Abstract
The immune and central nervous systems are functionally connected and interacting. The concept that the immune signaling to the brain which induces fever during infection and inflammation is mediated by circulating cytokines has been traditionally accepted. Administration of bacterial lipopolysaccharide (LPS) induces the appearance of a so-termed "cytokine cascade" in the circulation more or less concomitantly to the developing febrile response. Also, LPS-like fever can be induced by systemic administration of key cytokines (IL-1ß, TNF-alpha, and others). However, anti-cytokine strategies against IL-1ß or TNF-alpha along with systemic injections of LPS frequently lead to attenuation of the later stages of the febrile response but not of the initial phase of fever, indicating that cytokines are rather involved in the maintenance than in the early induction of fever. Within the last years experimental evidence has accumulated indicating the existence of neural transport pathways of immune signals to the brain. Because subdiaphragmatic vagotomy prevents or attenuates fever in response to intraperitoneal or intravenous injections of LPS, a role for vagal afferent nerve fibers in fever induction has been proposed. Also other sensory nerves may participate in the manifestation of febrile responses under certain experimental conditions. Thus, injection of a small dose of LPS into an artificial subcutaneous chamber results in fever and formation of cytokines within the inflamed tissue around the site of injection. This febrile response can be blocked in part by injection of a local anesthetic into the subcutaneous chamber, indicating a participation of cutaneous afferent nerve signals in the manifestation of fever in this model. In conclusion, humoral signals and an inflammatory stimulation of afferent sensory nerves can participate in the generation and maintenance of a febrile response.
fever; cytokines; neuroimmunomodulation; humoral factors; afferent nerves; central nervous system
Braz J Med Biol Res, March 2001, Volume 34(3) 301-314
Fever induction pathways: evidence from responses to systemic or local cytokine formation
J. Roth1 and G.E.P. de Souza2
1Department of Veterinary Physiology, School of Veterinary Medicine, University of Giessen, Giessen, Germany
2Laboratório de Farmacologia, Faculdade de Ciências Farmacêuticas, Universidade de São Paulo, Ribeirão Preto, SP, Brasil
Text
Abstract

The immune and central nervous systems are functionally connected and interacting. The concept that the immune signaling to the brain which induces fever during infection and inflammation is mediated by circulating cytokines has been traditionally accepted. Administration of bacterial lipopolysaccharide (LPS) induces the appearance of a so-termed "cytokine cascade" in the circulation more or less concomitantly to the developing febrile response. Also, LPS-like fever can be induced by systemic administration of key cytokines (IL-1ß, TNF-a, and others). However, anti-cytokine strategies against IL-1ß or TNF-a along with systemic injections of LPS frequently lead to attenuation of the later stages of the febrile response but not of the initial phase of fever, indicating that cytokines are rather involved in the maintenance than in the early induction of fever. Within the last years experimental evidence has accumulated indicating the existence of neural transport pathways of immune signals to the brain. Because subdiaphragmatic vagotomy prevents or attenuates fever in response to intraperitoneal or intravenous injections of LPS, a role for vagal afferent nerve fibers in fever induction has been proposed. Also other sensory nerves may participate in the manifestation of febrile responses under certain experimental conditions. Thus, injection of a small dose of LPS into an artificial subcutaneous chamber results in fever and formation of cytokines within the inflamed tissue around the site of injection. This febrile response can be blocked in part by injection of a local anesthetic into the subcutaneous chamber, indicating a participation of cutaneous afferent nerve signals in the manifestation of fever in this model. In conclusion, humoral signals and an inflammatory stimulation of afferent sensory nerves can participate in the generation and maintenance of a febrile response.
Key words: fever, cytokines, neuroimmunomodulation, humoral factors, afferent nerves, central nervous system
Humoral signals and fever: the relation between circulating cytokines and the febrile response
After a challenge with an infectious or inflammatory stimulus somewhere at the periphery of the body a number of responses are generated within the CNS. These brain-mediated signs of illness include changes in neuroendocrine activities including activation of the hypothalamic-pituitary-adrenal (HPA) axis, anorexia and adipsia, changes in sleep patterns, decreases in locomotor activity, libido, social contacts and exploration, symptoms which are collectively termed sickness behavior, and the development of fever, a regulated rise of body core temperature (1-4).
A number of exogenous substances are capable of evoking fever and the other listed brain-controlled sickness signs or symptoms when injected systemically into experimental animals. Such fever-inducing agents are called exogenous pyrogens. All these substances induce the production and release of immunoregulatory proteins which are generally termed cytokines. It has been proposed that circulating cytokines are the endogenous mediators of fever in response to administration of an exogenous pyrogen. This concept about the pathogenesis of fever (2) is mainly based on three experimental observations. Lipopolysaccharide (LPS) has been used as exogenous pyrogen in most experimental studies and therefore focus will be kept on the events that occur in response to systemic or local administration of this substance.
The first set of observations relate to the appearance of a number of cytokines in the bloodstream more or less simultaneously to the development of fever after injection of an exogenous pyrogen such as LPS from the cell wall of Gram-negative bacteria. Depending on the route and injected dose of LPS, tumor necrosis factor alpha (TNF-a) is the first cytokine which appears in the circulation (2,5,6), followed by traces of interleukin-1ß (IL-1ß) (5), and high amounts of IL-6 (6,7), IL-8 (8) as well as other cytokines such as, for example, macrophage inflammatory protein-1 (MIP-1) (9). A similar cytokine cascade can be monitored in response to other pyrogenic compounds of Gram-positive and Gram-negative bacteria such as superantigens, peptidoglycans or muramyl-dipeptides. Invading viruses or synthetic viral compounds activate a distinct pattern of cytokine production including interferons as initial mediators within the cytokine cascade (10). Among all cytokines which are measurable in blood plasma during LPS-induced fever, circulating levels of IL-6 show the best correlation with the febrile changes of body temperature (6,7).
