Abstracts
Tributyl[(phenylselanyl)aryl]stannanes were prepared in good yields by reaction between tributyl(phenylselanyl)stannane and 2-(trimethylsilyl)aryl triflates in the presence of potassium fluoride and18-crown-6 ether in THF at 0 ºC.
benzyne; arynes; insertion reaction; selenostannanes
Tributil[(fenilselanil)aril]estananas foram preparadas em bons rendimentos por reação entre tributil(fenilselanil)estanana e 2-(trimetilsilil)aril triflatos na presença de fluoreto de potássio e éter 18-coroa-6 em THF a 0 ºC.
SHORT REPORT
Selenostannylation of arynes produced by silylaryl triflates under mild reaction conditions
Fabiano T. ToledoI; João V. ComassetoI; Cristiano RaminelliII,* * e-mail: raminelli@ufgd.edu.br
IInstituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, Cidade Universitária, 05508-000 São Paulo-SP, Brazil
IIFaculdade de Ciências Exatas e Tecnologia, Universidade Federal da Grande Dourados, Rodovia Dourados-Itahum, km 12, Cidade Universitária, 79804-970 Dourados-MS, Brazil
ABSTRACT
Tributyl[(phenylselanyl)aryl]stannanes were prepared in good yields by reaction between tributyl(phenylselanyl)stannane and 2-(trimethylsilyl)aryl triflates in the presence of potassium fluoride and18-crown-6 ether in THF at 0 ºC.
Keywords: benzyne, arynes, insertion reaction, selenostannanes
RESUMO
Tributil[(fenilselanil)aril]estananas foram preparadas em bons rendimentos por reação entre tributil(fenilselanil)estanana e 2-(trimetilsilil)aril triflatos na presença de fluoreto de potássio e éter 18-coroa-6 em THF a 0 ºC.
Introduction
Benzyne is an important reactive intermediate in synthetic organic chemistry which has found application in several total syntheses1 and in the preparation of functional materials.2 Accordingly, a number of methods for benzyne formation have been reported in the literature.3-8 Among them we pay particular attention to the thermal decompositions of benzenediazonium-2-carboxylate4 and diphenyliodonium-2-carboxylate,5 the elimination reaction resulted from oxidative addition of magnesium in 2-bromofluorobenzene,6 the iodine-magnesium exchange reaction followed by elimination occurred in 2-iodophenyl 4-chlorobenzenesulfonate,7 and the oxidation of 1-aminobenzotriazole in the presence of lead tetraacetate.8
Although the methodologies for generating benzyne which were mentioned above are of great importance in the context of benzyne chemistry,3-8 they have employed harsh conditions, including high temperatures, strong bases or powerful oxidizing agents.3-8 In this scenario, 2-(trimethylsilyl)aryl triflates9 and phenyl[2-(trimethylsilyl)aryl]iodonium triflates10 arise as promising reagents to form arynes under mild reaction conditions.9,10 Once 2-(trimethylsilyl)phenyl triflate was transformed in a commercially available reagent and its derivatives can be easily prepared from phenols,9 2-(trimethylsilyl)aryl triflates have emerged as remarkable reagents for generating arynes recently employed in several insertion reactions into sigma bonds.11 Because of the highly electrophilic character of arynes that comes from their low-energy LUMOs, even neutral nucleophiles of diminished nucleophilicity readily attacks arynes to produce zwitterions, which act as key intermediates in the insertion reactions into, for example, C-C,12 N-H,13,14 O-H,14,15 Se-Se,16,17 Te-Te17 and S-Sn18 sigma bonds. These transformations lead to the formation of polysubstituted arenes and benzoannulated compounds, which would be hardly synthesized in just one step by other methods.
In this work, we report our results involving the insertion reaction of arynes generated from 2-(trimethylsilyl)aryl triflates under mild reaction conditions into Se-Sn sigma bond of tributyl(phenylselanyl)stannane,19 as an attractive alternative for the synthesis of tributyl[(phenylselanyl)aryl]stannanes20 in good yields.
The organometallic compounds produced by the selenostannylation process can be considered versatile intermediates in synthetic organic chemistry, finding application in cross-coupling reactions, transmetallations and iodolyses.18 In order to give some support to this fact, we subjected one of the aromatic selenostannanes produced in the course of our investigation to the iododestannylation reaction and the result obtained was enclosed herein.
Results and Discussion
Allowing the reaction of tributyl(phenylselanyl)stannane (1) with 1.5 equiv. of 2-(trimethylsilyl)phenyl triflate (2a) and 3 equiv. of CsF in acetonitrile at room temperature, we obtained tributyl[2-(phenylselanyl)phenyl]stannane (3a) in a very low yield (< 5%) (Table 1, entry 1). In an attempt to improve the obtained yield (entry 1), subsequent work focused on optimization of these reaction conditions (Table 1).
