar da presença de proteinúria e retinopatia grave. Entre as muitas variáveis examinadas, concentrações elevadas de colesterol e HAS foram preditivas de perda rápida da função renal (12). Também já se demonstrou a relação entre o tabagismo e o desenvolvimento de ND em DM1. Os fumantes têm risco 2,8 vezes maior de apresentar albuminúria em relação a não- fumantes (13). Sawicki e cols. mostraram que o tabagismo é fator de risco associado com a progressão da ND em DM1 hipertensos tratados (14).
No rastreamento da ND é importante salientar que, embora sua principal característica seja a presença de anormalidades na excreção urinária de albumina (EUA), um estudo derivado do EDIC (The Epidemiology of Diabetes Interventions and Complications)/DCCT demonstrou que um número significativo de DM1 desenvolve insuficiência renal sem a presença de microalbuminúria. Nesse estudo, a proporção de portadores de DM1 que tinha ritmo de filtração glomerular (RFG) estimado pela fórmula de Cockroft-Gault < 60 mL/min/1,73 m2 e EUA normal (< 30 mg/24h) variou de 29% aos 5 a 6 anos de estudo até 52% em 1 a 2 anos de estudo. No EDIC, aos 9-10 anos de estudo, 44% dos pacientes que tinham RFG < 60 ml/min/ 1,73m2 apresentavam EUA normal, o que ressalta a necessidade de se rastrear a ND não apenas com avaliações da EUA, mas também do RFG (15).
Retinopatia
Os estudos clínicos em diabéticos evidenciam variações significativas no início e na gravidade da retinopatia dia-bética (RD) que não são explicadas por fatores de risco conhecidos, como duração da doença e grau de controle glicêmico (16,17), sugerindo que a predisposição genética também influencie no aparecimento e na evolução desta microangiopatia. Outros achados que falam a favor da participação genética são o risco aumentado para desenvolvimento de RD grave em irmãos de pacientes afetados (18), a agregação familiar (17) e as diferenças na freqüência desta complicação entre diferentes populações (19).
A RD é clinicamente dividida em dois estágios principais: RD não-proliferativa, também chamada retinopatia background, e a RD proliferativa, cuja característica principal é a formação de neovasos retinianos. A duração (20) e a idade de início do DM são fatores relevantes para o desenvolvimento da RD e parece haver um risco aumentado de desenvolvimento das microangiopatias, de modo geral, em pacientes com DM de início na puberdade em relação aos pacientes cujo DM se iniciou antes da puberdade, talvez pelas alterações hormonais associadas a esse período da vida (21).
O Pittsburgh Epidemiology of Diabetes Complications Study II, publicado em 1990, encontrou retinopatia background presente em quase todos os pacientes após 20 anos de diabetes, enquanto a retinopatia proliferativa afetou 70% dos pacientes portadores de DM1 após 30 anos de diagnóstico (20). Estudos mais recentes têm mostrado diminuição na incidência de retinopatia proliferativa, mas faltam dados de grandes populações que demonstrem alterações na incidência de RD em seus estágios iniciais (10). Esta redução na freqüência da RD nas últimas décadas reflete a melhora do controle glicêmico dos pacientes, já que se sabe, desde os resultados do DCCT, que o controle intensivo reduz em 76% o risco de desenvolvimento de RD e, em pacientes com retinopatia leve a moderada, o tratamento intensivo diminui a progressão para retinopatia proliferativa ou retinopatia não-proliferativa grave (1).
Vários estudos demonstraram a influência da HAS no risco para aparecimento de RD, desenvolvimento de formas clínicas mais graves e progressão mais rápida da RD. A gravidez também é reconhecida como um fator de risco, já que a RD pode evoluir rapidamente durante ela, no entanto, a progressão é geralmente transitória e o risco da progressão no longo prazo não parece ser aumentado pela gravidez. Os fatores de risco para a progressão da RD durante a gestação são a gravidade da RD antes da concepção, controle glicêmico inadequado e a presença concomitante de HAS.
O tabagismo e a dislipidemia também já foram implicados no desenvolvimento da retinopatia, no entanto, as evidências são menos conclusivas que para a participação desses fatores de risco na ND. Existem estudos, ainda, que sugerem que a anemia possa aumentar o risco de progressão para formas mais graves de RD. Por outro lado, o uso de aspirina em diabéticos não está associado a risco aumentado de hemorragia ou progressão da RD ou do edema de mácula.
A inter-relação entre RD e ND é complexa e a freqüente coexistência das duas complicações pode refletir fatores predisponentes em comum. De qualquer modo, a presença e a gravidade da RD são indicadores de risco para proteinúria importante, assim como a proteinúria prediz RD proliferativa (22).
