Abstract
OBJECTIVE: Prader-Willi Syndrome is a common etiology of syndromic obesity that is typically caused by either a paternal microdeletion of a region in chromosome 15 (microdeletions) or a maternal uniparental disomy of this chromosome. The purpose of this study was to describe the most significant clinical features of 35 Brazilian patients with molecularly confirmed Prader-Willi syndrome and to determine the effects of growth hormone treatment on clinical outcomes. METHODS: A retrospective study was performed based on the medical records of a cohort of 35 patients diagnosed with Prader-Willi syndrome. The main clinical characteristics were compared between the group of patients presenting with microdeletions and the group presenting with maternal uniparental disomy of chromosome 15. Curves for height/length, weight and body mass index were constructed and compared between Prader-Willi syndrome patients treated with and without growth hormone to determine how growth hormone treatment affected body composition. The curves for these patient groups were also compared with curves for the normal population. RESULTS: No significant differences were identified between patients with microdeletions and patients with maternal uniparental disomy for any of the clinical parameters measured. Growth hormone treatment considerably improved the control of weight gain and body mass index for female patients but had no effect on either parameter in male patients. Growth hormone treatment did not affect height/length in either gender. CONCLUSION: The prevalence rates of several clinical features in this study are in agreement with the rates reported in the literature. Additionally, we found modest benefits of growth hormone treatment but failed to demonstrate differences between patients with microdeletions and those with maternal uniparental disomy. The control of weight gain in patients with Prader-Willi syndrome is complex and does not depend exclusively on growth hormone treatment.
Obesity; Body composition; Genetics; Prader-Willi syndrome
CLINICAL SCIENCE
A clinical follow-up of 35 Brazilian patients with Prader-Willi Syndrome
Caio Robledo D'Angioli Costa QuaioI; Tatiana Ferreira de AlmeidaI; Lilian Maria José AlbanoI; Israel GomyI; Debora Romeo BertolaI; Monica Castro VarelaII; Celia P. KoiffmannII; Chong Ae KimI
IInstituto da Criança da Faculdade de Medicina da Universidade de São Paulo, Genetics Unit, São Paulo/SP, Brazil
IIDepartment of Genetics and Evolutionary Biology, Instituto de Biosciências da Universidade de São Paulo, São Paulo/SP, Brazil
ABSTRACT
OBJECTIVE: Prader-Willi Syndrome is a common etiology of syndromic obesity that is typically caused by either a paternal microdeletion of a region in chromosome 15 (microdeletions) or a maternal uniparental disomy of this chromosome. The purpose of this study was to describe the most significant clinical features of 35 Brazilian patients with molecularly confirmed Prader-Willi syndrome and to determine the effects of growth hormone treatment on clinical outcomes.
METHODS: A retrospective study was performed based on the medical records of a cohort of 35 patients diagnosed with Prader-Willi syndrome. The main clinical characteristics were compared between the group of patients presenting with microdeletions and the group presenting with maternal uniparental disomy of chromosome 15. Curves for height/length, weight and body mass index were constructed and compared between Prader-Willi syndrome patients treated with and without growth hormone to determine how growth hormone treatment affected body composition. The curves for these patient groups were also compared with curves for the normal population.
RESULTS: No significant differences were identified between patients with microdeletions and patients with maternal uniparental disomy for any of the clinical parameters measured. Growth hormone treatment considerably improved the control of weight gain and body mass index for female patients but had no effect on either parameter in male patients. Growth hormone treatment did not affect height/length in either gender.
CONCLUSION: The prevalence rates of several clinical features in this study are in agreement with the rates reported in the literature. Additionally, we found modest benefits of growth hormone treatment but failed to demonstrate differences between patients with microdeletions and those with maternal uniparental disomy. The control of weight gain in patients with Prader-Willi syndrome is complex and does not depend exclusively on growth hormone treatment.
Keywords: Obesity; Body composition; Genetics; Prader-Willi syndrome.
INTRODUCTION
One of the most common causes of syndromic obesity is Prader-Willi syndrome (PWS), which has a peculiar evolution that is characterized by childhood-onset obesity, facial dysmorphisms, hypogonadism, short stature, intellectual handicap and an insatiable appetite, leading to significant clinical complications later in life. Interestingly, these symptoms are preceded by marked neonatal hypotonia, a poor suck reflex and a failure to thrive beginning at birth that typically improves within the first year of life (1-10).