In order to examine the effect of the fluoride ion sources on the transformation, we carried out the reaction of tributyl(phenylselanyl)stannane (1) with 1.5 equiv. of 2-(trimethylsilyl)phenyl triflate (2a), 1.5 equiv. of KF and 1.5 equiv. of 18-crown-6 ether in THF at 0 ºC for 24 h and obtained tributyl[2-(phenylselanyl)phenyl]stannane (3a) in a moderate yield of 52% (Table 1, entry 2). By performing the reaction of the stannyl selenide 1 with 1.5 equiv. of the benzyne precursor 2a in the presence of 1.8 equiv. of tetrabutylammonium fluoride (TBAF) in THF at room temperature for 3 h, the insertion product 3a was not produced (entry 3). No further attempts were made to optimize this TBAF procedure. Afterwards, we treated tributyl(phenylselanyl)stannane (1) with 2 equiv. of 2-(trimethylsilyl)phenyl triflate (2a), 2 equiv. of KF and 2 equiv. of 18-crown-6 ether in THF at 0 ºC for 24 h to give tributyl[2-(phenylselanyl)phenyl]stannane (3a) in a good 62% yield (entry 4). When the same reaction was carried out at room temperature, compound 3a was obtained in a slightly lower yield of 57% (entry 5). No significant improvement in the isolated yield of tributyl[2-(phenylselanyl)phenyl]stannane (3a) was obtained when we allowed the stannyl selenide 1 to react with 2.5 equiv. of the benzyne precursor 2a in the presence of 2.5 equiv. of KF and 2.5 equiv. of 18-crown-6 ether at 0 ºC for 24 h (entry 6). As can be seen in Table 1, entry 7, compound 3a was not obtained and the starting materials 1 and 2a were recovered when the reaction was carried out in the absence of KF. This experiment shows that the success of our reaction depends dramatically on the presence of a fluoride ions source.
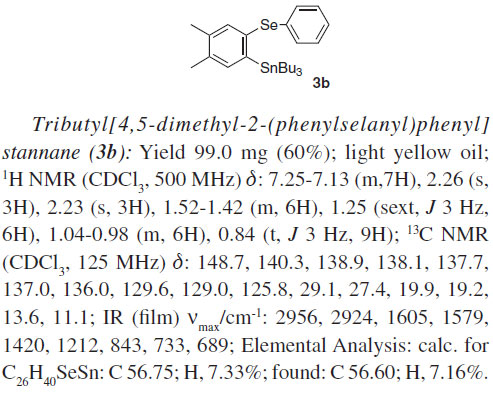
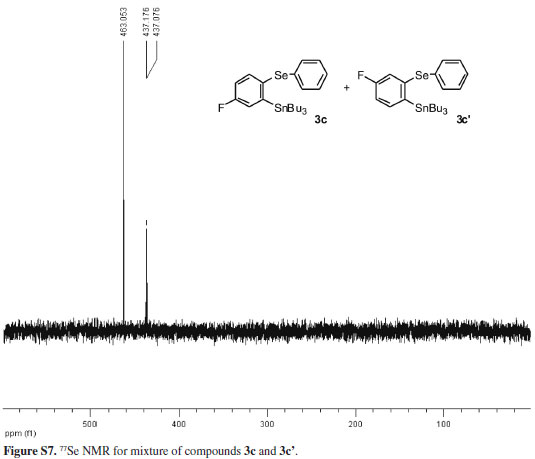
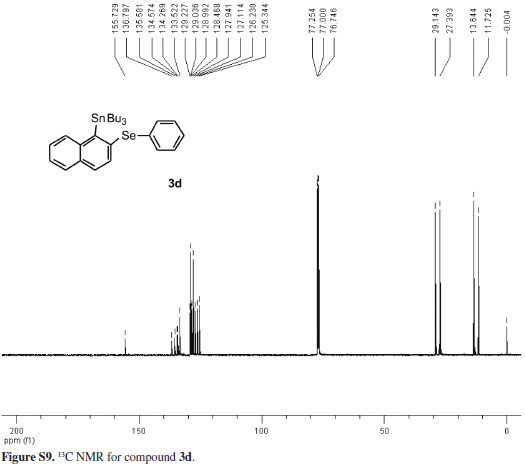
Employing the optimal conditions shown in Table 1, entry 4,21 we examine the scope and limitations of the process (Table 2). The reaction between tributyl(phenylselanyl)stannane (1) and the electron-rich aryne precursor 2b gave the aromatic selenostannane 3b in a 60% isolated yield (entry 2). To better understand the regioselectivity of the reaction when unsymmetrical arynes are employed, we treated tributyl(phenylselanyl)stannane (1) with the 4-fluoro aryne precursor 2c and obtained a mixture of isomers 3c and 3c' in a 2:1 ratio with a total yield of 34% (entry 3). In this case, the electron-withdrawing group promoted a more sluggish transformation leading to a regioisomeric mixture of products in a moderate yield. In addition, the major product 3c was obtained by the attack of the stannyl selenide 1 at the electronically most favorable position of the fluorinated aryne formed from compound 2c. This result indicates that our reaction involves the formation of benzyne and its derivatives as intermediates. Trying to give support to the formation of arynes as intermediates in this selenostannylation process, we allowed the reaction of tributyl(phenylselanyl)stannane (1) with the silylaryl triflates 2d and 2e, and the same aromatic selenostannane 3d was produced in 55 and 54% yields, respectively (Table 2, entries 4 and 5). The formation of the same insertion product 3d from the silylaryl triflates 2d (entry 4) and 2e (entry 5) suggests the generation of 1,2-naphthalyne as a common intermediate for the transformations. Besides, we have to mention that the exclusive formation of the regioisomer 3d in both reactions (Table 2, entries 4 and 5) can be rationalized by steric factors related to the attack of the stannyl selenide 1 to the 1,2-naphthalyne intermediate.22