Polineuropatia periférica
A implicação de fatores genéticos no desenvolvimento da ND deriva da observação da falta de correlação entre o grau e a duração do DM e o surgimento desta complicação, com grandes diferenças interindividuais. A maior incidência e gravidade da neuropatia em certas populações, por exemplo, em diabéticos originados do norte da África em comparação a diabéticos europeus, também fala a favor da predisposição genética (23).
A neuropatia é um grupo heterogêneo de desordens de patologia variada, o que sugere que, além dos fatores metabólicos, outros mecanismos patogênicos estejam envolvidos, como insuficiência microvascular e auto-imunidade (24). Estudos clínicos e experimentais sugerem que o estresse oxidativo tenha relação com o desenvolvimento da polineuropatia diabética (25,26).
Estima-se que a polineuropatia afete até 30% dos pacientes com DM1 de ascendência européia (27), embora esta prevalência possa variar consideravelmente entre os estudos, principalmente em razão dos diferentes critérios diagnósticos e da seleção de pacientes. Evidências sugerem que o controle glicêmico, a idade, a duração do DM e a presença de RD e ND, o tabagismo e a altura (a altura é explicada como fator de risco porque os pacientes mais altos têm axônios mais longos e mais susceptíveis ao dano celular) são fatores de risco para o desenvolvimento de polineuropatia diabética (28). Outros fatores de risco implicados no desenvolvimento da neuropatia foram níveis elevados de triglicerídeos de jejum, presença de microalbuminúria e consumo excessivo de álcool (29).
Assim como para as demais complicações, o controle intensivo e as intervenções para minimizar o impacto dos fatores de risco modificáveis são fundamentais. O DCCT (1) demonstrou que a terapia intensiva com insulina reduz o desenvolvimento de neuropatia clínica em 64% após cinco anos de seguimento quando comparada à terapia convencional.
PRINCIPAIS MECANISMOS IMPLICADOS NA PATOGÊNESE DAS COMPLICAÇÕES CRÔNICAS
Dano celular induzido pela hiperglicemia
Alguns mecanismos são propostos para explicar o dano celular induzido pela hiperglicemia (glicotoxicidade). Os principais estão resumidos a seguir (Figura 1) (30).
Aumento da ativação da via dos polióis
O aumento da glicose intracelular naqueles tecidos nos quais a captação de glicose não depende de insulina, incluindo retina, rins e nervos periféricos, resulta conversão dela a sorbitol pela aldose redutase, uma enzima que, em situações de normoglicemia, tem uma baixa afinidade pela glicose. O sorbitol, por sua vez, é oxidado à frutose pela enzima sorbitol desidrogenase. O sorbitol não se difunde facilmente por meio das membranas e isso pode resultar aumento do dano osmótico nas células. A redução da glicose a sorbitol consome nicotinamida adenina difosfato (NADPH). Como o NADPH é necessário para regeneração da glutationa reduzida pela enzima glutationa redutase, isso pode induzir ou exacerbar o estresse oxidativo intracelular. O uso de inibidores da aldose redutase in vivo diminuiu a incidência de ND (31).
Aumento da formação dos AGEs
A glicose possui um grupo aldeído reativo capaz de reagir não enzimaticamente com o grupo amino das proteínas, levando à formação dos produtos de Amadori, dos quais o mais conhecido é a hemoglobina glicada (HbA1c). Outras reações ocorrem a partir deste ponto para produzir um grupo de compostos denominados AGEs (advanced glycation end-products), que se ligam irreversivelmente às proteínas. Essas modificações químicas de proteínas teciduais, lipídios e DNA afetam as suas estruturas, funções e turnover, o que contribui para a patogênese das complicações diabéticas de inú-meras formas, por exemplo, promovendo alteração da composição da matriz extracelular nos vasos e nos glomérulos e da expressão de fatores de crescimento por macrófagos e células endoteliais vasculares e pelas células mesangiais renais. Entre as várias proteínas celulares capazes de se ligar aos AGEs e mediar seus efeitos deletérios, está o receptor de AGEs (RAGEs).
Aumento da ativação da proteína quinase C
A família da proteína quinase C (PKC) compreende várias isoformas, algumas ativadas pelo segundo mensageiro diacilglicerol (DAG). A ativação da PKC resulta diminuição da produção de óxido nítrico (NO), aumento da atividade da endotelina-1, alteração na expressão de fatores de crescimento, como fator de crescimento endotelial vascular (VEGF) e fator transformante de crescimento beta (TGF-b), além da ativação do NFkb. Essas alterações resultam vasoconstrição, aumento da permeabilidade vascular e comprometimento do fluxo sanguíneo que podem transformar-se em edema, hipóxia e estímulo para neovascularização.