The second set of observations relate to the fact that fever of similar pattern, extent and duration as that induced by LPS can also be induced by peripheral administration of the same cytokines which are endogenously released in response to systemic injection of LPS. Intraperitoneal, intravenous or intra-arterial injections or infusions of IL-1ß (2,11), TNF-a (2,12), IL-6 (13), IL-8 (14) or MIP-1 (15) evoke fever in various species of experimental animals. Whether the pyrogenic effects of these cytokines truly reflect the physiological conditions which are induced by peripheral injection of LPS is still a matter of debate (2). It has been argued that the fever-inducing properties of cytokines may rather represent pharmacological effects unrelated to LPS fever (2). Furthermore, it should be noted that only cytokines such as IL-6 which are circulating in large amounts during LPS fever are able to evoke moderate febrile responses (13), and that the pyrogenic effect does not manifest itself in all species of experimental animals as reported for IL-8 (14,16). However, the fact that cytokines circulating during LPS fever are themselves pyrogenic in most animal species still supports the hypothesis that these endogenous mediators can potentially act as humoral signals for the brain to induce fever and other sickness signs in response to infectious or inflammatory stimulation. The putative mechanisms of how circulating cytokines might influence brain structures which are involved in the manifestation of fever will be described in the chapter "Fever: what happens within the brain?".
The third body of evidence stems from experimental procedures which result in an attenuation of LPS-induced formation of circulating cytokines or in a neutralization of the biological activities of a given cytokine with a concomitant reduction of LPS fever. TNF-a and IL-1ß are regarded as initial mediators within the LPS-induced cytokine cascade. Therefore, most experimental "anti-cytokine strategies" are directed against TNF-a or IL-1ß.
As shown in Figure 1, intra-arterial injections of 10 µg/kg LPS cause a characteristic biphasic febrile response in guinea pigs (upper panel of Figure 1). Administration of a dimeric polyethylene glycol-linked form of the type 1 soluble receptor of TNF, a neutralizing TNF-binding protein, abolishes LPS-induced circulating bioactive TNF and causes a significant attenuation of the second phase of the febrile response (17). Intraperitoneal injection of 30 µg/kg LPS induces a similar biphasic fever in guinea pigs (lower panel of Figure 1). Again, the second fever phase can be depressed by administration of an IL-1 receptor antagonist (IL-1ra). Treatment with TNF-binding protein or IL-1ra not only attenuates the second phase of LPS fever but also suppresses circulating levels of IL-6, a result which confirms that TNF-a and IL-1ß both contribute to the LPS-induced formation of systemic IL-6. According to the results shown in Figure 1, the initial phase of LPS-induced fever is unimpaired by anti-TNF or anti-IL-1 strategies in guinea pigs. These findings suggest that circulating cytokines are not involved in the generation of the early phase of fever but rather provide signals which contribute to the maintenance of the later phase of fever. Such a role for circulating cytokines is compatible with the fact that the quick onset of LPS fever (3) or LPS-induced activation of the HPA axis (18) seems to precede the systemic formation of considerable amounts of these pyrogenic cytokines.
If the first phase of LPS fever develops independently of circulating TNF-a, IL-1ß, IL-6, IL-8 or MIP-1, what kind of signal then mediates the quick onset of the early stage of fever? One possibility which has been discussed in this context is the existence of a preformed pyrogenic factor (PFPF) which is continuously present in macrophages and can be released immediately after LPS stimulation (19). Also, pyrogenic substances which can be induced by LPS within seconds or minutes such as complement fragments have been proposed as endogenous humoral signals for induction of the early phase of fever (3,20). Finally, it has been speculated that a direct action of LPS on brain endothelial cells via activation of an ancient "toll-like receptor" which belongs to the IL-1 receptor family might contribute to the quick induction of fever (21).
An alternative and rapid communication pathway between the activated immune system and those parts of the brain which are involved in fever induction would be via stimulation of afferent nerves. Within the last 5 years a number of studies have focussed on this topic.
Afferent neural signals and fever: the role of the vagus nerve
Evidence for a role of the vagus nerve as a vehicle for transfer of peripheral immune signals to the brain derives from observations that transection of the abdominal trunks of the vagus nerve (subdiaphragmatic vagotomy) attenuates or even abrogates a number of illness responses to intraperitoneal or intravenous injections of moderate doses of bacterial LPS or proinflammatory cytokines. Sickness behavior induced by intraperitoneal injections of pyrogens is prevented in vagotomized animals (22). The LPS-induced activation of the HPA axis (23), hyperalgesia (24) or increase of slow-wave sleep (25) are all attenuated by subdiaphragmatic vagotomy. Even the LPS-induced expression of the c-Fos and IL-1ß genes within the brain is depressed in vagotomized rats or mice (26,27). Suppression of LPS fever after vagotomy has been reported for guinea pigs (3,28) and rats (29), but this phenomenon seems to depend on the injected dose of LPS (29) and on the route of LPS administration (28). One study even reported that subdiaphragmatic vagotomy failed to cause a significant suppression of fever (30). An example from experiments in vagotomized guinea pigs is shown in Figure 2.
The upper panel of Figure 2 shows the effect of intraperitoneal injection of LPS on the abdominal temperature of vagotomized and sham-operated guinea pigs. Subdiaphragmatic vagotomy results in a significant suppression of LPS fever as long as the pyrogen is injected by the intraperitoneal route. The lower panel of Figure 2 shows that intramuscular injection of LPS results in identical fever in both vagotomized and sham-operated guinea pigs. Vagal afferent fibers thus seem to play a role in the transduction of immune signals from the abdominal cavity to the brain. Apparently, however, it is possible to bypass the vagal communication pathway between the immune system and the brain, otherwise vagotomized guinea pigs would not develop unimpaired fever in response to intramuscular LPS injection. This finding is supported by another study showing that sickness behavior induced by IL-1 is suppressed in vagotomized rats only if the cytokine is injected by the intraperitoneal route but not by other routes (22).