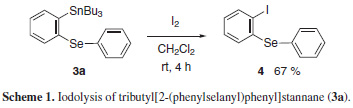
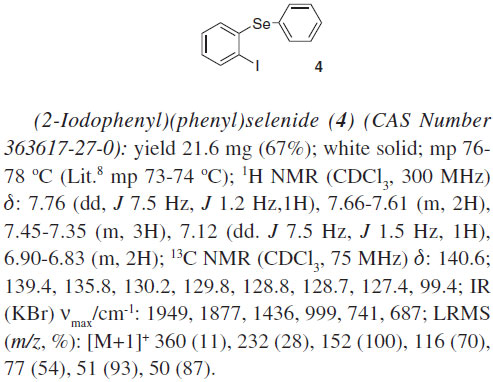
The products from the selenostannylation of arynes (3a-d) are versatile reagents in organic synthesis, which can find application in cross-coupling reations, homocouplings and in iodolyses. Accordingly, we subjected the stannane 3a to the reaction with iodine and obtained the diphenylselenide iodinated 4 in a good isolated yield of 67% (Scheme 1).
The structures of compounds 3a-d and 4 were assigned on the basis of a variety of spectroscopic techniques, namely, according to their IR, 1H, and 13C NMR spectra. The compounds 3a-d provided elemental analyses that agree with the proposed structures. Compound 4 presented LRMS that agrees with its structure and the mixture of regioisomers 3c and 3c', and compound 3d had their 77Se NMR spectra recorded.
Conclusions
In summary, a simple procedure which involves mild reaction conditions for the selenostannylation of arynes was developed and aromatic selenostannanes were produced in good yields. This attractive process provides an alternative for the preparation of organometallic compounds which are hardly synthesized in just one step by other methods and should find applications in the construction of molecules with interesting biological properties and in the preparation of functional materials.
Acknowledgments
We gratefully acknowledge the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and the Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT) for financial support.
Supplementary Information
Experimental data and NMR spectra are available free of charge at http://jbcs.sbq.org.br, as PDF file.
References
1. Buszek, K. R.; Brown, N.; Luo, D.; Org. Lett. 2009, 11, 201; Knight, D. W.; Qing, X.; Tetrahedron Lett. 2009, 50, 3534; Soorukram, D.; Qu T.; Barrett, A. G. M.; Org. Lett. 2008, 10, 3833; Sparks, S. M.; Chen, C.-L.; Martin, S. F.; Tetrahedron 2007, 63, 8619; Larrosa, I.; Da Silva, M. I.; Gómez, P. M.; Hannen, P.; Ko, E.; Lenger, S. R.; Linke, S. R.; White, A. J. P.; Wilton, D.; Barrett, A. G. M.; J. Am. Chem. Soc. 2006, 128, 14042; Chen, C.-L.; Sparks, S. M.; Martin, S. F.; J. Am. Chem. Soc. 2006, 128, 13696; Tambar, U. K.; Ebner, D. C.; Stoltz, B. M.; J. Am. Chem. Soc. 2006, 128, 11752; Sato, Y.; Tamura, T.; Mori, M.; Angew. Chem., Int. Ed. 2004, 43, 2436.
2. Lu, X.; Nikawa, H.; Tsuchiya, T.; Akasaka, T.; Toki, M.; Sawa, H.; Mizorogi, N.; Nagase, S.; Angew. Chem., Int. Ed. 2010, 49, 594; Shen, Y.-M.; Grampp, G.; Leesakul, N.; Hu, H.-W.; Xu, J.-H.; Eur. J. Org. Chem. 2007, 3718; Chen, Y.-L.; Wong, M.-S.; Wong, W.-Y.; Lee, A. W. M.; Tetrahedron Lett. 2007, 48, 2421; Guitián, E.; Pérez, D.; Peña, D.; Top. Organomet. Chem. 2005, 14, 109.
