Abstracts
Primary immunodeficiency diseases comprise a genetically heterogeneous group of disorders that affect distinct components of the innate and adaptive immune system, such as neutrophils, macrophages, dendritic cells, complement proteins and natural killer cells, as well as T and B lymphocytes. The study of these diseases has provided essential insights into the functioning of the immune system. Primary immunodeficiency diseases have been linked to over 120 different genes, abnormalities in which account for approximately 180 different forms of these diseases. Patients with primary immunodeficiency diseases are most often recognized because of their increased susceptibility to infections. However, these patients can also present with a variety of other manifestations, such as autoimmune diseases, inflammatory diseases and cancer. The purpose of this article is to update the main aspects of primary immunodeficiency diseases, especially regarding the clinical manifestations related to the diagnosis, emphasizing the need for the early recognition of warning signs for these diseases.
Respiratory tract infections; Complement activation; Immunologic deficiency syndromes; Phagocytes; Immunoglobulins
As imunodeficiências primárias são um grupo de doenças geneticamente heterogêneas que afetam diferentes componentes da imunidade inata e adaptativa, como neutrófilos, macrófagos, células dendríticas, proteínas do sistema complemento, células natural killer e linfócitos B e T. O estudo dessas doenças tem fornecido importantes entendimentos sobre o funcionamento do sistema imune. Mais de 120 diferentes genes já foram identificados, cujas anormalidades são responsáveis aproximadamente 180 diferentes formas de imunodeficiências primárias. Pacientes com imunodeficiências primárias são frequentemente reconhecidos pela sua elevada suscetibilidade a infecções; porém, esses pacientes podem apresentar também várias outras manifestações, como doenças autoimunes, doenças inflamatórias e câncer. O propósito deste artigo é atualizar os principais aspectos das imunodeficiências primárias, especialmente em relação às manifestações clínicas relacionadas ao diagnóstico, enfatizando a necessidade do reconhecimento precoce dos sinais de alerta para essas doenças.
Infecções respiratórias; Ativação do complemento; Síndromes de imunodeficiência; Fagócitos; Imunoglobulinas
REVIEW ARTICLE
Primary immunodeficiency diseases: relevant aspects for pulmonologists*
Pérsio Roxo Júnior
Assistant Professor. Immunology, Allergy and Rheumatology Section, Department of Pediatrics, University of São Paulo at Ribeirão Preto School of Medicine, Ribeirão Preto, Brazil
Correspondence to
ABSTRACT
Primary immunodeficiency diseases comprise a genetically heterogeneous group of disorders that affect distinct components of the innate and adaptive immune system, such as neutrophils, macrophages, dendritic cells, complement proteins and natural killer cells, as well as T and B lymphocytes. The study of these diseases has provided essential insights into the functioning of the immune system. Primary immunodeficiency diseases have been linked to over 120 different genes, abnormalities in which account for approximately 180 different forms of these diseases. Patients with primary immunodeficiency diseases are most often recognized because of their increased susceptibility to infections. However, these patients can also present with a variety of other manifestations, such as autoimmune diseases, inflammatory diseases and cancer. The purpose of this article is to update the main aspects of primary immunodeficiency diseases, especially regarding the clinical manifestations related to the diagnosis, emphasizing the need for the early recognition of warning signs for these diseases.
Keywords: Respiratory tract infections; Complement activation; Immunologic deficiency syndromes; Phagocytes; Immunoglobulins.
Introduction
Primary immune diseases (PIDs) comprise a genetically heterogeneous group of rare disorders that are generally caused by genetic defects or developmental defects of the immune system.(1) These disorders are widely studied in immunology, and the results of such studies have provided insights into the functioning of the immune system and into the interactions within this system, as well as into the relationships between the host and the pathogenic agent. For instance, between the 1950s and the 1960s, the discovery of congenital agammaglobulinemia, DiGeorge syndrome and severe combined immunodeficiency was a milestone for the division of specific immunity into antibody-mediated response (humoral) and cell-mediated response (cellular) 15 years before the discovery of T and B lymphocytes.(2) Recently, the importance of defects in the function of natural killer cells(3) and in the signaling of toll-like receptors(4,5) has become the focus of attention in PIDs.