Subdiaphragmatic vagotomy is a drastic procedure which interrupts both afferent and efferent fibers of the vagus nerve and may disturb vital homeostatic functions. Therefore, the question arises if there are additional arguments that support a role of the vagus nerve as a pathway for transmission of immune signals to the brain. Direct evidence for a stimulation of afferent fibers of the vagus nerve by immune signals derives from the observation that injection of IL-1ß into the portal vein at physiological doses of 10 or 100 pg per animal results in a significant increase of the firing rate of the vagal hepatic afferent nerve branch (31). Furthermore, glomus cells of vagal paraganglia are labeled with a biotinylated IL-1 receptor antagonist, indicating binding sites for IL-1 on the membranes of these cells (32). Finally, intraperitoneal administration of bacterial LPS (33) induces c-Fos immunoreactivity in primary afferent neurons of the vagus nerve. This protein product of the immediate early gene c-Fos indicates a rapid activation of these neurons in response to LPS. The activation of vagal afferent fibers (31) and of primary afferent neurons of the vagus nerve (33) by immune stimuli may thus provide direct evidence that vagal afferents, together with other mechanisms, transmit signals from the activated immune system to the brain to induce a number of brain-controlled sickness symptoms which accompany peripheral infection or inflammation.
Afferent neural signals and fever: a role for cutaneous sensory nerves?
Accepting that afferents from the vagus nerve serve to transport signals from the activated immune system from the peritoneum to the brain, the question then arises if afferents from cutaneous nerves may represent a neural route for immune-to-brain communication. The investigation of this question requires an experimental model which compromises local cytokine induction within a localized subcutaneous compartment and a developing febrile response or modifications of other brain-controlled functions. Such a model has been introduced into experimental fever research over the last few years. In rats, injection of LPS into a subcutaneous air pouch results in fever and a local formation of cytokines (34,35). Of the proinflammatory cytokines, which can be detected in the air pouch lavage in response to administration of LPS, only small amounts of IL-6 enter the systemic circulation from the site of inflammation (34). Recently, the subcutaneous air pouch model was evaluated in guinea pigs (36,37). Essentially the same results were obtained as those obtained with rats (34,35). In one of our studies on guinea pigs the air pouch was replaced with a subcutaneously placed Teflon chamber equipped with a catheter (37). The cylindric chambers, opened at both ends, were implanted into preformed subcutaneous cavities. The open sides of the chambers were in close contact with the skin tissue (for details, see 37). This approach allowed administration of drugs into the chamber through the catheter and repeated collections of lavage fluid from the chamber. An intra-arterial catheter allowed the collection of blood samples. Using this model, we analyzed the febrile responses to injections of high (100 µg/kg) and low (10 µg/kg) doses of LPS into the chamber. A local anesthetic, ropivacaine, was also administered into the chamber. Levels of proinflammatory cytokines in the systemic circulation and in lavage fluid collected from the chamber were monitored.
The magnitude of fever in different groups is represented in the columns of Figure 3 as the integrated areas between thermal responses of febrile and normothermic animals. The mean body temperature measured at 15-min intervals over the 2 h before injection of LPS or control solutions was calculated as the baseline temperature (Figure 1). The mean magnitude of the fever which develops in response to injection of 100 µg/kg LPS into the subcutaneous Teflon chamber was defined as 100% fever response. Co-administration of 10 mg/kg of the local anesthetic ropivacaine into the chamber apparently has no influence on the febrile response to this dose of LPS. A reduction of the LPS dose to 10 µg/kg caused a smaller fever when injected into the subcutaneous chamber. Interestingly, the febrile response to the low dose of LPS was markedly reduced by co-injection of ropivacaine (10 mg/kg) into the chamber. A systemic action of the local anesthetic may be ruled out since, when injected subcutaneously on the contralateral side of the animal (in Figure 3) in relation to the chamber at the same dose, it failed to alter the fever induced by the low dose of LPS. This result suggests that afferent neural signals which are blocked by the local anesthetic in the chamber may participate in the transmission of febrile signals from a localized site of peripheral inflammation, in this case the subcutaneous chamber, to parts of the brain which control the thermoregulatory set point and are involved in fever induction. Support for this hypothesis is given by other studies in which an impairment of afferent C fibers by capsaicin desensitization significantly attenuated pyrogen-induced fever and HPA axis activation (38,39).
Assuming that localized peripheral inflammation alters the activity of cutaneous afferent fibers, further questions arise: why was not the febrile response to the high dose of LPS altered by treatment with ropivacaine? Which kind of signal was responsible for the part of the febrile response to the low dose of LPS which was not blocked by the local anesthetic? Regarding the first question, it is possible that LPS when injected at high doses may leave the compartment (e.g., via circulation) and provoke systemic or local responses in other tissues. It must be remembered that in vagotomized rats the febrile effect of intraperitoneal LPS, mainly at high doses, is not always blocked (29) and that the pyrogen may induce fever when injected in other compartments of vagotomized animals. Using the Limulus amebocyte lysate assay we recently obtained evidence that, with the exception of few animals treated with a high LPS dose, LPS does not appear in the circulation of guinea pigs after its injection into the subcutaneous chamber. Excluding the possibility of LPS reaching the systemic circulation, other interpretations may be examined. A possible answer to both questions raised above is that neural signals can be activated by inflammatory stimuli but provide a moderate portion of the information which is transmitted to the brain to induce fever. The other portion of the febrile signal, probably provided by humoral signals, would totally override the neuronal component in the case of high LPS doses. The humoral component, in the case of a high dose of LPS, may be depicted as a spillover of endogenous mediators into the systemic circulation. The search for humoral mediators involved in fever induction in the subcutaneous chamber or the air pouch focusses on the cytokine IL-6. Measurements (19,37) confirming previous observations (34) showed a moderate increase of IL-6, but not of IL-1 or TNF in the systemic circulation after LPS injection into the chamber or air pouch. Recently, the authors of the latter study (34) provided convincing experimental evidence that the rise of circulating IL-6 in response to LPS injection in the subcutaneous air pouch in rats is due to a spillover of this cytokine from the local site of inflammation into the systemic circulation and that the increase of IL-6 in the bloodstream, albeit moderate, is involved in the manifestation of fever (40). However, it should be noted that a high LPS dose (100 µg/kg) was used in the rat air pouch (19,34). Fever in response to the same dose of LPS in the subcutaneous chamber of the guinea pig could not be antagonized by treatment with the local anesthetic ropivacaine (Figure 3). Thus, our findings support the view that fever in response to the high dose of LPS (100 µg/kg) is, indeed, mostly mediated by humoral signal transmission. IL-6 which enters the circulation from the local site of inflammation is a likely candidate for a humoral signal molecule for fever induction (40). The circulating cytokine could stimulate vascular sensory fibers throughout the circulatory compartment or directly stimulate the brain as discussed in the next chapter. The attenuation of fever induced by the lower dose of LPS (10 µg/kg) by administration of ropivacaine at the site of inflammation at present can be interpreted as modest evidence for the participation of a neural pathway in fever induction from a site of locally induced inflammation within the subcutaneous chamber to the thermoregulatory centers of the brain.
