Abstracts
We have evaluated a girl and a boy with the blepharophimosis, ptosis and epicanthus inversus syndrome (BPES). The girl presented cleft palate and the boy showed cleft lip and palate as additional clinical signs. Both showed familial recurrence in fourth and third generations, respectively. The other family members also presented blepharophimosis, ptosis, and epicanthus inversus, but without lip and palatal involvement. There were no additional clinical signs nor infertility in these patients. To our knowledge this is the first instance of cleft lip and palate reported as additional signs of the BPES syndrome.
Os autores descrevem uma menina e um menino com a síndrome de blefarofimose, ptose e epicanto inverso (BPES). A menina apresentou fissura de palato e o menino mostrou fissura de lábio e palato como sinais clínicos adicionais. Ambos mostraram recorrência familial em quatro e três gerações, respectivamente. Outros membros de ambas as famílias apresentaram também blefarofimose, ptose e epicanto inverso, mas sem envolvimento de lábio e palato. Não existem sinais clínicos adicionais nem infertilidade nestes pacientes. De acordo com o nosso conhecimento, este é o primeiro relato de fissura de lábio e palato registrada como sinal adicional na síndrome BPES.
Blepharophimosis, ptosis, epicanthus inversus syndrome (BPES) and cleft lip and palate. Report of two Brazilian families
N.M. Kokitsu-Nakata and A. Richieri-Costa
Serviço de Genética Clínica, Hospital de Pesquisa e Reabilitação de Lesões Lábio-Palatais, Universidade de São Paulo, Caixa Postal 620, 17043-070 Bauru, SP, Brasil. Send correspondence to A.R.-C
ABSTRACT
We have evaluated a girl and a boy with the blepharophimosis, ptosis and epicanthus inversus syndrome (BPES). The girl presented cleft palate and the boy showed cleft lip and palate as additional clinical signs. Both showed familial recurrence in fourth and third generations, respectively. The other family members also presented blepharophimosis, ptosis, and epicanthus inversus, but without lip and palatal involvement. There were no additional clinical signs nor infertility in these patients. To our knowledge this is the first instance of cleft lip and palate reported as additional signs of the BPES syndrome.
INTRODUCTION
Blepharophimosis occurring either per se or syndromally is part of over 100 conditions, but the combination with epicanthus inversus, ptosis and cleft lip and palate as phenotypic manifestations of a particular syndrome is very unusual (Oley and Baraitser, 1988). Here we reported two Brazilian families with blepharophimosis, ptosis and epicanthus inversus. Cleft lip and palate were present in two of the reported patients. Clinical, genetic, and differential diagnosis aspects are discussed.
CLINICAL REPORTS
Family A
Patient 1
LAVM (Figure 1), female, was born in 1993 to a 29-year-old G2P2 mother and her 41-year-old unrelated husband. Pregnancy was unremarkable, and there was no exposure to toxic or infectious agents or to X-rays. Delivery was at term by cesarean section. Neuropsychological development was normal. Examination at the age of 3 7/12 years showed: height of 102 cm (75th centile), weight of 17 kg (75th centile), occipitofrontal circumference (OFC) of 51 cm (50th-98th centile), inner canthal distance (ICD) of 2.6 cm (25th-50th centile), outer canthal distance (OCD) of 6.8 cm (3rd-25th centile), and palpebral fissure length (PFL) of 2.1 cm (<5th centile). She is an intelligent girl, with evident blepharophimosis, epicanthus inversus, ptosis, and cleft palate. There were no additional anomalies. Chromosomes were normal.
Patient 2
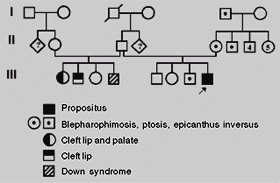
VVM (Figure 2), the proposita's mother, had an identical facial appearance to that of her daughter. Clinical examination at the age of 27 years showed: height of 156 cm (10th-25th centile), weight of 54 kg (25th-50th centile), OFC of 52.5 cm (2nd-50th centile), ICD of 3.0 cm (25th-50th centile), OCD of 7.9 cm (3rd-25th centile), and PFL of 2.3 cm (<5th centile). She is an intelligent woman, with evident blepharophimosis, ptosis and epicanthus inversus. There were no additional anomalies. The results of routine laboratory blood tests were normal. Familial history showed several affected individuals in four generations (Figure 3).
Family B
Patient 3
SBR (Figure 4), male, was born in 1996, at term, by normal and spontaneous vaginal delivery after uncomplicated prenancy. He was the fourth child of a 28-year-old G4P4 mother and her unrelated 53-year-old husband. Birth weight was 3,950 g (75th-90th centile) and length was not recorded. Neuropsychological development was normal. Evaluation at the age of 3 months showed height of 65 cm (50th centile), weight of 6.3 kg (50th centile), occipitofrontal circumference of 42.5 cm (50th-98th centile), inner canthal distance of 2.8 cm (>97th centile), outer canthal distance of 6.9 cm (50th-75th centile) and palpebral fissure length of 1.9 cm (25th-50th centile). He had ocular hypertelorism, blepharophimosis, epicanthus inversus, palpebral ptosis, telecanthus and cleft lip and palate. There were no additional anomalies. Cytogenetic investigation revealed a normal male karyotype with 46,XY.