Clinical phenotypes derived from a particular genotype can vary greatly, depending on a number of factors. Therefore, the relationship between genetic characteristics and clinical phenotypes is not linear but rather a complex expression of molecular defects regulated by endogenous and exogenous factors, which might justify the wide phenotypic heterogeneity observed.(6) In recent years, a substantial number of genes involved in immunity, as well as their functions, have been identified through the study of patients with PIDs. These diseases are traditionally considered predisposing factors for infections caused by a wide variety of pathogens, and a growing number of immunodeficiencies with high susceptibility to infections by specific germs has been described.(7)
These diseases are constant challenges for physicians who work with primary care services, in which recurrent infections are quite common reasons for seeking medical attention. This reinforces the observation that the diagnosis of PIDs is delayed, probably because physicians have little knowledge regarding these diseases, which increases the risk for complications and death secondary to infections and other comorbidities.(8) In addition, many cases are incorrectly diagnosed, resulting in the adoption of inappropriate therapeutic measures.(9)
Since 1952, when the first PID (X-linked agammaglobulinemia or congenital agammaglobulinemia) was described by Bruton,(10) over 180 different types of PIDs have been described, and over 120 genes have been linked to these diseases,(7) which makes the classification of PIDs increasingly complex. In this context, since 1970, an international committee of specialists in PIDs has gathered every two years in order to update the classification of this large group of diseases, focusing on clinical, genetic and molecular aspects.(2,7)
Despite this great complexity, PIDs can be didactically divided into five major groups, according to how the immune response is affected, as follows: humoral immunodeficiency or antibody deficiency; cellular immunodeficiency or T cell immunodeficiency; combined immunodeficiency, affecting humoral and cellular immunity; phagocytic disorders; and complement deficiency.
The purpose of the present review is to provide tools for the recognition of patients with suspected PID, so that these patients can rapidly be investigated and referred for treatment at specialized centers.
Epidemiology
Although PIDs are considered to have a low incidence, it is estimated that they affect more than 1 infant per 2,000 births.(8) According to Conley and Stiehm,(11) of the patients with recurrent respiratory infections in the pediatric age bracket, approximately 50% are healthy, 30% have allergies; 10% have chronic pathologies, and 10% might have immunodeficiency.
Great geographic and racial variations in the prevalence and distribution of PIDs have been demonstrated in various epidemiological studies, and many developed countries have well defined studies on this topic. Such variations can be related to the genetic characteristics of each population and to the differences regarding the availability of diagnostic laboratory resources, especially of methods for molecular identification.(12)
Studies conducted in various countries in the world have shown that humoral immunodeficiencies are the most frequent PIDs, accounting for approximately half of the cases,(13,14) and that complement deficiencies are the most rare PIDs.(15,16)
Since certain PIDs are X-linked diseases, PIDs, in general, primarily affect males (male/female ratio, 5:1). The incidence of certain PIDs is as follows: 1:1,000 for IgA deficiency (in Brazil); 1:66,000 to 1:75,000 for common variable immunodeficiency; 1:100,000 for X-linked agammaglobulinemia; 1:183,000 to 1:200,000 for chronic granulomatous disease; 1:30,000 to 1:100,000 for severe combined immunodeficiency; and 1:10,000 to 1:50,000 for hereditary angioedema.(15,17)
Pathogenesis
Most PIDs are determined by X-linked autosomal inheritance or autosomal recessive inheritance, although some PIDs have no defined inheritance pattern and can be observed in more than one family member. The identification of the genetic inheritance is essential for subsequent genetic counseling.
This group of diseases results from heterogeneous disorders, involving defects in various parts of the immune system or defects in one single protein produced by a specific cell lineage. These gene defects can affect enzymes, structural proteins, signal transduction molecules or DNA repair proteins.(18)
Diagnosis
The first stage for the diagnosis of PIDs is to recognize that, although these diseases are rare in the general population, they are a medical reality and not only a myth. The clinical spectrum of PIDs is very wide and heterogeneous. Usually, the clinical manifestations of PIDs occur during childhood, although some manifestations can be observed in the second or third decades of life, as seen in cases of common variable immunodeficiency.(18) Therefore, PIDs are not restricted to the pediatric age bracket.