Fever: what happens within the brain?
As illustrated above there is evidence for both humoral and neural signaling from the immune system to the brain in the presence of a peripheral inflammatory stimulus. The following topics are addressed in this chapter:
i) How do humoral or neural signals access those parts of the brain which are involved in the febrile shift of the thermoregulatory set point?
ii) Are endogenous brain-derived signal molecules the final mediators of fever and which molecules are the most likely candidates for this function?
Entry of humoral or neural signals into the brain
Accepting that circulating cytokines are important humoral mediators for the induction or at least maintenance of fever, the question arises of how these large hydrophilic peptides pass the relatively impermeable blood-brain barrier to stimulate relevant thermoregulatory brain structures. On the one hand, there is experimental evidence that peripheral cytokines can pass the blood-brain barrier by active and saturable transport systems which are specific for individual cytokines (41). An alternative humoral pathway is the access of cytokines through areas of the brain which lack a tight blood-brain barrier. These areas are the so-termed circumventricular organs. A role for the circumventricular organs in fever has especially been ascribed to the organum vasculosum laminae terminalis and the subfornical organ (3,4,42). At these sites circulating cytokines might enter the perivascular space and interact with receptors located at terminals of glial cells. Also, receptors on the surface of endothelial cells in brain vasculature are potential targets for circulating cytokines or even LPS (21). Glial cells and endothelial cells might then produce and release secondary mediators (see below) which are released into the brain and can thus obtain access to thermoregulatory structures within the preoptic area and hypothalamic nuclei which are involved in body temperature regulation.
The proposed pathway of how sensory nerves, namely afferents of the vagus nerve, transmit signals to the relevant hypothalamic areas has been recently described (3,20) and can be summarized as follows. The appearance of LPS in the bloodstream leads to a rapid complement activation within minutes. Components of the complement cascade stimulate Kupffer cells of the liver to produce further endogenous mediators, most likely cytokines, which are able to activate afferent fibers of the hepatic branch of the vagus nerve (3). The signals of hepatic vagal afferents are transported to the central projection areas of the vagus nerve within the nucleus of the solitary tract. The pyrogenic message is then passed to the noradrenergic A1/A2 cell groups which are located in this brainstem area and project directly to preoptic and hypothalamic areas via the ventral noradrenergic bundle (for further details of this hypothesis, see 3,20).
Final fever-inducing mediators in the thermoregulatory centers of the brain
A large number of neurons located in the rostral hypothalamus are thermosensitive. Several endogenous inflammatory mediators cause changes in the activity of these neurons which are consistent with the development of fever, i.e., an inhibition of heat-sensitive neurons or a stimulation of the few cold-sensitive neurons which exist within the preoptic/hypothalamic areas (43). These findings were obtained in ex vivo protocols by registering neuronal firing rates in rat brain slices at changing temperatures and in the presence of superfusion with cytokines. Considering that such neuronal mechanisms account for the febrile response to peripheral injection of LPS, it may be postulated that thermosensitive neurons of the brain are exposed to one or more of these endogenous mediators at the onset of the febrile response. Therefore, intensive research has been focussed on the question as to which putative fever-promoting substances are produced and released within the brain in response to peripheral administration of LPS. While peripheral injections of septic doses of LPS induce global expression of proinflammatory cytokines in the brain, subseptic - but fever-inducing - LPS doses seem to induce expression of IL-1ß and TNF-a only in the choroid plexus, the meninges and the circumventricular organs which are implicated in fever (44,45). The organum vasculosum laminae terminalis at the anterior wall of the third ventricle has been identified as a site of production of IL-1ß and TNF-a in response to peripheral injection of a moderate dose of LPS (44,45). The organum vasculosum laminae terminalis is located close to the surrounding preoptic area so that a diffusion of pyrogenic mediators to the thermosensitive structures is apparently possible. Alternatively, the endothelium of the cerebral vasculature may be the site where circulating cytokines or LPS could interact with receptors and stimulate the endothelial cells to release additional cytokines or other secondary mediators abluminally into the brain. For example, IL-6 (5-7), IL-8 (16) and MIP-1 (46) are produced and released within the brain in response to peripheral LPS and a central pyrogenic activity of these cytokines has been demonstrated (2,7,13,16,19,47). Especially, the appearance and biological action of IL-6 within the brain seem to be a critical component of fever generation as convincingly demonstrated in a study using IL-6-deficient ("IL-6 knockout") mice (48). In these mice neither peripheral injections of LPS and IL-1ß nor central administration of IL-1ß cause fever while intracerebroventricular injection of IL-6 evokes a pronounced febrile response. The results of this study indicate that central IL-6 is a crucial component of fever which acts downstream from peripheral and central IL-1ß (48).