Aumento da ativação da via da hexosamina
A glicose intracelular em excesso é desviada para a via da hexosamina, onde é convertida à frutose-6-fosfato, que, por sua vez, é transformada a N-acetilglicosamina 6-fosfato pela enzima GFAT. A N-acetilglicosamina 6-fosfato é, posteriormente, convertida a UDP-N-acetilglicosamina que, por mecanismos não totalmente esclarecidos, modula a expressão de proteínas que funcionam como fatores de transcrição e que acabam por alterar a expressão de várias proteínas, como o PAI-I e o fator de crescimento TGF-b, envolvidos na vasculopatia diabética.
Recentemente, o estresse oxidativo foi proposto como o elemento unificador de todas essas vias na instalação das complicações diabéticas, já que as quatro diferentes vias descritas anteriormente determinam um mesmo processo induzido pela hiperglicemia: a superprodução de radical superóxido (O2) pela cadeia de transporte de elétron mitocondrial. O excesso de O2 inibe parcialmente o GAPDH, uma enzima da via glicolítica, o que resulta aumento dos metabólitos formados antes da ação do GAPDH. Esses metabólitos são desviados para utilização nas quatro vias metabólicas responsáveis pelo dano celular da hiperglicemia, em um ciclo vicioso que aumenta ainda mais a geração de O2 (Figura 1).
Estresse oxidativo
Durante o metabolismo normal, o oxigênio é reduzido à água e, neste processo, os produtos intermediários são o radical superóxido (O2), o peróxido de hidrogênio (H2O2) e o radical hidroxil (OH), que conjuntamente são denominadas de espécies reativas de oxigênio (ROS: reactive oxigen species). Esses compostos têm meia-vida ultracurta, pois a presença de um elétron não pareado os tornam extremamente reativos e capazes de causar danos a moléculas de DNA, proteínas e lipídios. O estresse oxidativo se estabelece quando as defesas intracelulares antioxidantes são insuficientes para detoxificar as ROS ou, também, quando há produção excessiva de ROS.
Os sistemas enzimáticos e não-enzimáticos de defesa celular contra o estresse oxidativo envolvem vários compostos, entre eles as enzimas superóxido dismutase (SOD), catalase e o sistema glutationa, que engloba a glutationa e as diferentes enzimas envolvidas em seu metabolismo.
Superóxido dismutases (SOD)
Pertencem à família da metaloenzimas e convertem o radical superóxido em peróxido de hidrogênio e oxi-gênio molecular. Há três principais formas de SOD: cobre-zinco-SOD (SOD1) no espaço intracelular, a manganês-SOD (SOD2) na mitocôndria e a SOD extracelular (SOD3). Estas enzimas são as mais extensivamente estudadas no processo do estresse oxidativo.
Catalase (CAT)
Enzima presente no citosol e nos peroxisomos que converte o peróxido de hidrogênio em água.
Sistema glutationa
A glutationa é um tripeptídeo composto pelos aminoácidos glutamina, cisteína e glicina e representa um dos principais compostos antioxidantes. A síntese da glutationa requer a ação consecutiva de duas enzimas, a primeira (etapa limitante) é a g-glutamilcisteína sintetase (g-CGS) e a seguir a glutationa sintetase. A glutationa detoxifica uma série de compostos por meio da enzima glutationa S-transferase e das glutationas pero-xidases (GPX) (32).
Os níveis de todos os marcadores do estresse oxidativo estão modificados em pacientes portadores de DM1 (33). Um estudo comparou a expressão das enzimas SOD1, SOD2, catalase e GPX em sangue periférico de pacientes DM1 com ND, sem ND e indivíduos controle em condições de normo e hiperglicemias. Houve aumento na expressão da SOD1, catalase e da GPX nos indivíduos normais e nos pacientes com DM sem nefropatia e diminuição da expressão destas enzimas nos pacientes com ND (34). Outro estudo avaliou a resposta dessas enzimas, desta vez em fibroblastos, observando que a expressão da GPX era significativamente maior em pacientes com DM sem nefropatia e controles normais comparados aos pacientes diabéticos com ND (35). Esses dados sugerem que a menor capacidade de aumentar as defesas antioxidantes esteja relacionada ao desenvolvimento da ND.
GENES CANDIDATOS PARA A MICROANGIOPATIA
Duas principais estratégias são usadas para explorar o genoma humano e buscar evidências de alterações genéticas que contribuam para o desenvolvimento de doenças monogênicas (causadas por mutações em um único gene) e poligênicas (causadas por polimorfismos em inúmeros genes e por fatores ambientais).