The most typical manifestations of PIDs are recurrent infections. This high predisposition is seen in one or more clinical dimensions of infections, such as pathogen virulence, infection site (local or generalized), severity (degree of tissue lesion), persistence or resistance to therapy and frequency of recurrence or reinfection.(19) Infections caused by specific microorganisms or low-virulence pathogens are predominant. Although they can have a mild clinical expression, most infections have severe and prolonged evolution, inadequate response to the antibiotic therapy routinely used and high risk for complications and hospitalizations.(20) Regarding these aspects, it is often difficult to distinguish between normal and abnormal. Hygiene, the prevalence of certain pathogens and the availability of vaccines should be considered.(19) The criteria for normality are generally based on laboratory tests for immunocompetence; however, clinical and biological correlation is not always observed. Therefore, patients with specific infectious diseases who have no detectable immunological alterations are frequently neglected.(21)
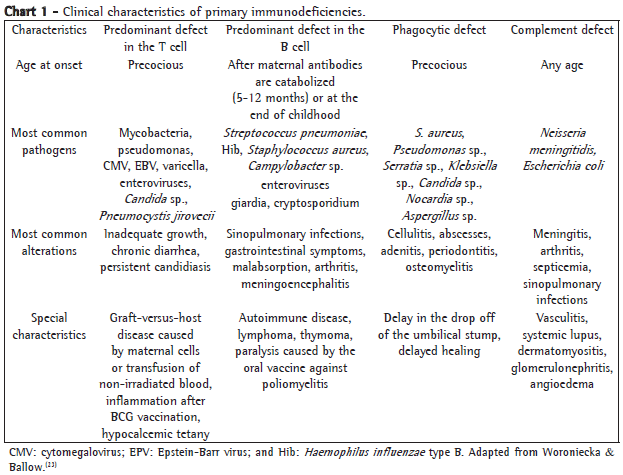
The age at onset, the type of pathogen and the location of the infections can suggest the nature of the immunological disorder (Chart 1).
The immune response mediated by antibodies is the principal defense mechanism against respiratory pathogens.(22) Therefore, humoral immunodeficiencies are predominantly accompanied by sinopulmonary infections caused by extracellular encapsulated bacteria and secondarily accompanied by gastrointestinal infections caused by enteroviruses and Giardia lamblia. Severe forms of congenital agammaglobulinemia or variable common immunodeficiency can evolve with complications such as bronchiectasis, gastrointestinal diseases, malignancies and autoimmune diseases.(23,24) The association between humoral immunity alterations and asthma is also frequent. The patterns of immune dysfunction in these cases are variable. High levels of IgG4,(25,26) IgG2 deficiency(27) and IgA deficiency(28) have been described. In our experience, we believe there is also an association between severe asthma and congenital agammaglobulinemia, common variable immunodeficiency and specific polysaccharide antibody deficiency with normal levels of immunoglobulins, with clinical improvement of the asthma after the replacement of gamma globulin i.v. (unpublished data). It is likely that this improvement in the clinical parameters is due to the significant reduction in respiratory infections, which play a key role in the triggering of exacerbations and in the intensification of the bronchial inflammatory process in these patients.
Specific cellular immunodeficiencies cause severe infections by pathogens of intracellular replication, such as viruses, fungi, mycobacteria and salmonellae.(8) Children with respiratory infections caused by Pneumocystis jirovecii can have severe cellular immunodeficiencies, such as hyper-IgM syndrome or severe combined immunodeficiency.(29) Specific deficiencies in the quantity or cytotoxic activity of natural killer cells can be particularly associated with fatal infections or disseminated infections caused by the herpes zoster virus,(30,31) although other viral infections, such as recurrent vulvar condylomata associated with cervical carcinoma and pulmonary infiltrate, have also been reported.(32)
Phagocytic disorders should be considered in patients with cutaneous and deep abscesses, as well as respiratory infections, neurological infections and infections of the reticuloendothelial system caused by staphylococci, gram-negative bacteria and fungi.(8)
Individuals who have terminal complement deficiencies generally present infections caused by bacteria of the genus Neisseria.(13)
Therefore, four aspects are essential for the clinician to suspect of a PID, as follows:
History and physical examination suggestive of PID
Infections caused by specific pathogens or low-virulence pathogens
Association with genetic syndromes
Family history of PID
The following are key aspects in the clinical history: age at onset, location probable etiology, frequency and severity of the infections, as well as the presence of postinfection complications, hospitalizations and severe post-vaccination reactions caused principally by live attenuated vaccines, such as BCG, oral poliomyelitis, rotavirus, yellow fever, measles/mumps/rubella and varicella vaccines. For instance, patients with chronic granulomatous disease can present systemic infections caused by Mycobacterium bovis after BCG vaccination, and patients with congenital agammaglobulinemia can develop poliomyelitis after taking the Sabin vaccine.(33)