Not only cytokines but also other putative endogenous mediators which are involved in fever are produced in the brain in response to peripheral injection of LPS. Bacterial LPS and proinflammatory cytokines induce or up-regulate a number of enzymes in the brain which catalyze the formation of small signal molecules such as nitric oxide (NO), carbon monoxide (CO) and prostaglandin E2 (PGE2). There is experimental evidence that these substances modulate neuronal activity and are thereby able to alter thermoregulatory characteristics in a direction which is consistent with the generation of fever. NO as well as CO are produced by constitutive and inducible forms of specific enzymes, NO synthases and heme oxygenases, and both act on the cellular level via activation of soluble guanylate cyclase leading to increased cGMP levels. In some experimental models NO and CO have been shown to promote the development of fever (49,50). However, the described effects of NO on thermoregulation and fever are not consistent in all investigated species of experimental animals (49). In contrast to the comparatively novel central nervous messenger molecules NO and CO, PGE2 is traditionally regarded as a centrally acting mediator of fever. The formation of PGE2 depends on the activity of cyclooxygenase (COX) which exists in two isoforms, the constitutively expressed COX-1 and the inducible form COX-2. The induction of COX-2, measured by mRNA or protein expression, in response to peripheral injection of a fever-inducing dose of LPS can be demonstrated in brain endothelial cells, perivascular microglia and meningeal macrophages (51,52). The production of PGE2 via induction of COX-2 or via constitutive COX-1 may be an important step in the manifestation of the febrile response to LPS. Such a view is supported by the following findings. Central injection of prostaglandins evokes fever (2-4). The levels of PGE2 in the cerebrospinal fluid from the third ventricle (53) and in microdialysis samples from the preoptic area/anterior hypothalamus (3) rise more or less in parallel to the LPS-induced changes of body temperature. Drugs which block prostaglandin synthesis also effectively inhibit the febrile response (2-4). Finally, knockout mice which are lacking one of the four PGE receptors, the EP-3 receptor (54), show impaired fever in response to peripheral injection of LPS. Because of the strong evidence for an important role of central PGE2 in fever, the following chain of events has been frequently suggested: LPS appearing in the blood induces circulating cytokines. Then, circulating cytokines and LPS induce another source of cytokines as well as COX-2 within the brain. Centrally produced cytokines are further triggers for COX-2 induction and thereby for prolonged formation of PGE2 within the preoptic area and the hypothalamus. However, it should be noted that there are some exceptions in which fever develops independently from prostaglandins. Thus, prostaglandins are obviously not involved in MIP-1- (15), IL-8- (16), PFPF- (19), substance P- (55) or endothelin-1 (56)-induced fever.
There is also evidence for the recruitment of final brain-derived pyrogenic mediators of fever which are produced and released in response to stimulation of afferent fibers of the vagus nerve. Thus, subdiaphragmatic vagotomy blocks the LPS-induced induction of IL-1ß mRNA in the brain (27). In turn, electrical stimulation of the vagus nerve induces IL-1ß expression within the hypothalamus (57). IL-1ß is not the only molecule the LPS-induced expression of which is blocked in the hypothalamus of vagotomized animals. It was recently shown that intraperitoneally injected LPS (50 µg/kg) induced fever and kinin B1 receptor mRNA expression in the rat hypothalamus (58). Subdiaphragmatic vagotomy blocked both fever and B1, but not B2 kinin receptor or ß-actin mRNA expression, suggesting a participation of central hypothalamic kinins in the pyrogenic response to LPS (Figure 4, Ref. 58).
The upper panel of Figure 4 shows that intraperitoneal injection of 50 µg/kg LPS induced the expression of mRNA for B1 kinin receptors in the rat hypothalamus. mRNA was detected by RT-PCR in the rat hypothalamus 6 h after intraperitoneal injection of LPS. The hypothalami were dissected and stored at -80oC. cDNA was obtained by reverse transcriptase reaction with a poly-T primer from purified hypothalamic mRNA. Kinin B1 and B2 receptor cDNA and ß-actin cDNA were amplified with specific primers and the reaction products were visualized in ethidium bromide-stained gels. mRNA for B2 receptors (and ß-actin), which is constitutively expressed, was present in sham and vagotomized animals whether they received saline or LPS. However, the kinin B1 receptor mRNA after LPS was present in sham-operated but not in vagotomized animals. These results suggest that ascending vagal inputs mediate kinin B1 receptor mRNA expression in the hypothalamus after peripheral injection of LPS and that newly expressed hypothalamic B1 receptors may participate in the central mechanism for fever development (58). These and other findings are important evidence for an ascending vagal pathway in the mediation of fever and the expression of receptors and/or mediators in the hypothalamus.
Once more prostaglandins seem to be critical final mediators in the vagally activated fever pathway since bradykinin-induced fever is blocked by indomethacin, a COX-1 and COX-2 inhibitor (Figure 4). So, one can suppose that local mediators, cytokines and possibly PFPF, released in the peritoneum stimulate the vagus nerve, which in turn would promote the expression of B1 receptors. By acting on its constitutive B2 and newly expressed B1 receptors, bradykinin could trigger prostaglandin synthesis, which would act as one of the final mediators of fever. Moreover, as already mentioned above (3), the inflammatory stimulation of afferent parts of the vagus nerve to their central projection areas seems to result in an activation of the ventral noradrenergic bundle which results in an increased release of noradrenaline within the preoptic/hypothalamic area. Indeed, an increased intrahypothalamic release of noradrenaline is measurable in response to peripheral administration of LPS (59). Based on the observation that microdialysis of noradrenaline into the hypothalamus augments the local intrahypothalamic production of PGE2 and that the LPS-induced rise of hypothalamic PGE2 is blocked by subdiaphragmatic vagotomy (3), the proposition that hypothalamic PGE2 is also an important endogenous mediator in the vagally mediated fever pathway is reinforced.
The ability of some cytokines (except TNF-a and IL-1a) and PGF2a to induce fever seems to be related to the generation of corticotropin-releasing hormone (CRH) within the brain since the increase in body temperature induced by these substances is abolished by the CRH antagonist, a-helical CRH9-41 (19,60). Moreover, the increase in noradrenaline turnover in the rat hypothalamus caused by intraperitoneally injected IL-1ß is blocked by a polyclonal anti-CRH antibody (61). These findings show that IL-1ß can activate CRH neurons that connect with the noradrenergic neurons projecting to the hypothalamus (61). Interestingly, subdiaphragmatic vagotomy suppressed c-Fos expression in CRH neurons and the plasma ACTH and corticosterone response after intraperitoneal injection of a low, but not a high, dose of LPS, showing that vagal afferents are involved in the activation of the HPA axis (23), a parallel characteristic signal of fever in the acute phase response.