The underlying genetic cause of this pleiotropic disorder is the lack of expression of paternal genes in the critical chromosome region 15q11-13. This lack of expression is due to a de novo paternal microdeletion of this region in 75% of cases (microdeletions), a maternal uniparental disomy (mUPD) of chromosome 15 in 20% of cases and either structural chromosomal aberrations or imprinting center defects in roughly 5% of cases (1-13).
The most important complications of PWS are related to the cardiovascular and respiratory involvement caused by obesity. These complications are directly responsible for the high incidence of death among children and adults with PWS, close to 3% per year (2-4). However, the literature lacks comprehensive information on the long-term survival of PWS patients.
There is no specific treatment for PWS. The clinical followup is based on constant surveillance and the treatment of common complications. In addition, growth hormone replacement therapy (GHt) is often used to improve final stature and to control weight gain (1,9,10).
The purpose of this study was to describe the most significant clinical features during a long-term follow-up of 35 Brazilian patients with molecularly confirmed PWS and to determine the effects of GHt on the clinical outcomes.
METHODS
A retrospective study was performed based on the medical records of a cohort of 35 patients (18 males and 17 females) diagnosed with PWS and followed in our service. Twenty-three patients had undergone regular follow-up visits in our service, six patients had incomplete clinical information, and six patients died from complications related to PWS.
All subjects included in this study had a typical DNA methylation pattern of the PWS critical region. The DNA was modified by bisulfite treatment, and SNURF-SNRPN exon 1 was amplified by PCR (14). The characteristic PWS pattern is defined by the presence of only the 313 bp maternal band. Three microsatellite markers within the critical region of 15q11-q13 (D15S11, D15S113 and GABRB3) and at least one marker outside this region (D15S984, D15S131, D15S117, D15S115 and CYP19) were studied in 24 patients and their parents to distinguish between deletions and maternal uniparental disomy; 16 patients had microdeletions (eleven males and five females), and eight had mUPD (two males and six females). All patients presented with normal peripheral blood karyotypes. In 11 cases (five males and six females), molecular tests to differentiate between mUPD and microdeletions were not performed.
The clinical features for which the data were not normally distributed were compared between the mUPD and microdeletion groups using Fisher's exact test. Variables with normal distributions, including weight, BMI and height, were compared with Student's t test.
To determine how GHt affected body composition, curves for height/length, weight and body mass index (BMI) were visually compared between PWS patients treated with and without GHt. These growth curves were also compared with the reference growth charts for normal populations from the World Health Organization and the National Center for Health Statistics (WHO/NCHS; referred to as the normal population curve).
The means and standard deviations (SDs) of all ages (from 0 to 19 y; calculated using the mean of each individual for each age) were adjusted using the distance-weighted least squares method to calculate the growth curves (Lam, 1983; McLain, 1974) for our patents and for the normal population. Each patient contributed between 1 and 22 measurements at different ages, ranging from birth to 23 y 5 mo. Graphical and statistical analyses were performed using STATISTICA 7.0 and Microsoft Excel.
RESULTS
The most relevant clinical features of our cohort of PWS patients and the comparisons between patients with microdeletions and mUPD are summarized in Table 1.
Some of the main clinical findings in our cohort were as follows: preterm birth (3%), birth by Cesarean section (90%), hypotonia (100%), poor suck reflex and feeding problems (92%), developmental delay (97%), cryptorchidism in males (43%) and hypogonadism (57%).
The birth weight ranged from 1,020 g to 3,650 g (mean: 2,717 g), and the birth length ranged from 34.5 cm to 54.0 cm (mean: 47.7 cm). The mean age at walking was 2.4 years (range: 1.2 y-4.5 y). Hyperphagia presented between 1 y and 5 y (mean: 2.8 y) of age, and obesity developed between 1 y and 6 y (mean: 2.6 y) of age.
GHt was started in 11 patients (eight male and three female patients) between 1.2 y and 11.5 y (mean: 6.5 y) of age, with the length of therapy varying from 0.5 y to 6.6 y (mean: 3.8 y).