Although recurrent infections are the most frequent manifestations associated with PIDs, other associated conditions can also be observed, such as severe allergic reactions, asthma, lymphoid hematopoietic neoplasia, autoimmune diseases, chronic inflammatory bowel disease and endocrinopathies.(34,35)
With regard to the family history, it is necessary to investigate the presence of parent consanguinity; the history of recurrent infections; deaths by severe infections, neoplasia or autoimmune diseases in other family members and maternal history of miscarriage with unknown cause. Patients with common variable immunodeficiency or IgA deficiency frequently present a family history of autoimmune diseases. The presence of parent consanguinity increases the possibility of recessive autosomal diseases, such as certain severe combined immunodeficiencies and certain forms of chronic granulomatous disease. However, a negative family history does not exclude the possibility of PID in a patient, since the disease might have been caused by a new mutation.(36)
Physical examination must be complete, meticulous and systematic. Growth in height and weight is frequently affected due to the chronic and recurrent infections or to the PID itself, according to the severity of the disease. Therefore, the growth in height and weight can be normal in mild cases of PID, such as IgA deficiency and specific polysaccharide antibody deficiency with normal levels of immunoglobulins. The clinician must be on the alert for the presence of abnormal phenotypic characters (face, type of hair, presence of cutaneous alterations). Chronic non-atopic eczema in patients with a prominent forehead and larger nasal bridge is suggestive of hyper-IgE).(37) Petechiae and eczema in male children are suggestive of Wiskott-Aldrich syndrome. Oral ulcers and recurrent gingivostomatitis can be observed in phagocytic disorders. Syndromic alterations, such as low-set ears, micrognathia, hypertelorism and bifid uvula, associated with congenital cardiopathy are suggestive of DiGeorge syndrome.(36) Clinical examination of the lymphoid tissue is essential. The absence of palatine tonsils (in the absence of surgery) or of lymph nodes, even in the presence of severe infections, provides strong evidence of congenital agammaglobulinemia or severe combined immunodeficiency. However, the excessive development of lymphoid tissue with hepatosplenomegaly is suggestive of chronic granulomatous disease.(8)
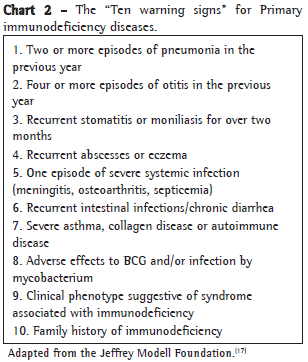
In 1999, the Jeffrey Modell Foundation, together with the American Red Cross, published the "Ten warning signs" for PID in order to facilitate clinical reasoning regarding patients who required laboratory evaluation. As illustrated in Chart 2, these warning signs were adapted for use in Brazil by the Brazilian Immunodeficiency Group.(17) The presence of one or more of the ten warning signs for PID makes laboratory evaluation mandatory.
Pulmonologists should consider the hypothesis of PID in the following situations:
Two or more episodes of pneumonia in the previous year
Chronic bronchitis with no history of smoking
Severe respiratory manifestations secondary to BCG vaccination
Respiratory infections caused by low-virulence pathogens (typical or atypical mycobacteria, fungi and protozoa)
Postinfection pulmonary complications (empyema, fistulae, bronchiectasis, pneumatoceles, abscesses, pulmonary fibrosis)
Severe asthma or difficult-to-control asthma
Evaluation of immunocompetence
The evaluation of immunocompetence is essential to define the diagnosis of PIDs. However, the evaluation of immunocompetence has two major limitations, namely the high cost of laboratory examinations and the small number of specialized laboratories available to perform the tests. Therefore, it is recommended that the laboratory investigation begin with screening tests, which are inexpensive and easy, according to the clinical history and the physical examination.(38) To that end, it is essential that the economic status of each center be considered.
The principal screening tests for the investigation of PIDs in Brazil are shown in Chart 3. The results should be interpreted very carefully and always compared with the reference values for individuals of the corresponding age bracket.