O desenvolvimento de mapas do genoma permitiu a identificação de genes relacionados a doenças mesmo que não se tenha um conhecimento avançado sobre elas. Esta estratégia, conhecida como rastreamento genômico (genome scan) identifica genes não com base em suas funções, mas sim em sua posição no genoma. Esta posição é determinada pelo estudo da co-segregação de uma doença com marcadores polimórficos distribuídos no DNA de várias famílias acometidas. Estes polimorfismos não têm significado funcional, mas estão em posições conhecidas em cada um dos cromossomas e por isso funcionam como marcadores. Se um determinado marcador estiver muito próximo do gene causador da doença que se está investigando, eles serão herdados juntos. Análises estatísticas complexas determinam a probabilidade dos diferentes marcadores estarem associados ao gene causador da doença e permitem que se conheça a região que alberga o gene. Como essas regiões em geral são de grande extensão, é necessário que se avalie que gene presente naquela região pode ser relevante na doença em questão e o mesmo deve ser seqüenciado nos pacientes acometidos para rastreamento de mutações.
A segunda estratégia, mais simples que o rastreamento genômico, é aquela que pressupõe algum conhecimento sobre a fisiopatologia da doença: a abordagem do gene candidato, na qual se investigam genes envolvidos em vias biológicas específicas relacionadas ao que se conhece da fisiopatologia da doença que está sendo estudada.
Aproximadamente 0,1% da seqüência do genoma difere entre os seres humanos e a maioria dessas diferenças corresponde a polimorfismos (ou seja, variações presentes em mais de 1% da população). A forma mais comum de polimorfismo é conhecida por SNP (single nucleotide polymorphism), na qual há troca, inserção ou deleção de um único nucleotídeo na molécula de DNA (Figura 2).
A predisposição a doenças poligênicas, como obesidade, DM e suas complicações, dislipidemias e osteoporose, entre outras, são determinadas por esses polimorfismos e a investigação de polimorfismos que participam da susceptibilidade a essas doenças têm se mostrado muito mais difícil do que a identificação de genes causadores de doenças monogênicas, por inúmeras razões, tais como: 1. grande número de SNPs no genoma humano (aproximadamente 10 milhões); 2. múltiplos genes influenciando a expressão da doença, cada um deles com um efeito relativamente fraco; 3. interação entre os genes; e 4. importante participação dos fatores ambientais modulando o efeito genético (36). Além disso, características étnicas e geográficas influenciam a freqüência dos SNPs, de maneira que a associação de polimorfismo com um traço fenotípico em determinada população pode não se reproduzir em outras (37).
A maioria dos polimorfismos não tem repercussão funcional e auxilia na identificação de genes da mesma forma que os marcadores polimórficos mencionados anteriormente, ou seja, por proximidade física com o gene. Alguns polimorfismos, no entanto, são considerados funcionais por serem capazes de influenciar a expressão gênica, aumentando ou diminuindo a quantidade final da proteína codificada por aquele gene (38).
Os genes candidatos que têm sido investigados como potenciais genes de susceptibilidade para o desenvolvimento da microangiopatia diabética incluem: 1. aqueles envolvidos nas vias metabólicas que medeiam o dano celular induzido pela hiperglicemia, quais sejam, genes relacionados à via dos polióis, à formação e ação dos AGEs e à ativação da PKC; 2. genes envolvidos na formação e na defesa contra ROS; 3. genes que codificam fatores de crescimento modulados pelas vias metabólicas desencadeadas pela hiperglicemia; 4. genes relacionados ao transporte de glicose; 5. genes relacionados à inflamação e à coagulação; 6. genes relacionados ao metabolismo lipídico; e 7. genes relacionados a alterações hemodinâmicas.