No significant differences were found when comparing the clinical parameters between patients with microdeletions and those with mUPD.
Six patients died between 1.5 y and 19 y (mean: 11.6 y) of age. The cause of death was determined in four cases; three patients died due to cardiovascular/respiratory complications from obesity, and one died from endocarditis at 1.5 y of age. In the two other cases, no autopsy was performed, and the cause of death was not determined. Of the six deceased patients, three had microdeletions, one had mUPD, and in the remaining two, the molecular test to determine the genetic mechanism of deletion was not performed. Comparing the death rate between the mUPD and microdeletion groups showed no significant difference (p<0.05).
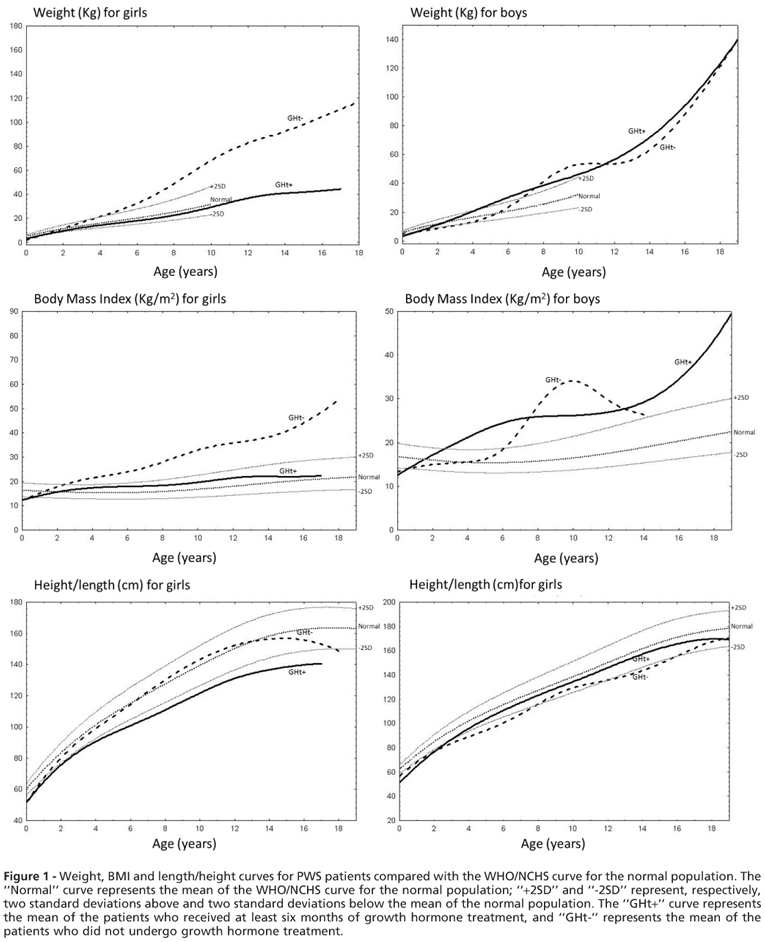
The weight, BMI and length/height curves for PWS patients were compared with the WHO/NCHS curves for the normal population (Figure 1).
Weight/BMI curves
Males: The use of GHt in males did not decrease weight gain. The mean weight for PWS patients treated with growth hormone exceeded +2 SDs of the mean weight of the normal population by age 4, and the mean weight of PWS patients not treated with growth hormone exceeded 2 SDs of the normal mean weight by the age of 7. Thereafter, both remained groups above +2 SDs compared with the WHO/NCHS curve for the normal population. When comparing the BMI curves of male patients with or without GHt, both groups started below -2 SDs and later exceeded +2 SDs of the WHO/NCHS curve for the normal population during infancy. This increase in BMI occurred earlier in patients previously treated with growth hormone, reflecting rapid weight gain and the inability of the length/height increase to counterbalance the weight gain.