Patient 4
RBR (Figure 5), the propositus's mother, showed an identical facial appearance of her son. Clinical examination at the age of 29 years showed: ICD 3.3 cm (75th-97th centile), OCD of 9.1 cm (75th-97th centile), and PFL of 2.7 cm (25th-50th centile). She is an intelligent woman, with evident blepharophimosis, palpebral ptosis, telecanthus and epicanthus inversus. There were no additional anomalies. Familial history showed several affected individuals in three generations (Figure 6). In all of them a similar facial appearance was referred.
DISCUSSION
The BPES is an autosomal dominant condition with more than 150 families reported. Main clinical features are: blepharophimosis, blepharoptosis, epicanthus inversus and telecanthus. Occasional findings include microphthalmos, anophthalmos, microcornea, hypermetropia, strabismus, nystagmus, trichiasis, flat broad nasal bridge, high arched palate, minor ear anomalies, cardiac defects, mild mental retardation and infertility in females (Oley and Baraitser, 1988). The transmission, usually, is mainly through affected males than affected females (Fraser et al., 1988; Oley and Baraitser, 1988), and female infertility can be responsible for this discordance. The existence of two types of BPES was proposed by Zlotogora et al., 1983: type I, the most common, totally penetrant, only transmitted by males, and the affected females are infertile, and type II transmitted both by affected males and females, with a penetrance of 96.5%. This subdivision has been subject of controversy, since without informative pedigrees a plausible classification is not possible. Genetic counselors must be alert to non-penetrance or very minimal expression of the gene in some patients concerning the very low offspring risks to unaffected members of these families (Temple and Baraitser, 1989).
In the family reported here, the transmission was through maternal line, and there is no information about infertility. The occurrence of cleft palate in the proposita gives rise to some questions concerning differential diagnosis, since only in few cases of BPES high arched palate has been reported (Oley and Baraitser, 1988). Krieble and Bixler (1984) reported on a boy and his father, both presenting blepharophimosis, epicanthus, ear anomalies, cleft palate, minor hand/foot anomalies, and inguinal hernia. Tetralogy of Fallot was diagnosed shortly after birth in the boy, and surgically corrected within the first year of life. The whole clinical picture of these patients clearly distinguishes them from the present one, and the same is true for the autosomal dominant syndrome of blepharophimosis and craniosynostosis described by Pueschel and Barsel-Bowers (1979) (Table I). There is evidence that terminal deletion of chromosome 3p is related with the blepharophimosis-mental retardation syndrome and 3q23 microdeletion is related to the blepharophimosis-ptosis-epicanthus inversus syndrome (Fujita et al., 1992; Moncla et al., 1995). In the patient reported here, chromosomes were normal, but a non-detectable microdeletion by G-banding (minimum 550 band level) at 3q23 cannot be excluded. Finally, cleft lip and palate may represent a very low frequency sign within the spectrum of the BPES, or be present in our patients only by chance, especially in family 2 where two half-sibs presented cleft lip/palate.
ACKNOWLEDGMENTS
This study was supported by FAPESP (No. 95/4768-0).
RESUMO
Os autores descrevem uma menina e um menino com a síndrome de blefarofimose, ptose e epicanto inverso (BPES). A menina apresentou fissura de palato e o menino mostrou fissura de lábio e palato como sinais clínicos adicionais. Ambos mostraram recorrência familial em quatro e três gerações, respectivamente. Outros membros de ambas as famílias apresentaram também blefarofimose, ptose e epicanto inverso, mas sem envolvimento de lábio e palato. Não existem sinais clínicos adicionais nem infertilidade nestes pacientes. De acordo com o nosso conhecimento, este é o primeiro relato de fissura de lábio e palato registrada como sinal adicional na síndrome BPES.
(Received April 26, 1996)
- Fraser, I.S., Sherman, R.P., Smith, A. and Russel, P. (1988). An association between blepharophimosis, resistant ovary syndrome and true premature menopause. Fertil. Steril. 50: 747-751.
- Fujita, H., Meng, J., Kawamura, M., Tozuka, N., Ishii, F. and Tanaka, N. (1992). Boy with a chromosome del (3)(q12q23) and blepharophimosis syndrome. Am. J. Med. Genet. 44: 434-436.
- Krieble, B.F. and Bixler, D. (1984). Autosomal dominant blepharophimosis with multiple congenital anomalies. J. Clin. Dysmorphol. 2: 24-29.
- Moncla, A., Philip, N. and Mattei, J.F. (1995). Blepharophimosis-mental retardation syndrome and terminal deletion of chromosome 3p. J. Med. Genet. 32: 245-246.
- Oley, C. and Baraitser, M. (1988). Blepharophimosis, ptosis, epicanthus inversus syndrome (BPES syndrome). J. Med. Genet. 25: 47-51.
- Pueschel, S.M. and Barsel-Bowers, G. (1979). A dominantly inherited congenital anomaly syndrome with blepharo-phimosis. J. Pediatr. 95: 1010-1012.
- Temple, I.K. and Baraitser, M. (1989). Pitfalls in counselling of the blepharophimosis, ptosis, epicanthus inversus syndrome (BPES). J. Med. Genet. 26: 517-519.
- Zlotogora, J., Sagi, M. and Cohen, T. (1983). The blepharophimosis, ptosis, and epicanthus inversus syndrome: delineation of two types. Am. J. Hum. Genet. 35: 1020-1027.
Publication Dates
-
Publication in this collection
06 Jan 1999 -
Date of issue
June 1998
History
-
Received
26 Apr 1996