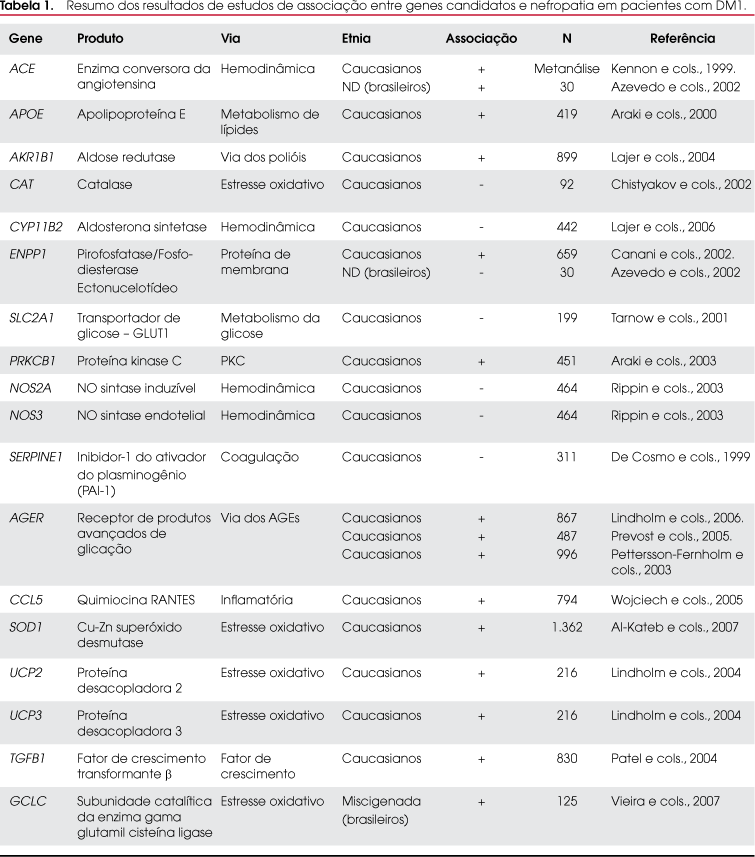
Nefropatia
A utilização do rastreamento genômico já identificou algumas regiões cromossômicas associadas ao desenvolvimento da ND em DM1, tal como a região onde se localiza o gene que codifica o receptor tipo 1 da angiotensina (AGTR1). Acredita-se que este local possa conter algum gene de susceptibilidade para essa complicação, já que associações não foram encontradas com o próprio gene AGTR1 neste mesmo estudo (39).
A Tabela 1 demonstra alguns genes candidatos que já foram avaliados quanto à associação com a ND. O gene ACE, que codifica a enzima conversora da angiotensina (ECA), foi o mais estudado em várias populações, tanto pelas evidências de participação do sistema renina-angiotensina (SRA) na lesão renal induzida pela glicose quanto pelo fato de existir uma variante genética neste gene que influencie diretamente as concentrações plasmáticas da ECA o polimorfismo I/D (40). Este polimorfismo envolve a presença (inserção, I) ou a ausência (deleção, D) de um fragmento de DNA de 286 pares de bases no intron 16 do gene. Os níveis de atividade da ECA em indivíduos homozigotos para o alelo D (DD) são aproximadamente duas vezes maiores que os encontrados nos homozigotos para o alelo I (II), porquanto os indivíduos heterozigotos I/D apresentam atividade intermediária (41). Acredita-se que na região de 286 pares de bases exista uma seqüência que regule negativamente a expressão do gene, de modo que o alelo D, por não possuir esse elemento regulador negativo, favoreça maior expressão gênica com conseqüente aumento dos níveis da ECA (42).
A associação do polimorfismo I/D com a predisposição para nefropatia foi demonstrada em uma coorte de 1.365 pacientes com DM1 (DCCT/EDIC Genetics Study), em que o genótipo II conferiu menor risco para desenvolvimento de microalbuminúria persistente (razão de risco de 0,62 [IC 95% 0,43-0,89], p = 0,009) e de nefropatia grave (0,56 [0,32-0,96], p = 0,033) (43).
O polimorfismo I/D também já foi avaliado em uma população de 30 DM1 normoalbuminúricos brasileiros seguidos prospectivamente por 10,2 ± 2,0 anos, na qual se observou que a presença do alelo D foi o único fator preditivo para o declínio do RFG. Adicionalmente, um aumento significativo nos casos de hipertensão e retinopatia foi observado nos pacientes com genótipos I/D e DD, mas não nos pacientes com genótipo II (44).
O estudo EURODIAB Controlled Trial of Lisinopril in IDDM foi um estudo prospectivo, multicêntrico, randomizado, controlado por placebo, que avaliou se o polimorfismo I/D modularia a progressão da doença renal em 530 pacientes com DM1 e se os inibidores da ECA influenciariam esta relação. Não havia diferenças significativas nas características basais dos pacientes distribuídos entre os três genótipos (15% II, 58% I/D e 27% DD). Uma interação significativa foi observada no grupo com genótipo II e o tratamento com lisinopril na evolução da albuminúria, pois os pacientes homozigotos para a inserção apresentaram maior renoproteção com o lisinopril em relação aos pacientes com os genótipos I/D e DD. Este é um exemplo de farmacogenômica, em que o conhecimento de um genótipo tem valor na determinação do impacto de uma terapêutica medicamentosa (45).