Final conclusions
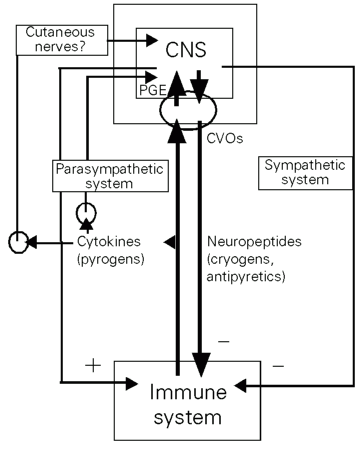
The current evidence for distinct pathways of fever induction in response to peripheral inflammatory stimulation was summarized. Induction and regulation of fever result from complex bilateral communication between the immune system and the peripheral and central nervous system. Figure 5 schematically shows the communication pathways for which experimental evidence was presented (see Figures 1-4).
Immune cells which are activated by exogenous pyrogens, i.e., LPS, release a number of soluble mediators including proinflammatory cytokines which coordinate the immune defense responses. Cytokines can stimulate afferent sensory nerves or when released into the bloodstream can activate the CNS via circumventricular organs through their fenestrated capillaries or directly stimulate brain vascular endothelial cells. The formation of peripheral cytokines in response to LPS as well as the humoral signal transfer into the CNS requires some time. The quick onset of LPS fever seems to precede the formation of sufficiently high amounts of circulating cytokines, at least in some experimental fever models (3). In addition, as shown in Figure 1, systemic anti-cytokine strategies predominantly attenuate the second phase of LPS fever. Thus, it seems that the peripheral release of cytokines rather contributes to the maintenance of fever than to the induction of the early phase of fever. These observations and the fact that fever can sometimes occur in the absence of circulating cytokines support the view that also a localized formation of cytokines or other rapidly inducible pyrogenic factors can activate alternative fever pathways. Postulated neuronal mechanisms of pyrogen signaling to the brain could explain the quick onset of fever. Two examples of fever pathways in which neuronal routes are involved are also indicated in Figure 5. For the stimulation of afferent parts of the parasympathetic system (vagus nerve) by inflammatory signals a lot of experimental evidence has accumulated. The results shown in Figure 3 provide a first modest amount of evidence that also cutaneous afferent nerves might participate in the fever response which develops after subcutaneously localized tissue inflammation. These results can be interpreted as a participation of afferent nerves in a "sensory host-monitoring system" that rapidly informs the brain about a challenge to the immune system somewhere at the periphery of the body.
Acknowledgments
J. Roth wishes to express his deep gratitude for the kind invitation to the meeting in Brazil and for the support by FAPESP. The authors gratefully acknowledge the helpful comments and the linguistic revision of the manuscript by Dr. Charles Lindsey (Departamento de Biofísica, Escola Paulista de Medicina, UNIFESP, São Paulo, Brazil).
Address for correspondence: J. Roth, Department of Veterinary Physiology, School of Veterinary Medicine, University of Giessen, Frankfurter Strasse 100, 35392 Giessen, Germany. Fax: +49-641-993-8159. E-mail: joachim.roth@vetmed.uni-giessen.de
Parts of this paper have been presented at the XIV Annual Meeting of the Federação de Sociedades de Biologia Experimental, Caxambu, MG, Brazil, August 25-28, 1999, and at the Satellite Symposium to the 1999 Meeting of the Society for Neuroscience, "Neuropeptides at the Millennium", Miami, FL, USA, October 21-23, 1999. Research supported by FAPESP (No. 1997/9837-6). Received November 21, 2000. Accepted January 19, 2001.
- 1. Dantzer R, Bluthe RM, Gheusi G, Cremona S, Laye S, Parnet P & Kelley K (1998). Molecular basis of sickness behavior. Annals of the New York Academy of Sciences, 856: 132-138.
- 2. Kluger MJ (1991). Fever: role of pyrogens and cryogens. Physiological Reviews, 71: 93-127.
- 3. Blatteis CM & Sehic E (1997). Fever: how may circulating cytokines signal the brain? News in Physiological Sciences, 12: 1-9.
- 4. Zeisberger E (1999). From humoral fever to neuroimmunological control of fever. Journal of Thermal Biology, 24: 287-326.
- 5. Jansky L, Vybiral S, Pospisilowa D, Roth J, Dornand J, Zeisberger E & Kaminkowa J (1995). Production of systemic and hypothalamic cytokines during the early phase of the endotoxin fever. Neuroendocrinology, 62: 55-61.
- 6. Roth J, Conn CA, Kluger MJ & Zeisberger E (1993). Kinetics of systemic and intrahypothalamic IL-6 and tumor necrosis factor during endotoxin fever in the guinea pig. American Journal of Physiology, 265: R653-R658.
- 7. LeMay LG, Vander AJ & Kluger MJ (1990). Role of interleukin-6 in fever in the rat. American Journal of Physiology, 258: R798-R803.
- 8. Van Zee KJ, DeForge LE, Fischer E, Marano MA, Kenney JS, Remick DG, Lowry SF & Moldawer LL (1991). IL-8 in septic shock, endotoxemia, and after IL-1 administration. Journal of Immunology, 146: 3478-3482.
- 9. Ziegler SF, Tough TW, Franklin TL, Armitage RJ & Alderson M (1991). Induction of macrophage inflammatory protein-1 gene expression in human monocytes by lipopolysaccharide and IL-7. Journal of Immunology, 147: 2234-2239.
- 10. Dinarello CA, Bernheim HA, Duff GS, Le HV, Nagabhushan TL, Hamilton NC & Coceani F (1984). Mechanisms of fever induced by recombinant human interferon. Journal of Clinical Investigation, 74: 906-913.
- 11. Anforth HR, Bluthe RM, Bristow A, Hopkins SJ, Lenczowski MJP, Luheshi GN, Lundkvist J, Michaud B, Mistry Y, Van Dam AM, Zhen C, Dantzer R, Poole S, Rothwell NJ, Tilders FJH & Wollmann EE (1998). Biological activity and brain actions of recombinant rat interleukin-1a and interleukin-1ß. European Cytokine Network, 9: 279-288.