The blood workup with differential cell count is essential for all patients with suspected PID; it provides important information regarding suspected cytopenias (neutropenia, lymphopenia or thrombocytopenia) or qualitative alterations of cells, such as the presence of gigantic cytoplasmic inclusions associated with the Golgi complex and lysosomes in neutrophils and platelets of patients with Chediak-Higashi syndrome(39) or reduction in the size and function of platelets, suggestive of Wiskott-Aldrich syndrome, which also causes thrombocytopenia.(40) A sharp reduction in all cell series can be observed in certain severe combined immunodeficiencies, such as reticular dysgenesis. Persistent lymphopenia (less than e 3,000 lymphocytes/mm3 in children younger than 2 years of age) is suggestive of cellular immunodeficiency or combined immunodeficiency.(36,41)
The determination of serum immunoglobulins (IgG, IgM, IgA and IgE) is the first step for the evaluation of humoral immunity and allows the diagnosis of quantitative immunoglobulin deficiencies, such as congenital agammaglobulinemia, common variable immunodeficiency and IgA deficiency, as well as of humoral alterations associated with other defects, such as hyper-IgE syndrome and hyper-IgM syndrome.(36) Some patients might not produce antibodies against specific antigens, although they present normal immunoglobulin levels. Therefore, patients who remain seronegative when there is evidence of infection should be investigated.(42) A cavum X-ray is also useful for the initial evaluation of humoral immunity, since it allows the visualization of adenoid tissue, which can be absent in certain PIDs, such as congenital agammaglobulinemia and common variable immunodeficiency.(43)
Cellular immunity can be initially assessed through a blood workout, which can show lymphopenia; through a chest X-ray, to visualize the thymus; and through intradermal skin testing for delayed hypersensitivity. The antigens most frequently used are Candida, PPD, trichophyton, streptokinase-streptodornase and mumps. These tests have no diagnostic utility in children younger than 1 year due to the great possibility of false-negative results; such tests are considered negative when a papule greater than 2 mm in diameter is formed.(44)
Phagocytic disorders can be initially evaluated through a blood workup, which provides tools for the diagnosis of neutropenia; through the nitroblue tetrazolium reduction test, which assesses the oxidative metabolism of neutrophils and is extremely altered in patients with chronic granulomatous disease.(45)
The test most frequently used for the evaluation of the total hemolytic activity of the classical complement pathway is CH50.(36)
Considering the high prevalence of AIDS, it is recommended that all patients with recurrent infections be submitted to HIV serology.
It is also noteworthy that laboratory screening can be normal in some PIDs, such as in specific cellular immunodeficiencies or natural killer cell dysfunction.(13) However, patients with high level of clinical suspicion should be referred for specialized immunological evaluation.
Differential diagnosis
The differential diagnosis of recurrent infections should be comprehensive, considering that, in addition to PIDs, various conditions cause increased susceptibility to infections(11). These diseases are listed in Chart 4.
Treatment
Treatment should be started immediately after the diagnosis is confirmed, thus avoiding possible complications. A multidisciplinary approach is essential, involving physicians (especially pediatric immunologists, clinical immunologists, pulmonologists, infectious disease specialists, rheumatologists, endocrinologists, gastroenterologists and hematology oncologists), nurses, nutritionists, physiotherapists, psychologists, social workers and speech therapists. Treatment can be divided into general and specific.
General therapeutic measures include the following(44)
To adopt high standards of environmental and personal hygiene.
To educate patients and family members regarding the disease.
To reestablish nutritional and micronutrient conditions.
To adopt a diet without raw or undercooked foods.
To avoid crowded areas.
To perform frequent nasal lavages using saline solution.
To drain the secretions through respiratory physical therapy. Mucolytic drugs, such as N-acetylcysteine, can sometimes be inhaled, depending on the degree of viscosity of the secretions.
To avoid live attenuated vaccines (BCG, Sabin, rotavirus, measles/mumps/rubella, yellow fever and varicella) in certain PIDs, especially in cases of severe cellular immunodeficiencies and agammaglobulinemia. In these cases, family members and other people who live with the patients should not take the Sabin vaccine because of the risk of transmitting the vaccine strains. However, inactivated or subunit vaccines can be safely administered to immunocompromised patients,(8) although the efficacy of these vaccines is reduced. Patients with terminal complement deficiencies can benefit from immunization against encapsulated bacteria, especially Neisseria meningitidis.
Whenever necessary, to infuse blood products only when these have been previously irradiated, in order to avoid graft versus host disease.
To undergo aggressive and precocious treatment for infections using antimicrobials, if possible based on the previous isolation of pathogens in cultures from blood and other body fluids and antibiogram. The options and doses should be similar to those used for immunocompromised patients, but the period of treatment should be generally longer. Severe cases should be preferentially treated in hospitals, opting for the administration of antibiotic therapy i.v.(43) The use of prophylactic antibiotics (with quarterly rotations) is indicated for certain PIDs, in patients who present susceptibility to infections by specific agents.(13,46) For instance, patients with hyper-IgM syndrome and severe cellular immunodeficiencies should be submitted to prophylaxis with trimethoprim-sulfamethoxazole, since these patients are more likely to develop pneumonia by P. jirovecii.(8)
To treat the comorbidities and their complications.