Retinopatia
Poucos estudos empregaram a abordagem do rastreamento genômico para avaliar a predisposição genética à RD, e os que o fizeram analisaram populações de pacientes com DM2, em que se demonstrou fraca associação da RD com marcadores localizados nos cromossomas 3 e 9 e, mais recentemente, com uma região localizada no braço curto do cromossoma 1 (46).
A Tabela 2 demonstra alguns genes candidatos já estudados quanto à associação com a RD. Apesar de vários genes já terem sido avaliados, poucos demonstraram uma forte associação com a freqüência ou a gravidade da retinopatia. A maioria dos estudos publicados foi fundamentada em um número pequeno de pacientes e parece não haver a replicação de alguns achados em diferentes populações, o que é atribuído, entre outras coisas, a diferenças na forma de se classificar a RD (47).
Vários estudos sugerem que polimorfismo que consiste de um número variável de repetições do dinucleotídeo AC (AC(n)) localizado a aproximadamente 2.000 pares de base do local de início da transcrição do gene da aldose redutase (ALR2) esteja relacionado à ND e à RD. Um outro polimorfismo localizado na região promotora deste mesmo gene (-106 C/T) também já foi correlacionado com a susceptibilidade para o desenvolvimento da RD isoladamente ou em combinação com o polimorfismo AC(n). Esses polimorfismos foram estudados em 64 pacientes brasileiros com DM1 com pelo menos 10 anos de diagnóstico, divididos conforme o grau de RD: sem RD, com RD não-proliferativa e com RD proliferativa. O alelo contendo 24 repetições AC (denominado alelo Z) foi significativamente associado com o desenvolvimento de RD proliferativa, enquanto o polimorfismo -106C/T não demonstrou associação. Observou-se, ainda, uma correlação positiva entre o desenvolvimento de RD proliferativa e a presença concomitante dos alelos Z e C (48). Este estudo ilustra a importância da validação dos polimorfismos em cada população, pois estudos realizados em outras populações correlacionaram a susceptibilidade para o desenvolvimento de RD com a presença do alelo contendo 23 repetições AC (alelo Z-2) (49,50).
Polineuropatia periférica
Os estudos até o momento realizados para avaliar a associação de fatores genéticos com a polineuropatia diabética utilizaram a abordagem do gene candidato. A maior parte dos genes investigados pertence à via de estresse oxidativo (Tabela 3) e quase todos os estudos resultaram associação positiva com a polineuropatia, no entanto, esses dados precisam ser validados em outras populações e em casuísticas maiores.
Em conclusão, para melhor compreensão dos fatores genéticos associados ao desenvolvimento das complicações microvasculares são necessários estudos com grande número de pacientes incluídos, que estes apresentem fenótipos bem caracterizados e que formem grupos bem estratificados. Com a evolução da tecnologia e a popularização de métodos capazes de rastrear milhares de polimorfismos simultaneamente, os estudos de associação ganharão maior importância e poderão permitir que se conheça mais profundamente a patogênese das complicações e que se desenvolvam medidas preventivas específicas a serem adotadas no tratamento daqueles pacientes predispostos.
Recebido em 10/12/2007
Aceito em 18/12/2007
- 1. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977-86.
- 2. Monti MC, Lonsdale JT, Montomoli C, Montross R, Schlag E, Greenberg DA. Familial risk factors for microvascular complications and differential male-female risk in a large cohort of american families with type 1. J Clin Endocrinol Metab. 2007; Sep 18 [Epub ahead of print&
- 3. Girach A, Manner D, Porta M. Diabetic microvascular complications: can patients at risk be identified? A review. Int J Clin Pract. 2006;60:1471-83.
- 4. Seaquist ER, Goetz FC, Rish S, Barbosa J. Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med. 1989;320:1161-5.
- 5. Quinn M, Angelico MC, Warram JH, Krolewski AS. Familial factors determine the development of nephropathy in patients with IDDM. Diabetologia. 1996;39:940-5.
- 6. Fioretto P, Steffes MW, Barbosa J, Rich SS, Miller ME, Mauer M. Is diabetic nephropathy inherited? Studies of glomerular structure in type 1 diabetic sibling pairs. Diabetes. 1999;48:865-9.
- 7. Roglic G, Colhoun HM, Stevens LK, Lemkes HH, Manes C, Fuller JH. Parental history of hypertension and parental history of diabetes and microvascular complications in insulin dependent diabetes mellitus: the EURODIAB complications study. Diabet Med. 1998;15:418-26.