- 12. Goldbach JM, Roth J, Störr B & Zeisberger E (1996). Repeated infusions of TNF-a cause attenuation of the thermal response and influence LPS fever in guinea pigs. American Journal of Physiology, 270: R749-R754.
- 13. Blatteis CM, Quan N, Xin L & Ungar AL (1990). Neuromodulation of acute phase responses to interleukin 6 in guinea pigs. Brain Research Bulletin, 25: 895-901.
- 14. Zampronio AR, Silva CAA, Cunha FQ, Ferreira SH, Pela IR & Souza GEP (1995). Indomethacin blocks the febrile response induced by interleukin-8 in rabbits. American Journal of Physiology, 269: R1469-R1474.
- 15. Davatelis G, Wolpe SD, Sherry B, Dayer JM, Chicheportiche R & Cerami A (1989). Macrophage inflammatory protein-1: a prostaglandin-independent endogenous pyrogen. Science, 243: 1066-1068.
- 16. Zampronio AR, Souza GEP, Silva CAA, Cunha FQ & Ferreira SH (1994). Interleukin-8 induces fever by a prostaglandin-independent mechanism. American Journal of Physiology, 266: R1670-R1674.
- 17. Roth J, Martin D, Störr B & Zeisberger E (1998). Neutralization of bacterial pyrogen-induced circulating tumour necrosis factor by its type 1 soluble receptor in guinea pigs: effects on fever and endogenous formation of interleukin-6. Journal of Physiology, 509: 267-275.
- 18. Givalois L, Dornand J, Mekaouche M, Solier MD, Bristow AF, Ixart G, Siaud P, Assenmacher I & Barbanel G (1994). Temporal cascade of plasma level surges in ACTH, corticosterone, and cytokines in endotoxin-challenged rats. American Journal of Physiology, 267: R164-R170.
- 19. Zampronio AR, Melo MCC, Silva CAA, Pela IR, Hopkins SJ & Souza GEP (1994). A pre-formed pyrogenic factor released by lipopolysaccharide stimulated macrophages. Mediators of Inflammation, 3: 365-373.
- 20. Li S, Sehic E, Wang Y, Ungar AL & Blatteis CM (1999). Relation between complement and the febrile response of guinea pigs to systemic endotoxin. American Journal of Physiology, 277: R1635-R1645.
- 21. Dinarello CA, Gatti S & Bartfai T (1999). Fever: Links with an ancient receptor. Current Biology, 9: R147-R150.
- 22. Bluthe RM, Michaud B, Kelley KW & Dantzer R (1996). Vagotomy blocks behavioral effects of interleukin-1 injected via the intraperitoneal route but not via other systemic routes. NeuroReport, 7: 2823-2827.
- 23. Gaykema RPA, Dijkstra I & Tilders FJH (1995). Subdiaphragmatic vagotomy suppresses endotoxin-induced activation of hypothalamic corticotropin-releasing hormone neurons and ACTH secretion. Endocrinology, 136: 4717-4720.
- 24. Watkins LR, Wiertelak EP, Goehler LE, Mooney-Heiberger K, Martinez J, Furness L, Smith KP & Maier SF (1994). Neurocircuitry of illness-induced hyperalgesia. Brain Research, 639: 283-299.
- 25. Hansen MK & Krueger JM (1997). Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1ß. American Journal of Physiology, 273: R1246-R1253.
- 26. Wan W, Wetmore L, Sorensen CM, Greenberg AH & Nance DM (1994). Neural and biochemical mediators of endotoxin and stress-induced c-fos expression in the rat brain. Brain Research Bulletin, 34: 7-14.
- 27. Laye S, Bluthe RM, Kent S, Combe C, Medina C, Parnet P, Kelley K & Dantzer R (1995). Subdiaphragmatic vagotomy blocks induction of IL-1ß mRNA in mice brain in response to peripheral LPS. American Journal of Physiology, 268: R1327-R1331.
- 28. Goldbach JM, Roth J & Zeisberger E (1997). Fever suppression by subdiaphragmatic vagotomy in guinea pigs depends on the route of pyrogen administration. American Journal of Physiology, 272: R675-R681.
- 29. Romanovsky AA, Simons CT, Szekely M & Kulchitsky VA (1997). The vagus nerve in the thermoregulatory response to systemic inflammation. American Journal of Physiology, 273: R407-R413.
- 30. Luheshi GN, Bluthe RM, Rushforth D, Mulcahy N, Konsman JP, Goldbach M & Dantzer R (2000). Vagotomy attenuates the behavioral but not the pyrogenic effects of interleukin-1 in rats. Autonomic Neuroscience, 85: 127-132.
- 31. Niijima A (1996). The afferent discharges from sensors for interleukin 1ß in the hepatoportal system in the anesthetized rat. Journal of the Autonomic Nervous System, 61: 287-291.
- 32. Goehler LE, Relton JK, Dripps D, Kiechle R, Tartaglia N, Maier SF & Watkins LR (1997). Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Research Bulletin, 43: 357-364.
- 33. Gaykema RPA, Goehler LE, Tilders FJH, Bol JGJM, McGorry M, Fleshner M, Maier SF & Watkins LR (1998). Bacterial endotoxin induces Fos immunoreactivity in primary afferent neurons of the vagus nerve. Neuroimmunomodulation, 5: 234-240.
- 34. Miller AJ, Luheshi GN, Rothwell NJ & Hopkins SJ (1997). Local cytokine induction by LPS in the rat air pouch and its relationship to the febrile response. American Journal of Physiology, 272: R857-R861.
- 35. Miller AJ, Hopkins SJ & Luheshi GN (1997). Sites of action of IL-1 in the development of fever and cytokine responses to tissue inflammation in the rat. British Journal of Pharmacology, 120: 1274-1279.
- 36. Roth J, Störr B, Martin D, Voigt K & Zeisberger E (2000). The role of local induction of tumor necrosis factor by LPS within a subcutaneous air pouch in the development of a febrile response in guinea pigs. Neuroimmunomodulation, 7: 169-176.