3. Pellissier, H.; Santelli, M.; Tetrahedron 2003, 59, 701; Hart, H. In The Chemistry of Triple-Bonded Functional Groups, Supplement C2; Patai, S., ed.; Wiley: Chichester, 1994, cap. 18; Gilchrist, T. L. In The Chemistry of Functional Groups, Supplement C; Patai, S.; Rappoport, Z., eds.; Wiley: Chichester, 1983, cap. 11; Hoffmann, R. W. Dehydrobenzene and Cycloalkynes, Academic Press: New York, 1967.
4. Logullo, F. M.; Seitz, A. H.; Friedman, L.; Organic Syntheses, Coll. Vol. 5, Willey: New York, 1973, p. 54; Stiles, M.; Miller, R. G.; Burckhardt, U.; J. Am. Chem. Soc. 1963, 85, 1792.
5. Fieser, L. F.; Haddadin, M. J.; Organic Syntheses, Coll. Vol. 5, Willey: New York, 1973, p. 1037; Le Goff, E.; J. Am. Chem. Soc. 1962, 84, 3786.
6. Wittig, G.; Organic Syntheses, Coll. Vol. 4, Willey: New York, 1963, p. 964; Wittig, G.; Erhard, K.; Chem. Ber. 1958, 91, 895.
7. Lin, W.; Chen, L.; Knochel, P.; Tetrahedron 2007, 63, 2787; Lin, W.; Ilgen, F.; Knochel, P.; Tetrahedron Lett. 2006, 47, 1941; Lin, W.; Sapountzis, I.; Knochel, P.; Angew. Chem., Int. Ed. 2005, 44, 4258; Sapountzis, I.; Lin, W.; Fisher, M.; Knochel, P.; Angew. Chem., Int. Ed. 2004, 43, 4364.
8. Campbell, C. D.; Rees, C. W.; J. Chem. Soc. (C), 1969, 752; Campbell, C. D.; Rees, C. W.; J. Chem. Soc. (C), 1969, 748; Campbell, C. D.; Rees, C. W.; J. Chem. Soc. (C), 1969, 742.
9. Atkinson, D. J.; Sperry, J.; Brimble, M. A.; Synthesis 2010, 911; Bronner, S. M.; Garg, N. K.; J. Org. Chem. 2009, 74, 8842; Peña, D.; Iglesias, B.; Quintana, I.; Pérez, D.; Guitián, E.; Castedo, L.; Pure Appl. Chem. 2006, 78, 451; Peña, D.; Cobas, A.; Pérez, D.; Guitián, E.; Synthesis 2002, 1454; Wu, Q.-C.; Himeshima, Y.; Sonoda, T.; Kobayashi, H.; Chem. Lett. 1983, 1211.
10. Xue, J.; Yang, Y.; Huang, X.; Synllett 2007, 1533; Kitamura, T.; Aoki, Y.; Isshiki, S.; Wasai, K.; Fujiwara, Y.; Tetrahedron Lett. 2006, 47, 1709; Kitamura, T.; Yamane, M.; Inoue, K.; Todaka, M.; Fukatsu, N.; Meng, Z.; Fujiwara, Y.; J. Am. Chem. Soc. 1999, 121, 11674; Kitamura, T.; Yamane, M.; J. Chem. Soc., Chem. Commun. 1995, 983.
11. For reviews, see: Gallo, R. D. C.; Rezende, H. V.; Muzzi, R. M.; Raminelli, C.; Quim. Nova, 2009, 32, 2437; Peña, D.; Pérez, D.; Guitián, E.; Angew. Chem., Int. Ed. 2006, 45, 3579.
12. Yoshida, H.; Kishida, T.; Watanabe, M.; Ohshita, J.; Chem. Commun. 2008, 5963; Ni, C.; Zhang, L.; Hu, J.; J. Org. Chem. 2008, 73, 5699; Xue, J.; Wu, L. L.; Huang, X.; Chin. Chem. Lett. 2008, 19, 631; Yoshida, H.; Watanabe, M.; Morishita, T.; Ohshita, J.; Kunai, A.; Chem. Commun. 2007, 1505; Huang, X.; Xue, J.; J. Org. Chem. 2007, 72, 3965; Tambar, U. K.; Stoltz, B. M.; J. Am. Chem. Soc. 2005, 127, 5340; Yoshida, H.; Watanabe, M.; Ohshita, J.; Kunai, A.; Chem. Commun. 2005, 3292; Yoshida, H.; Watanabe, M.; Ohshita, J.; Kunai, A.; Tetrahedron Lett. 2005, 46, 6729; Biehl, E. R.; Khanapure, S. P.; Acc. Chem. Res. 1989, 22, 275; Caubere, P.; Acc. Chem. Res. 1974, 7, 301.