Females: Female patients treated with growth hormone showed improvement in weight gain. Female patients treated with growth hormone had a weight curve that remained close to the mean of the normal population, whereas female patients not treated with growth hormone had a curve that exceeded +2 SDs by the age of four, with a continuing upward trend. When comparing BMI curves, a similar trend was observed. Female patients previously treated with growth hormone remained between the mean and +2 SDs of the normal population, whereas female patients not treated with growth hormone exceeded +2 SDs at the age of two and continued a rapid upward trend.
Length/Height
Males: When comparing the length/height curves for male patients, we observed that patients who received GHt started below -2 SDs in the WHO/NCHS curve of normal children and, by the age of six, were closer to the mean, whereas those without GHt fell below the -2 SD curve. Despite these early differences, when male patients with or without GHt reached eighteen years old, the curves crossed at a point close to -2 SDs.
Females: When analyzing the length/height curves for female patients, female patients without GHt started below 2 SDs and, by the age of six, approached the mean of the WHO/NCHS curve of the normal population and stayed at the same level until the age of 15, when a negative slope was noted. The GHt group always remained below -2 SDs.
DISCUSSION
We have presented the most relevant clinical features of a large case series of Prader-Willi syndrome patients in Brazil. Several findings in our retrospective analysis are consistent with the results published in the literature, such as the frequencies of microdeletion/mUPD, hypotonia, facial dysmorphisms, developmental milestone delays, feeding difficulties, hypogonadism and cryptorchidism (1,11,12).
Clinical findings
Hypotonia was present in the first year of life for all patients, leading to feeding problems in the great majority of patients. In this study, only six patients were diagnosed with PWS before 12 months of age. This low number of early diagnoses reflects the difficulty in correctly diagnosing PWS and the lack of proper diagnostic methods available to the public health system in Brazil. The late diagnoses may have skewed our results. Interestingly, we observed that hyperphagia onset and the time of PWS diagnosis were positively correlated. The presence of severe hypotonia or significant feeding problems should indicate the possibility of PWS and warrants genetic testing to confirm the diagnosis.
Obesity and hyperphagia began early in life. This observation suggests that the pathophysiology of obesity in PWS is complex and is not only related to high caloric intake but also to hypothalamic and hormonal dysregulation and low energy expenditure (5,6).
Our study showed a tendency for children with PWS to have birth lengths and weights below the -2 SD curve for the normal population. Children with PWS then exhibit an increase in weight at approximately two years of age, when they exceed the +2 SD curve. The height curve also showed an increase, but this increase was modest and not sufficient to pull the patients above the -2 SD curve for height. These results are concordant to those in the literature (1,13).
The mortality rate (17%) observed in our study was high for a young population. It is remarkable that all six patients died prematurely before adulthood (mean age: 11.6 y).
Microdeletions and mUPD
We did not find significant differences in any of the other clinical parameters measured between patients with microdeletions and mUPD. We believe that our sample, though considerable for a genetic disorder, was not large enough to reach statistical significance.
Growth hormone replacement therapy
In our study, GHt considerably improved the control of weight gain and the BMI for female patients but not for male patients. GHt did not improve height/length in either gender. The control of weight gain in patients with PWS is complex and does not depend exclusively on GHt; other factors, such as food-intake control, exercise and active participation from the family, were not addressed and may have overcome the benefits of GHt in male patients (1,8).
The benefits of GHt in individuals with PWS have been demonstrated in multiple studies. GHt improves linear growth velocity and ultimate height, body composition (i.e., increased lean body mass and decreased fat mass), muscle function and the level of activity (9,10). However, in the present study, there was a large heterogeneity among patients receiving GHt, and the variability in the length of treatment may have directly influenced the efficacy of GHt, especially for the male patients. Furthermore, there may have been a selection bias because fewer data were available for older patients.
The treatment of PWS patients involves identifying and managing symptoms. A multidisciplinary approach, including controlling food access, hormone replacement therapies, special education, and psychological follow-up, is crucial for the successful management of the disease (1).
We presented our observations from a clinical follow-up of a large cohort of Brazilian PWS patients to raise awareness in the medical community about PWS as an important cause of syndromic obesity and to characterize the most serious clinical outcomes in Brazil. We observed a high mortality rate in a young population. Moreover, GHt did not improve the control of the majority of the clinical parameters that were analyzed, demonstrating that the control of weight gain in patients with PWS is complex and does not depend exclusively on GHt.