- 37. Ross G, Roth J, Störr B, Voigt K & Zeisberger E (2000). Afferent nerves are involved in the febrile response to injection of LPS into artificial subcutaneous chambers in guinea pigs. Physiology and Behavior, 71: 305-313.
- 38. Szekely M, Balasko M & Romanovsky AA (1997). Peripheral neural inputs: their role in fever development. Annals of the New York Academy of Sciences, 813: 427-434.
- 39. Watanabe T, Morimoto A, Tan N, Makisumi T, Shimada SG, Nakamori T & Murakami N (1994). ACTH response induced in capsaicin-desensitized rats by intravenous injection of interleukin-1 or prostaglandin E. Journal of Physiology, 475: 139-145.
- 40. Cartmell T, Poole S, Turnbull AV, Rothwell NJ & Luheshi GN (2000). Circulating interleukin-6 mediates the febrile response to localised inflammation in rats. Journal of Physiology, 526: 653-661.
- 41. Banks WA & Kastin A (1991). Blood to brain transport of interleukin links the immune and central nervous systems. Life Sciences, 48: L117-L121.
- 42. Cartmell T, Luheshi GN & Rothwell NJ (1999). Brain sites of action of endogenous interleukin-1 in the febrile response to localized inflammation in the rat. Journal of Physiology, 518: 585-594.
- 43. Shibata M & Blatteis CM (1991). Differential effects of cytokines on thermosensitive neurons in guinea pig preoptic area slices. American Journal of Physiology, 261: R1096-R1103.
- 44. Nakamori T, Morimoto A, Yamaguchi K, Watanabe T, Long NC & Murakami N (1993). Organum vasculosum laminae terminalis is a brain site to produce interleukin-1ß during fever. Brain Research, 618: 155-159.
- 45. Quan N, Stern EL, Whiteside MB & Herkenham M (1999). Induction of pro-inflammatory cytokine mRNA in the brain after peripheral injection of subseptic doses of lipopolysaccharide in the rat. Journal of Neuroimmunology, 93: 72-80.
- 46. Minano FJ, Fernandez-Alonso A, Benamar K, Myers RD, Sancibrian M, Ruiz RM & Armengol JA (1996). Macrophage inflammatory protein-1 beta (MIP-1 beta) produced endogenously in brain during E. coli fever in rats. European Journal of Neuroscience, 8: 424-428.
- 47. Minano FJ, Fernandez-Alonso A, Myers RD & Sancibrian M (1996). Hypothalamic interaction between macrophage inflammatory protein-1 alpha (MIP-1 alpha) and MIP-1 beta in rats: a new level for fever control? Journal of Physiology, 491: 209-217.
- 48. Chai Z, Gatti S, Toniatti C, Poli V & Bartfai T (1999). Interleukin (IL)-6 gene expression in the central nervous system is necessary for fever response to lipopolysaccharide or IL-1ß: a study on IL-6-deficient mice. Journal of Experimental Medicine, 183: 311-316.
- 49. Gerstberger R (1999). Nitric oxide and body temperature control. News in Physiological Sciences, 14: 30-36.
- 50. Steiner AA, Colombari E & Branco LGS (1999). Carbon monoxide as a novel mediator of the febrile response in the central nervous system. American Journal of Physiology, 277: R499-R507.
- 51. Cao C, Matsumura K, Yamagata K & Watanabe Y (1995). Induction by lipopolysaccharide of cyclooxygenase-2 mRNA in rat brain; its possible role in the febrile response. Brain Research, 697: 187-196.
- 52. Elmquist JK, Breder CD, Sherin JE, Scammel TE, Hickey WF, Dewitt D & Saper CB (1997). Intravenous lipopolysaccharide induces cyclooxygenase 2-like immunoreactivity in rat brain perivascular microglia and meningeal macrophages. Journal of Comparative Neurology, 381: 119-129.
- 53. Coceani F, Lees J & Bishai I (1988). Further evidence implicating prostaglandin E2 in the genesis of pyrogen fever. American Journal of Physiology, 254: R463-R469.
- 54. Ushikubi F, Segi E, Sugimoto Y, Murata T, Matsuoka T, Kobayashi T, Hizaki H, Tuboi K, Katsuyama M, Ichikawa A, Tanaka T, Yoshida N & Narumiya S (1998). Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature, 395: 281-284.
- 55. Szelenyi Z, Szekely M & Balasko M (1997). Role of substance P (SP) in the mediation of endotoxin (LPS) fever in rats. Annals of the New York Academy of Sciences, 813: 316-323.
- 56. Fabricio ASC, Silva CAA, Rae GA, D'Orleans-Juste P & Souza GEP (1998). Essential role for endothelin ETB receptors in fever induced by LPS (E. coli) in rats. British Journal of Pharmacology, 125: 542-548.
- 57. Hosoi T, Okuma Y & Nomura Y (2000). Electrical stimulation of afferent vagus nerve induces IL-1ß expression in the brain and activates HPA-axis. American Journal of Physiology, 279: R141-R147.
- 58. Souza GEP, Piza AM, Peroza EA, Couture R & Lindsey CJ (2000). Vagotomy blocks LPS-induced fever and bradykinin B1 receptor gene expression in rat hypothalamus. Brain Research, 848: 39 (Abstract).
- 59. Linthorst ACE, Flachskamm C, Holsboer F & Reul JMHM (1995). Intraperitoneal administration of bacterial endotoxin enhances noradrenergic neurotransmission in the rat preoptic area: relationship with body temperature and hypothalamic-pituitary-adrenocortical axis activity. European Journal of Neuroscience, 7: 2418-2430.
- 60. Strijbos PJ, Hardwick AJ, Relton JK, Carey F & Rothwell NJ (1992). Inhibition of central actions of cytokines on fever and thermogenesis by lipocortin-1 involves CRF. American Journal of Physiology, 263: E632-E636.
- 61. Terao A, Oikawa M & Saito M (1993). Cytokine-induced change in hypothalamic norepinephrine turnover: involvement of corticotropin releasing hormone and prostaglandins. Brain Research, 622: 257-261.
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
16 Mar 2001 -
Date of issue
Mar 2001
History
-
Accepted
19 Jan 2001 -
Received
21 Nov 2000