13. Liu, Z.; Larock, R. C.; Org. Lett. 2003, 5, 4673.
14. Liu, Z.; Larock, R. C.; J. Org. Chem. 2006, 71, 3198.
15. Liu, Z.; Larock, R. C.; Org. Lett. 2004, 6, 99.
16. Toledo, F. T.; Marques, H.; Comasseto, J. V.; Raminelli, C.; Tetrahedron Lett. 2007, 48, 8125.
17. Bonilha, J. B. S.; Petragnani, N.; Toscano, V. G.; Chem. Ber. 1978, 111, 2510; Petragnani, N.; Toscano, V. G.; Chem. Ber. 1970, 103, 1652.
18. Yoshida, H.; Terayama, T.; Ohshita, J.; Kunai, A.; Chem. Commun. 2004, 1980.
19. For the synthesis of tributyl(phenylselanyl)stannane, see: Nishiyama, Y.; Kawamatsu, H.; Funato, S.; Tokunaga, K.; Sonoda, N.; J. Org. Chem. 2003, 68, 3599; Nishiyama, Y.; Tokunaga, K.; Sonoda, N.; Org. Lett. 1999, 1, 1725.
20. For recent reviews about organoselenium chemistry, see: Perin, G.; Lenardão, E. J.; Jacob, R. G.; Panatieri, R. B.; Chem. Rev. 2009, 109, 1277; Freudendahl, D. M.; Shahzad, S. A.; Wirth, T.; Eur. J. Org. Chem. 2009, 1649.
21. General procedure for selenostannylation of arynes: To a vial (10 mL) were added the tributyl(phenylselanyl)stannne 1 (0.3 mmol), the appropriate aryne precursor 2a-e (0.6 mmol), 18-crown-6 ether (158 mg, 0.6 mmol), and THF (3 mL). The mixture was cooled to 0 ºC and potassium fluoride (34.8 mg, 0.6 mmol) was added. The vial was sealed using a cap, and the reaction mixture was stirred for 24 h at 0 ºC. Afterwards, the reaction was added to a saturated solution of ammonium chloride (30 mL), which was extracted with ethyl acetate (3 × 30 mL). The organic phase was dried over MgSO4. After filtration, the solvent was evaporated under reduced pressure. The residue was purified by preparative TLC using silica gel as stationary phase and hexane as eluent unless otherwise indicated, affording the desired products 3a-d. Tributyl[2-(phenylselanyl)phenyl]stannane (3a): yield 97.1 mg (62%); light yellow oil; 1H NMR (CDCl3, 300 MHz) δ: 7.52 (dd, J 7.5 Hz, J 1.3 Hz, 1H), 7,45 (dd, J 7.5 Hz, J 1.6 Hz, 1H), 7.30-7.13 (m, 7H), 1.55-1.44 (m, 6H), 1.27 (sext, J 7.3 Hz, 6H), 1.08-1.03 (m, 6H), 0.84 (t, J 7.3 Hz, 9H); 13C NMR (CDCl3, 75 MHz) δ: 151.4, 139.2, 137.5, 135.8, 134.0, 130.5, 129.3, 129.1, 127.3, 126.3, 29.1, 27.4, 13.7, 11.2; IR (film) nmax/cm-1: 2956, 2923, 1926, 1849, 1793, 1732, 1578, 1470, 1441, 1070, 736, 689; Elemental Analysis: calc. for C24H36SeSn: C 55.20; H 6.95%; found: C 55.20; H 6.85%.
22. The structure proposed for regioisomer 3d is according to the pattern of regioselectivity followed by a number of insertion reactions of 1,2-naphthalyne into nucleophile-electrophile sigma bonds, see: Yoshida, H.; Shirakawa, E.; Honda, Y.; Hiyama, T.; Angew. Chem., Int. Ed. 2002, 41, 3247; Yoshida, H.; Fukushima, H.; Morishita, T.; Ohshita, J.; Kunai, A.; Tetrahedron 2007, 63, 4793; Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A.; Angew. Chem., Int. Ed. 2004, 43, 3935; Yoshida, H.; Minabe, T.; Ohshita, J.; Kunai, A.; Chem. Commun. 2005, 3454.
References(Supplementary Information)
1. Perrin, D. D.; Armarego, W. L. F.; Purification of Laboratory Chemicals, Pergamon: Oxford, 1980.
2. Nishiyama, Y.; Kawamatsu, H.; Funato, S.; Tokunaga, K.; Sonoda, N.; J. Org. Chem. 2003, 68, 3599.
3. Himeshima, Y.; Sonoda, T.; Kobayashi, H.; Chem. Lett. 1983, 1211.
4. Yoshida, H.; Sugiura, S.; Kunai, A.; Org. Lett. 2002, 4, 2767.
5. Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A.; Angew. Chem., Int. Ed. 2004, 43, 3935; Peña, D.; Cobas, A.; Pérez, D.; Guitián, E.; Synthesis 2002, 1454.