- 8. De Cosmo S, Bacci S, Piras GP, Cignarelli M, Placentino G, Margaglione M, et al. High prevalence of risk factors for cardiovascular disease in parents of IDDM patients with microalbuminuria. Diabetologia. 1997;40:1191-6.
- 9. Conway BR, Savage DA, Maxwell AP. Identifying genes for diabetic nephropathycurrent difficulties and future directions. Nephrol Dial Transplant. 2006;21:3012-7.
- 10. Rossing P. The changing epidemiology of diabetic microangiopathy in type 1 diabetes. Diabetologia. 2005;48:1439-44.
- 11. The Diabetes Control and Complications Trial Research Group. Effect of intensive therapy on the development and progression of diabetic nephropathy in the Diabetes Control and Complications Trial. Kidney Int. 1995;47:1703-20.
- 12. Krolewski AS, Warram JH, Christlieb AR. Hypercholesterolemia: a determinant of renal function loss and deaths in IDDM patients with nephropathy. Kidney Int. 1994;(Suppl 45):S125-31.
- 13. Chase HP, Garg SK, Marshall G, Berg CL, Harris S, Jackson WE, et al. Cigarette smoking increases the risk of albuminuria among subjects with type 1 diabetes. JAMA. 1991;265:614-7.
- 14. Sawicki PT, Didjurgeit U, Muhlhauser I, Bender R, Heinemann L, Berger M. Smoking is associated with progression of diabetic nephropathy. Diabetes Care. 1994;17:126-31.
- 15. Molitch ME, Rutledge B, Steffes M, Cleary P. Renal insufficiency in the absence of albuminuria among adults with type 1 diabetes in the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) Study. ADA Annual Meeting 2006 (Abstract 23-OR)
- 16. Moss SE, Klein R, Klein BEK. 10-year incidence of visual loss in a diabetic population. Ophthalmology. 1994;101:1061-70.
- 17. Diabetes Control and Complications Trial Research Group. Clustering of long term complications in families with diabetes in the diabetes control & complication trial. Diabetes. 1997;46:1829-39.
- 18. Leslie RDG, Pyke DA. Diabetic retinopathy in identical twins. Diabetes. 1982;31:19-21.
- 19. Guillausseau PJ, Tielmans D, Virally-Monod M, Assayag M. Diabetes: from phenotypes to genotypes. Diabetes Metab. 1997;23:14-21.
- 20. Orchard TJ, Dorman JS, Maser RE, Becker DJ, Drash AL, Ellis D, et al. Prevalence of complications in IDDM by sex and duration. Pittsburgh Epidemiology of Diabetes Complications Study II. Diabetes. 1990;39:1116-24.
- 21. Vogt L, Jutzi E, Michaelis D. Different frequencies of diabetic complications in insulin-treated patients with diabetes of comparable duration, in relation to age at onset of diabetes. Soz Praventivmed. 1992;37:231-6.
- 22. Aiello LP, Cahill MT, Wong JS. Systemic considerations in the management of diabetic retinopathy. Am J Ophthalmol. 2001;132:760-76.
- 23. Vague P, Brunetti O, Valet AM, Attali I, Lassmann-Vague V, Vialettes B. Increased prevalence of neurologic complications among insulin dependent diabetic patients of Algerian origin. Diabete Metab. 1988;14:706-11.
- 24. Strokov IA, Bursa TR, Drepa OI, Zotova EV, Nosikov VV, Ametov AS. Predisposing genetic factors for diabetic polyneuropathy in patients with type 1 diabetes: a population-based case-control study. Acta Diabetol. 2003;40 Suppl 2: S375-9.
- 25. Low PA, Nickander KK, Tritschler HJ. The roles of oxidative stress and antioxidant treatment in experimental diabetic neuropathy. Diabetes.1997;46 Suppl 2:S38-42.
- 26. Rösen P, Nawroth PP, King G, Möller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev. 2001;17:189-212.
- 27. Siemionow M, Demir Y. Diabetic neuropathy: pathogenesis and treatment, J Reconstr Microsurg. 2004;20:241-52.
- 28. Tesfaye S, Stevens LK, Stephenson JM, Fuller JH, Plater M, Ionescu-Tirgoviste C, et al. Prevalence of diabetic peripheral neuropathy and its relation to glycaemic control and potential risk factors: the EURODIAB IDDM Complications Study. Diabetologia. 1996;39:1377-84
- 29. Swade TF, Emanuele NV. Alcohol & diabetes. Compr Ther. 1997;23:135-40.
- 30. Brownlee M. Biochemistry and molecular cell biology of diabetic complication. Nature. 2001;414:813-20.