Specific therapeutic measures should only be employed when the diagnosis is well-established; such measures vary according to the PID. The principal procedures available and their indications are as follows:
Immunoglobulin replacement therapy is the treatment of choice for patients with certain humoral immunodeficiencies, especially congenital agammaglobulinemia, common variable immunodeficiency and severe cases of specific antibody deficiency with normal levels of immunoglobulins. Immunoglobulin replacement therapy is also indicated for patients with severe combined immunodeficiencies. This therapy is not indicated in IgA deficiency, except in selected cases in which patients also present specific polysaccharide antibody deficiency. The route of administration can be intravenous (most frequent) or subcutaneous. The preparations contain neutralizing antibodies against a wide variety of bacterial and viral pathogens, reflecting the immunological memory of the donors.(47) This treatment modality has proved very effective, significantly reducing the incidence of respiratory infections (especially pneumonia) and the rates of hospitalization due to infections, which in turn reduces morbidity and mortality.(48)
Bone-marrow or stem-cell transplantation is the treatment of choice for severe combined immunodeficiencies and severe cellular immunodeficiencies, although it can be an alternative therapy for other PIDs, such as phagocytic disorders (e.g., X-linked chronic granulomatous disease), Wiskott-Aldrich syndrome and Chediak-Higashi syndrome.(8,49)
Immunomodulators are cytokines with great clinical applicability for certain immunodeficiencies, such as IFN-γ in cases of chronic granulomatous disease and the granulocyte-colony stimulating factor in cases of congenital neutropenia.(44)
Enzyme replacement therapy is successfully used in one form of severe combined immunodeficiency, known as adenosine deaminase (ADA) deficiency.(50)
Gene therapy is the most promising procedure for most cases of severe PIDs. There are reports of patients with X-linked severe combined immunodeficiency, ADA deficiency and X-linked chronic granulomatous disease who benefited from this treatment modality.(51,52)
Prognosis and follow up
The survival and quality of life of patients with certain PIDs has improved significantly, especially due to the better clinical management of infections and other comorbidities, to the development of highly potent antimicrobial agents and to the development of specific therapeutic alternatives,(35) such as the infusion of immunoglobulins and other therapies previously discussed.
The reduction in the number of infections and hospitalizations usually leads to the disruption of regular clinical monitoring, which can bring serious consequences. The decision regarding the frequency of evaluations of patients with PID depends on a number of aspects, such as the type of PID, clinical conditions and age.(35) In general, patients should be evaluated, including the complete clinical history and physical examination, at least once every 6-12 months; if possible, this evaluation should be carried out by a clinical immunologist specializing in PIDs.(13) Patients with pulmonary complications should be submitted to spirometry and serial imaging studies. Infectious diseases, autoimmune diseases and neoplasia should be constantly monitored due to the high risk of association of PIDs with these diseases.(53)
Final considerations
Despite the developments obtained in PIDs, the diagnosis and treatment of this group of diseases is still a major challenge. Considering that many of these diseases are primarily associated with recurrent infections of the respiratory tract, it is crucial that pulmonologists be on the alert for the warning signs for PID, so that suspected cases can be adequately referred to specialized centers. Precise diagnosis and the early adoption of appropriate therapeutic measures can improve the clinical conditions, prevent complications and increase the survival of patients with PIDs.
References
- 1. Guzman D, Veit D, Knerr V, Kindle G, Gathmann B, Eades-Perner AM, et al. The ESID Online Database network. Bioinformatics. 2007;23(5):654-5.
- 2. Notarangelo L, Casanova JL, Conley ME, Chapel H, Fischer A, Puck J, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee Meeting in Budapest, 2005. J Allergy Clin Immunol. 2006;117(4):883-96.
- 3. Orange JS. Human natural killer cell deficiencies and susceptibility to infection. Microbes Infect. 2002;4(15):1545-58.
- 4. Orange JS, Levy O, Brodeur SR, Krzewski K, Roy RM, Niemela JE, et al. Human nuclear factor kappa B essential modulator mutation can result in immunodeficiency without ectodermal dysplasia. J Allergy Clin Immunol. 2004;114(3):650-6.
- 5. Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299(5615):2076-9.
- 6. Buckley RH. Variable phenotypic expression of mutations in genes of the immune system. J Clin Invest. 2005;115(11):2974-6.
- 7. Geha RS, Notarangelo LD, Casanova JL, Chapel H, Conley ME, Fischer A, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120(4):776-94.