6. Peña, D.; Pérez, D.; Guitián, E.; Castedo, L.; J. Org. Chem. 2000, 65, 6944.
7. Yoshida, H.; Shirakawa, E.; Honda, Y.; Hiyama, T.; Angew. Chem., Int. Ed. 2002, 41, 3247.
8. Nakanishi, W.; Hayashi, S.; Heteroat. Chem. 2001, 12, 369.
Submitted: February 22, 2010
Published online: July 8, 2010
FAPESP has sponsored the publication of this article.
Supplementary Information
General methods
1H and 13C NMR spectra were obtained on a Varian INOVA 300 spectrometer (1H at 300 MHz and 13C at 75 MHz) and on a Bruker DRX-500 spectrometer (1H at 500 MHz and 13C at 125 MHz). The spectra were taken in deuterated solvents and the chemical shifts were given in ppm using residual undeuterated solvents or tetramethylsilane (TMS) as internal standard. 77Se NMR spectra were taken on a Bruker DRX-500 spectrometer at 95 MHz using CDCl3 as solvent. The chemical shifts were given in ppm using diphenyl diselenide (PhSeSePh) diluted in CDCl3 (1 mol L-1) as an external standard (d 463 ppm at 25oC). Low-resolution mass spectra were obtained on a Shimadzu CG-17A/CGMS-QP5050A instrument. Elemental analyses were obtained on a Perkin-Elmer CHN 2400 equipment. Near-IR spectra were obtained on a Bomem MB-100 spectrometer. The melting point value obtained is uncorrected. Reagents and solvents were used as obtained commercially or when necessary were purified and/or dried using procedures described in the literature.1 THF was distilled under nitrogen from sodium/benzophenone and CH3CN was distilled from calcium hydride. Both solvents were distilled prior to use. Oxygen and moisture sensitive materials were manipulated under a nitrogen atmosphere. Column chromatography and preparative thin layer chromatography separations were carried out using silica gel 60. Tributyl(phenylselanyl) stannane (1),2 2-(trimethylsilyl)phenyl triflate (2a),3 4,5-dimethyl-2-(trimethylsilyl)phenyl triflate (2b),4 4-fluoro-2-(trimethylsilyl)phenyl triflate (2c),5 1-(trimethylsilyl)-2- naphtyl triflate (2d),6 and 2-(trimethylsilyl)-1-naphtyl triflate (2e)7 were prepared according to literature procedures.
General procedure for selenostannylation of arynes
To a vial (10 mL) were added the tributyl(phenylselanyl) stannne 1 (0.3 mmol), the appropriate aryne precursor 2a-e(0.6 mmol), 18-crown-6 ether (158 mg, 0.6 mmol), and THF (3 mL). The mixture was cooled to 0 ºC and potassium fluoride (34.8 mg, 0.6 mmol) was added. The vial was sealed using a cap, and the reaction mixture was stirred for 24 h at 0 ºC. Afterwards, the reaction was added to a saturated solution of ammonium chloride (30 mL), which was extracted with ethyl acetate (3 × 30 mL). The organic phase was dried over MgSO4. After filtration, the solvent was evaporated under reduced pressure. The residue was purified by preparative TLC using silica gel as stationary phase and hexane as eluent unless otherwise indicated, affording the desired products 3a-d. Iododestannylation of aromatic selenostannane To a vial (10 mL) were added the tributyl[2- (phenylselanyl)phenyl]stannane 3a (47.5 mg, 0.09 mmol), dichloromethane (1 mL), and iodine (23 mg, 0.09 mmol). The vial was sealed using a cap, and the mixture was stirred for 4 h at room temperature. Afterwards, the reaction was diluted with ethyl acetate (10 mL) and washed with a saturated solution of sodium thiosulfate (10 mL). The organic phase was stirred with an 1 mol.L-1 aqueous solution of potassium fluoride (10 mL) for 30 min at room temperature. The insoluble materials were filtered off through a celite pad. The organic phase was dried over MgSO4. After filtration, the solvent was evaporated under reduced pressure. The residue was purified by preparative TLC using silica gel as stationary phase and hexane as eluent, affording the desired product 4.
Characterization data



Copies of 1H and 13C NMR spectra







- 1. Buszek, K. R.; Brown, N.; Luo, D.; Org. Lett. 2009, 11, 201;
- Knight, D. W.; Qing, X.; Tetrahedron Lett. 2009, 50, 3534;
- Soorukram, D.; Qu T.; Barrett, A. G. M.; Org. Lett. 2008, 10, 3833;
- Sparks, S. M.; Chen, C.-L.; Martin, S. F.; Tetrahedron 2007, 63, 8619;
- Larrosa, I.; Da Silva, M. I.; Gómez, P. M.; Hannen, P.; Ko, E.; Lenger, S. R.; Linke, S. R.; White, A. J. P.; Wilton, D.; Barrett, A. G. M.; J. Am. Chem. Soc. 2006, 128, 14042;
- Chen, C.-L.; Sparks, S. M.; Martin, S. F.; J. Am. Chem. Soc. 2006, 128, 13696;
- Tambar, U. K.; Ebner, D. C.; Stoltz, B. M.; J. Am. Chem. Soc. 2006, 128, 11752;
- Sato, Y.; Tamura, T.; Mori, M.; Angew. Chem., Int. Ed. 2004, 43, 2436.