ACKNOWLEDGMENTS
The publication was supported by FAPESP, Brazil (2012/50300-8).
AUTHOR CONTRIBUTIONS
Quaio CR, Almeida TF, Albano LM, Bertola DR and Kim CA designed the study and were responsible for the patient data collection and manuscript writing. Almeida TF was also responsible for statistical analysis. Varela MC and Koiffmann CP performed the molecular analysis. Gomy I revised the manuscript.
Received for publication on May 19, 2012
First review completed on June 6, 2012
Accepted for publication on June 8, 2012
No potential conflict of interest was reported.
E-mail: chong.kim@icr.usp.br
Tel.: 55 11 2661-8671
- 1. Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet. 2009;17(1):3-13, http://dx.doi.org/10.1038/ejhg.2008.165
- 2. Schrander-Stumpel CT, Curfs LM, Sastrowijoto P, Cassidy SB, Schrander JJ, Fryns JP. Prader-Willi syndrome: causes of death in an international series of 27 cases. Am J Med Genet A. 2004;1;124A(4):333-8, http://dx.doi.org/10.1002/ajmg.a.20371
- 3. Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, Webb T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study. Dev Med Child Neurol. 2002;44(4):248-55, http://dx.doi.org/10.1017/S001216220100202X
- 4. Whittington JE, Holland AJ, Webb T, Butler J, Clarke D, Boer H. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J Med Genet. 2001;38(11):792-8, http://dx.doi.org/10.1136/jmg.38.11.792
- 5. Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J Clin Endocrinol Metab. 1995;80(2):573-9, http://dx.doi.org/10.1210/jc.80.2.573
- 6. Purtell L, Sze L, Loughnan G, Smith E, Herzog H, Sainsbury A, et al. In adults with Prader-Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-1 levels. Neuropeptides. 2011;45(4):301-7, http://dx.doi.org/10.1016/j.npep.2011.06.001
- 7. Crinò A, Schiaffini R, Ciampalini P, Spera S, Beccaria L, Benzi F, et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr. 2003;162(5):327-33.
- 8. Holland AJ, Treasure J, Coskeran P, Dallow J, Milton N, Hillhouse E. Measurement of excessive appetite and metabolic changes in Prader-Willi syndrome. Int J Obes Relat Metab Disord. 1993;17(9):527-32.
- 9. Hoybye C. Five-years growth hormone (GH) treatment in adults with Prader-Willi syndrome. Acta Paediatr 2007;96(3):410-3, http://dx.doi.org/10.1111/j.1651-2227.2006.00051.x
- 10. Festen DA, de Lind van Wijngaarden R, van Eekelen M, Otten BJ, Wit JM, Duivenvoorden HJ, et al. Randomized controlled GH trial: effects on anthropometry, body composition and body proportions in a large group of children with Prader-Willi syndrome. Clin Endocrinol (Oxf). 2008;69(3):443-51, http://dx.doi.org/10.1111/j.1365-2265.2008. 03228.x
- 11. Lin H-Y, Lin S-P, Chuang C-K, Chen M-R, Yen J-L, Lee Y-J, et al. Genotype and phenotype in patients with Prader-Willi Syndrome in Taiwan. Acta Paediatrica 2007;96(6):902-5, http://dx.doi.org/10.1111/j.1651-2227.2007.00284.x
- 12. Gunay-Aygun M, Schwartz S, Heeger S, O'Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001;108(5):E92, http://dx.doi.org/10.1542/peds.108.5.e92",-1,"xxx/5.e92
- 13. Butler MG. Prader-Willi Syndrome: Obesity due to Genomic Imprinting. Curr Genomics. 2011;12(3):204-15.
- 14. Zeschnigk M, Lich C, Buiting K, Doerfler W, Horsthemke B. A single tube PCR test for the diagnosis of Angelman and Prader-Willi syndrome based on allelic methylation differences at the SNRPN locus. Eur J Hum Genet. 1997;5(2):94-8.
Publication Dates
-
Publication in this collection
30 Aug 2012 -
Date of issue
Aug 2012
History
-
Received
19 May 2012 -
Accepted
08 June 2012 -
Reviewed
06 June 2012