- 8. Morimoto Y, Routes JM. Immunodeficiency overview. Prim Care. 2008;35(1):159-73, viii.
- 9. Buckley RH. Primary immunodeficiency or not? Making the correct diagnosis. J Allergy Clin Immunol. 2006;117(4):756-8.
- 10. Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9(6):722-8.
- 11. Conley ME, Stiehm ER. Immunodeficiency disorders: general considerations. In: Stiehm ER, editor. Immunologic disorders in infants and children. Philadelphia: W B Saunders; 1996. p. 201-49.
- 12. Lee WI, Jaing TH, Hsieh MY, Kuo ML, Lin SJ, Huang JL. Distribution, infections, treatments and molecular analysis in a large cohort of patients with primary immunodeficiency diseases (PIDs) in Taiwan. J Clin Immunol. 2006;26(3):274-83.
- 13. Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94(5 Suppl 1):S1-63. Erratum in: Ann Allergy Asthma Immunol. 2006;96(3):504.
- 14. Smith CIE, Ochs HD, Puck JM. Genetically determined immunodeficiency diseases: A perspective. In: Ochs HD, Smith CI, Puck J, editors. Primary immunodeficiency diseases: a molecular and genetic approach. New York: Oxford University Press; 1999. p. 3-11.
- 15. Grumach AS, Silva Duarte AJ. Imunodeficiências primárias - noções gerais. In: Grumach AS, editor. Alergia e imunologia na infância e na adolescência. São Paulo: Atheneu; 2009. p. 519-32.
- 16. Roxo Júnior P, Menezes UP, Ferriani. VPL. Relative frequency between primary immunodeficiency groups in the primary immunodeficiency ward of Ribeirão Preto Clinics Hospital-USP, from 1994 to 2005. Clinics. 2005;60:55.
- 17. Sociedade Brasileira de Pediatria [homepage on the Internet]. Rio de Janeiro: Sociedade Brasileira de Pediatria. [cited 2008 May 6]. Quando pensar em Imunodeficiência Primária; [about 8 screens]. Available from: http://www.sbp.com.br/show_item2.cfm?id_categoria=89&id_detalhe=2667&tipo_detalhe=s
- 18. Shearer WT, Fischer A. The last 80 years in primary immunodeficiency: how far have we come, how far need we go? J Allergy Clin Immunol. 2006;117(4):748-52.
- 19. Bonilla FA, Geha RS. Are you immunodeficient? J Allergy Clin Immunol. 2005;116(2):423-5.
- 20. Roxo Júnior P, Costa Carvalho BT, Tavares FS. Infecções de repetição: o que é importante para o pediatra. Rev Paul Pediatr. In press 2009.
- 21. Casanova JL, Fieschi C, Bustamante J, Reichenbach J, Remus N, von Bernuth H, et al. From idiopathic infectious diseases to novel primary immunodeficiencies. J Allergy Clin Immunol. 2005;116(2):426-30.
- 22. de Moraes Lui C, Oliveira LC, Diogo CL, Kirschfink M, Grumach AS. Immunoglobulin G subclass concentrations and infections in children and adolescents with severe asthma. Pediatr Allergy Immunol. 2002;13(3):195-202.
- 23. Woroniecka M, Ballow M. Office evaluation of children with recurrent infection. Pediatr Clin North Am. 2000;47(6):1211-24.
- 24. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92(1):34-48.
- 25. Schur PH. IgG subclasses--a review. Ann Allergy. 1987;58(2):89-96, 99.
- 26. Heiner DC. Significance of immunoglobulin G subclasses. Am J Med. 1984;76(3A):1-6.
- 27. Hoeger PH, Niggemann B, Haeuser G. Age related IgG subclass concentrations in asthma. Arch Dis Child. 1994;70(3):179-82.
- 28. Plebani A, Monafo V, Ugazio AG, Monti C, Avanzini MA, Massimi P, et al. Comparison of the frequency of atopic diseases in children with severe and partial IgA deficiency. Int Arch Allergy Appl Immunol. 1987;82(3-4):485-6.
- 29. Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore). 2003;82(6):373-84.
- 30. Etzioni A, Eidenschenk C, Katz R, Beck R, Casanova JL, Pollack S. Fatal varicella associated with selective natural killer cell deficiency. J Pediatr. 2005;146(3):423-5.
- 31. Levy O, Orange JS, Hibberd P, Steinberg S, LaRussa P, Weinberg A, et al. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. J Infect Dis. 2003;188(7):948-53.