- 2. Lu, X.; Nikawa, H.; Tsuchiya, T.; Akasaka, T.; Toki, M.; Sawa, H.; Mizorogi, N.; Nagase, S.; Angew. Chem., Int. Ed. 2010, 49, 594;
- Shen, Y.-M.; Grampp, G.; Leesakul, N.; Hu, H.-W.; Xu, J.-H.; Eur. J. Org. Chem. 2007, 3718;
- Chen, Y.-L.; Wong, M.-S.; Wong, W.-Y.; Lee, A. W. M.; Tetrahedron Lett. 2007, 48, 2421;
- Guitián, E.; Pérez, D.; Peña, D.; Top. Organomet. Chem. 2005, 14, 109.
- 3. Pellissier, H.; Santelli, M.; Tetrahedron 2003, 59, 701;
- Hart, H. In The Chemistry of Triple-Bonded Functional Groups, Supplement C2; Patai, S., ed.; Wiley: Chichester, 1994, cap. 18;
- Gilchrist, T. L. In The Chemistry of Functional Groups, Supplement C; Patai, S.; Rappoport, Z., eds.; Wiley: Chichester, 1983, cap. 11;
- Hoffmann, R. W. Dehydrobenzene and Cycloalkynes, Academic Press: New York, 1967.
- 4. Logullo, F. M.; Seitz, A. H.; Friedman, L.; Organic Syntheses, Coll. Vol. 5, Willey: New York, 1973, p. 54;
- Stiles, M.; Miller, R. G.; Burckhardt, U.; J. Am. Chem. Soc. 1963, 85, 1792.
- 5. Fieser, L. F.; Haddadin, M. J.; Organic Syntheses, Coll. Vol. 5, Willey: New York, 1973, p. 1037;
- Le Goff, E.; J. Am. Chem. Soc. 1962, 84, 3786.
- 6. Wittig, G.; Organic Syntheses, Coll. Vol. 4, Willey: New York, 1963, p. 964;
- Wittig, G.; Erhard, K.; Chem. Ber. 1958, 91, 895.
- 7. Lin, W.; Chen, L.; Knochel, P.; Tetrahedron 2007, 63, 2787;
- Lin, W.; Ilgen, F.; Knochel, P.; Tetrahedron Lett. 2006, 47, 1941;
- Lin, W.; Sapountzis, I.; Knochel, P.; Angew. Chem., Int. Ed. 2005, 44, 4258;
- Sapountzis, I.; Lin, W.; Fisher, M.; Knochel, P.; Angew. Chem., Int. Ed. 2004, 43, 4364.
- 8. Campbell, C. D.; Rees, C. W.; J. Chem. Soc. (C), 1969, 752;
- Campbell, C. D.; Rees, C. W.; J. Chem. Soc. (C), 1969, 748;
- Campbell, C. D.; Rees, C. W.; J. Chem. Soc. (C), 1969, 742.
- 9. Atkinson, D. J.; Sperry, J.; Brimble, M. A.; Synthesis 2010, 911;
- Bronner, S. M.; Garg, N. K.; J. Org. Chem. 2009, 74, 8842;
- Peña, D.; Iglesias, B.; Quintana, I.; Pérez, D.; Guitián, E.; Castedo, L.; Pure Appl. Chem. 2006, 78, 451;
- Peña, D.; Cobas, A.; Pérez, D.; Guitián, E.; Synthesis 2002, 1454;
- Wu, Q.-C.; Himeshima, Y.; Sonoda, T.; Kobayashi, H.; Chem. Lett. 1983, 1211.
- 10. Xue, J.; Yang, Y.; Huang, X.; Synllett 2007, 1533;
- Kitamura, T.; Aoki, Y.; Isshiki, S.; Wasai, K.; Fujiwara, Y.; Tetrahedron Lett. 2006, 47, 1709;
- Kitamura, T.; Yamane, M.; Inoue, K.; Todaka, M.; Fukatsu, N.; Meng, Z.; Fujiwara, Y.; J. Am. Chem. Soc. 1999, 121, 11674;
- Kitamura, T.; Yamane, M.; J. Chem. Soc., Chem. Commun. 1995, 983.
- 11. For reviews, see: Gallo, R. D. C.; Rezende, H. V.; Muzzi, R. M.; Raminelli, C.; Quim. Nova, 2009, 32, 2437;
- Peña, D.; Pérez, D.; Guitián, E.; Angew. Chem., Int. Ed. 2006, 45, 3579.