- 31. Greene DA, Arezzo JC, Brown MB. Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Zenarestat Study Group. Neurology. 1999;53:580-91.
- 32. Scandalios JG. Oxidative stress: molecular perception and transduction of signals triggering antioxidant gene defenses. Braz J Med Biol Res. 2005;38:995-1014.
- 33. Varvarovska J, Racek J, Stozicky F, Soucek J, Trefil L, Pomahacova R. Parameters of oxidative stress in children with type 1 diabetes mellitus and their relatives. J Diabetes Complications. 2003;17:7-10.
- 34. Hodgkinson AD, Bartlett T, Oates PJ, Millward BA, Demaine AG. The response of antioxidant genes to hyperglycemia is abnormal in patients with type 1 diabetes and diabetic nephropathy. Diabetes. 2003;52:846-51.
- 35. Ceriello A, Morocutti A, Mercuri F, Quagliaro L, Moro M, Damante G, et al. Defective intracellular antioxidant enzyme production in type 1 diabetic patients with nephropathy. Diabetes. 2000;49:2170-7.
- 36. Polychronakos C. Genetic testing in clinical endocrinology. Hormones. 2003;2:201-10.
- 37. Salisbury BA, Pungliya M, Choi JY, Jiang R, Sun XJ, Stephens JC. SNP and haplotype variation in the human genome. Mutat Res. 2003;526:53-61.
- 38. Shastry BS. Genetic markers for disease and drug response. Int J Mol Med. 2003;11:379-8.
- 39. Moczulski DK, Rogus JJ, Antonellis A, Warram JH, Krolewski AS. Major susceptibility locus for nephropathy in type 1 diabetes on chromosome 3q: of novel discordant sib-pair analysis. Diabetes. 1998;47:1164-9.
- 40. Jacobsen PK. Preventing end stage renal disease in diabetic patients genetic aspect (part I). Renin Angiotensin Aldosterone Syst. 2005;6:1-14.
- 41. Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343-6.
- 42. Hunley TE, Julian BA, Phillips JA 3rd, Summar ML, Yoshida H, Horn RG et al. Angiotensin converting enzyme gene polymorphism: potential silencer motif and impact on progression in IgA nephropathy. Kidney Int. 1996;49:571-7.
- 43. Boright AP, Paterson AD, Mirea L, Bull SB, Mowjoodi A, Scherer SW, et al. Genetic variation at the ACE gene is associated with persistent microalbuminuria and severe nephropathy in type 1 diabetes: the DCCT/EDIC Genetics Study. Diabetes. 2005;54:1238-44.
- 44. De Azevedo MJ, Dalmáz CA, Caramori ML, Pecis M, Esteves JF, Maia AL, et al. ACE and PC-1 gene polymorphisms in normoalbuminuric type 1 diabetic patients: a 10-year prospective study. J Diabetes Complications. 2002;16:255-62.
- 45. Penno G, Chaturvedi N, Talmud PJ, Cotroneo P, Manto A, Nannipieri M, et al. Effect of angiotensin-converting enzyme (ACE) gene polymorphism on progression of renal disease and the influence of ACE inhibition in IDDM patients. Findings from the EUCLID randomized controlled trial. Diabetes. 1998;47:1507-1.
- 46. Looker HC, Nelson RG, Chew E, Klein R, Klein BEK, Knowler WC, et al. Genome-wide linkage analyses to identify loci for diabetic retinopathy. Diabetes. 2007;56:1160-6.
- 47. Warpeha KM and Chakravarthy U. Molecular genetics of microvascular disease in diabetic retinopathy. Eye. 2003;17:305-11.
- 48. Richeti F, Noronha RM, Waetge RTL, Vasconcellos JPC, Souza OF, Kneipp B, et al. Evaluation of AC(n) and C(-106)T polymorphisms of the aldose reductase gene in brazilian patients with DM1 and susceptibility to diabetic retinopathy. Mol Vis. 2007;13:740-5.
- 49. Kao YL, Donaghue K, Chan A, Knight J, Silink M. A novel polymorphism in the aldose reductase gene promoter region is strongly associated with diabetic retinopathy in adolescents with type 1 diabetes. Diabetes. 1999;48:1338-40.
- 50. Demaine A, Cross D, Millward A. Polymorphisms of the aldose reductase gene and susceptibility to retinopathy in type 1 diabetes mellitus. Invest Ophthalmol Vis Sci. 2000;41:4064-8.
Datas de Publicação
-
Publicação nesta coleção
25 Abr 2008 -
Data do Fascículo
Mar 2008
Histórico
-
Aceito
18 Dez 2007 -
Recebido
10 Dez 2007