- 32. Ballas ZK, Turner JM, Turner DA, Goetzman EA, Kemp JD. A patient with simultaneous absence of "classical" natural killer cells (CD3-, CD16+, and NKH1+) and expansion of CD3+, CD4-, CD8-, NKH1+ subset. J Allergy Clin Immunol. 1990;85(2):453-9.
- 33. Sáfadi MA, Roxo Júnior P. Imunizações em pacientes imunocomprometidos. In: Roxo Júnior P, editor. Alergia e imunodeficiências em pediatria - abordagem prática. Ribeirão Preto: Tecmedd; 2006. p. 374-401.
- 34. Arkwright PD, Abinun M, Cant AJ. Autoimmunity in human primary immunodeficiency diseases. Blood. 2002;99(8):2694-702.
- 35. Chinen J, Anmuth D, Franklin AR, Shearer WT. Long-term follow-up of patients with primary immunodeficiencies. J Allergy Clin Immunol. 2007;120(4):795-7.
- 36. Roxo Júnior P, Ferriani VP, Grumach AS. Imunodeficiências primárias. In: Voltarelli JC, editor. Imunologia clínica na prática médica. São Paulo: Atheneu; 2009. p. 101-40.
- 37. Condino-Neto A, Costa Carvalho BT, Roxo Júnior P. Imunodeficiências de fagócitos. In: Roxo Júnior P, editor. Alergia e imunodeficiências em pediatria - abordagem prática. Ribeirão Preto: Tecmedd; 2006. p. 330-51.
- 38. Folz RJ, Routes JM. Pulmonary complications of organ transplantation and primary immunodeficiencies. In: Mason RJ, Murray JF, Broaddus VC, Nadel JA, editors. Textbook of respiratory medicine. Philadelphia: Saunders; 2005. p. 2163-99.
- 39. Nurden AT. Qualitative disorders of platelets and megakaryocytes. J Thromb Haemost. 2005;3(8):1773-82.
- 40. Roberts I, Stanworth S, Murray NA. Thrombocytopenia in the neonate. Blood Rev. 2008;22(4):173-86.
- 41. Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625-55.
- 42. Padeh YC, Rubinstein A, Shliozberg J. Common variable immunodeficiency and testing for HIV-1. N Engl J Med. 2005;353(10):1074-5.
- 43. Roxo Júnior P, Sorensen RU. Imunodeficiências predominantemente humorais. In: Roxo Júnior P, editor. Alergia e imunodeficiências em pediatria - abordagem prática. Ribeirão Preto: Tecmedd; 2006. p. 237-70.
- 44. Vasconcelos DM, Nudelman V. Imunodeficiências predominantemente celulares. In: Roxo Júnior P, editor. Alergia e imunodeficiências em pediatria-abordagem prática. Ribeirão Preto: Tecmedd; 2006. p. 271-307.
- 45. Prando-Andrade C, Agudelo-Florez P, Lopez JA, Paiva MA, Costa-Carvalho BT, Condino-Neto A. Doença granulomatosa crônica autossômica: relato de caso a análise genético-molecular de dois irmãos brasileiros. J. Pediatr (Rio J). 2004; 80(5):425-8.
- 46. Cunningham-Rundles C. Immune deficiency: office evaluation and treatment. Allergy Asthma Proc. 2003;24(6):409-15.
- 47. Simon HU, Späth PJ. IVIG--mechanisms of action. Allergy. 2003;58(7):543-52.
- 48. Pourpak Z, Aghamohammadi A, Sedighipour L, Farhoudi A, Movahedi M, Gharagozlou M, et al. Effect of regular intravenous immunoglobulin therapy on prevention of pneumonia in patients with common variable immunodeficiency. J Microbiol Immunol Infect. 2006;39(2):114-20.
- 49. Trigg ME, Schugar R. Chédiak-Higashi syndrome: hematopoietic chimerism corrects genetic defect. Bone Marrow Transplant. 2001;27(11):1211-3.
- 50. Booth C, Hershfield M, Notarangelo L, Buckley R, Hoenig M, Mahlaoui N, et al. Management options for adenosine deaminase deficiency; proceedings of the EBMT satellite workshop (Hamburg, March 2006). Clin Immunol. 2007;123(2):139-47.
- 51. Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296(5577):2410-3.
- 52. Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12(4):401-9.
- 53. Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117(4):725-38; quiz 739.
Publication Dates
-
Publication in this collection
06 Nov 2009 -
Date of issue
Oct 2009
History
-
Accepted
31 Mar 2009 -
Received
03 Feb 2009