- 12. Yoshida, H.; Kishida, T.; Watanabe, M.; Ohshita, J.; Chem. Commun. 2008, 5963;
- Ni, C.; Zhang, L.; Hu, J.; J. Org. Chem. 2008, 73, 5699;
- Xue, J.; Wu, L. L.; Huang, X.; Chin. Chem. Lett. 2008, 19, 631;
- Yoshida, H.; Watanabe, M.; Morishita, T.; Ohshita, J.; Kunai, A.; Chem. Commun. 2007, 1505;
- Huang, X.; Xue, J.; J. Org. Chem. 2007, 72, 3965;
- Tambar, U. K.; Stoltz, B. M.; J. Am. Chem. Soc. 2005, 127, 5340;
- Yoshida, H.; Watanabe, M.; Ohshita, J.; Kunai, A.; Chem. Commun. 2005, 3292;
- Yoshida, H.; Watanabe, M.; Ohshita, J.; Kunai, A.; Tetrahedron Lett. 2005, 46, 6729;
- Biehl, E. R.; Khanapure, S. P.; Acc. Chem. Res. 1989, 22, 275;
- Caubere, P.; Acc. Chem. Res. 1974, 7, 301.
- 13. Liu, Z.; Larock, R. C.; Org. Lett. 2003, 5, 4673.
- 14. Liu, Z.; Larock, R. C.; J. Org. Chem. 2006, 71, 3198.
- 15. Liu, Z.; Larock, R. C.; Org. Lett. 2004, 6, 99.
- 16. Toledo, F. T.; Marques, H.; Comasseto, J. V.; Raminelli, C.; Tetrahedron Lett. 2007, 48, 8125.
- 17. Bonilha, J. B. S.; Petragnani, N.; Toscano, V. G.; Chem. Ber. 1978, 111, 2510;
- Petragnani, N.; Toscano, V. G.; Chem. Ber. 1970, 103, 1652.
- 18. Yoshida, H.; Terayama, T.; Ohshita, J.; Kunai, A.; Chem. Commun. 2004, 1980.
- 19. For the synthesis of tributyl(phenylselanyl)stannane, see: Nishiyama, Y.; Kawamatsu, H.; Funato, S.; Tokunaga, K.; Sonoda, N.; J. Org. Chem. 2003, 68, 3599;
- Nishiyama, Y.; Tokunaga, K.; Sonoda, N.; Org. Lett. 1999, 1, 1725.
- 20. For recent reviews about organoselenium chemistry, see: Perin, G.; Lenardão, E. J.; Jacob, R. G.; Panatieri, R. B.; Chem. Rev. 2009, 109, 1277;
- Freudendahl, D. M.; Shahzad, S. A.; Wirth, T.; Eur. J. Org. Chem. 2009, 1649.
- 22. The structure proposed for regioisomer 3d is according to the pattern of regioselectivity followed by a number of insertion reactions of 1,2-naphthalyne into nucleophile-electrophile sigma bonds, see: Yoshida, H.; Shirakawa, E.; Honda, Y.; Hiyama, T.; Angew. Chem., Int. Ed. 2002, 41, 3247;
- Yoshida, H.; Fukushima, H.; Morishita, T.; Ohshita, J.; Kunai, A.; Tetrahedron 2007, 63, 4793;
- Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A.; Angew. Chem., Int. Ed. 2004, 43, 3935;
- Yoshida, H.; Minabe, T.; Ohshita, J.; Kunai, A.; Chem. Commun. 2005, 3454.
- 1. Perrin, D. D.; Armarego, W. L. F.; Purification of Laboratory Chemicals, Pergamon: Oxford, 1980.
- 2. Nishiyama, Y.; Kawamatsu, H.; Funato, S.; Tokunaga, K.; Sonoda, N.; J. Org. Chem. 2003, 68, 3599.
- 3. Himeshima, Y.; Sonoda, T.; Kobayashi, H.; Chem. Lett. 1983, 1211.
- 4. Yoshida, H.; Sugiura, S.; Kunai, A.; Org. Lett. 2002, 4, 2767.
- 5. Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A.; Angew. Chem., Int. Ed. 2004, 43, 3935;
- Peña, D.; Cobas, A.; Pérez, D.; Guitián, E.; Synthesis 2002, 1454.
- 6. Peña, D.; Pérez, D.; Guitián, E.; Castedo, L.; J. Org. Chem. 2000, 65, 6944.
- 7. Yoshida, H.; Shirakawa, E.; Honda, Y.; Hiyama, T.; Angew. Chem., Int. Ed. 2002, 41, 3247.
- 8. Nakanishi, W.; Hayashi, S.; Heteroat. Chem. 2001, 12, 369.
Publication Dates
-
Publication in this collection
15 Dec 2010 -
Date of issue
2010
History
-
Accepted
08 July 2010 -
Received
22 Feb 2010