Abstract
In this paper, we provide evidence that both the mRNA and protein levels of the cyclin-dependent kinase (CDK) inhibitor p21WAF1/CDK-interacting protein 1 (Cip1) increase upon infection of A431 cells with Vaccinia virus (VACV). In addition, the VACV growth factor (VGF) seems to be required for the gene expression because infection carried out with the mutant virus VACV-VGF- revealed that this strain was unable to stimulate its transcription. Our findings are also consistent with the notion that the VGF-mediated change in p21WAF1/Cip1 expression is dependent on tyrosine kinase pathway(s) and is partially dependent on mitogen-activated protein kinase/extracellular-signal regulated kinase 1/2. We believe that these pathways are biologically significant because VACV replication and dissemination was drastically affected when the infection was carried out in the presence of the relevant pharmacological inhibitors.
Vaccinia virus; virus-host cell interaction; p21WAF1/Cip1; MEK/ERK1/2; signal transduction
ARTICLES
Vaccinia virus regulates expression of p21WAF1/Cip1 in A431 cells
Anderson A AndradeI, II; Bruno SAF BrasilI, II; Anna CTC PereiraIII; Paulo CP FerreiraII; Erna G KroonII; Cláudio A BonjardimI, II, + + Corresponding author: claudio.bonjardim@pq.cnpq.br
IGrupo de Transdução de Sinal
IILaboratório de Vírus, Departamento de Microbiologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos 6627, 31270-901 Belo Horizonte, MG, Brasil
IIIUniversidade Federal do Piauí, Parnaíba, PI, Brasil
ABSTRACT
In this paper, we provide evidence that both the mRNA and protein levels of the cyclin-dependent kinase (CDK) inhibitor p21WAF1/CDK-interacting protein 1 (Cip1) increase upon infection of A431 cells with Vaccinia virus (VACV). In addition, the VACV growth factor (VGF) seems to be required for the gene expression because infection carried out with the mutant virus VACV-VGF- revealed that this strain was unable to stimulate its transcription. Our findings are also consistent with the notion that the VGF-mediated change in p21WAF1/Cip1 expression is dependent on tyrosine kinase pathway(s) and is partially dependent on mitogen-activated protein kinase/extracellular-signal regulated kinase 1/2. We believe that these pathways are biologically significant because VACV replication and dissemination was drastically affected when the infection was carried out in the presence of the relevant pharmacological inhibitors.
Key words: Vaccinia virus - virus-host cell interaction - p21WAF1/Cip1 - MEK/ERK1/2 - signal transduction
Vaccinia virus (VACV) is one of the largest animal double-stranded DNA viruses and replicates in the cytoplasm of host cells. It is the prototypic member of the Poxviridae family and its genome has the capacity to encode more than 250 gene products (Moss 2007). About half of the VACV genes encode immediate-early or early genes associated with the establishment of infection, regulatory proteins involved in transcription or DNA replication and proteins implicated in immune evasion or the spread of infection (Seet et al. 2003, Moss 2007). During viral replication, a phenomenon of localized cellular proliferation (hyperplasia) is observed (Burnet 1936) and is associated with the release of a soluble growth factor by the infected cells (Twardzik et al. 1985, Buller et al. 1988). This effect is attributed to the VACV growth factor (VGF), which shares amino acid sequence homology and functional properties with the cellular growth factors (EGF) and transforming growth factor beta (TGF-β) (Brown et al. 1985, Twardzik et al. 1985). VGF is highly conserved among different members of the Poxviridae family and it is sometimes associated with their oncogenicity (Tzahar et al. 1998). The mitogenic activity of VGF has been considered beneficial for viral replication because the VACV-VGF-deficient virus (VACV-VGF-) replicates better in proliferating cells than in quiescent ones (Buller et al. 1988), which is in line with the requirement of mitogen-activated protein kinase (MAPK)/extracellular-signal regulated kinase (ERK kinase) (MEK)/ERK for maximal viral replication (Andrade et al. 2004). Mitogenic signals triggered in A431 cells by VGF are mediated by the EGFR, which is tyrosine-phosphorylated (Y1173) (King et al. 1986, Tzahar et al. 1998) and activates phospholipase C-γ1 (Kim et al. 1995). Upon tyrosine-phosphorylation of phospholipase C-γ1, it promotes phosphoinositide breakdown to generate diacylglycerol and inositol triphosphate.
Exposure of A431 cells to EGF leads to phosphorylation of EGF receptor (EGFR) at Y1173 (Downward et al. 1984); coincidentally, the same tyrosine residue is activated upon VACV infection or exposure to VGF (King et al. 1986). Downstream targets of EGF signalling in A431 cells include protein kinase C (PKC), which is activated on threonine 654 (T654) as a consequence of phospholipid turnover (Hunter et al. 1984). VGF is also able to phosphorylate PKC on T654 (King et al. 1986). Thus, both EGF and VGF seem to share common signalling molecules to deliver intracellular signals.
Although EGF acts as a mitogen for a number of normal or transformed cell lines, it appears to arrest cell growth under certain circumstances, as is the case for A431 cells (Gill & Lazar 1981, Barnes 1982). These cells express a high number of EGFRs, which, upon EGF stimulation, undergo receptor hyper-phosphorylation followed by cell growth arrest (Gill & Lazar 1981). The mechanism associated with cell growth arrest appears to require the cyclin-dependent kinase (CDK) inhibitor (CKI) p21WAF1/cyclin-dependent kinase (CDK)-interacting protein (Cip1), which is regulated by both EGF and interferon-# (Chin et al. 1996, Zhou et al. 2002) and controls the transition from the G0/G1 to the S-phase during the cell cycle (el-Deiry et al. 1993, Sherr & Roberts 1999).
This study was undertaken to investigate whether p21WAF1/Cip1 expression is regulated upon VACV-wild-type VACV (WR) infection. Our findings are consistent with a role played by VACV in the regulation of gene expression. In addition, VGF of VACV appeared to control the expression of p21WAF1/Cip1 because infection with VACV-VGF- was unable to induce its expression. We further demonstrated that the same pathways associated with the expression of p21WAF1/Cip1 were also required for viral replication, emphasizing their biological role.
MATERIALS AND METHODS
Cells and reagents - Human squamous cell carcinoma A431 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 7.5% (v/v) heat-inactivated foetal bovine serum (FBS) (Cultilab, Campinas, SP, Brazil) and antibiotics in 5% CO2 at 37ºC. Cells were serum-starved in 0.5% FBS after reaching 80-90% confluence and incubated for 12-24 h. In order to collect supernatant from virus-infected cells [conditioned medium (CM)], serum-starved cells were infected at a multiplicity of infection (MOI) of 1 for 4 h. After the adsorption/penetration step (about 1 h at 37ºC), cultures were extensively washed with PBS to remove any remaining virus in the medium. The medium was replaced with 0.5% FBS and infection was allowed for additional 4 h, duration sufficient for the secretion of VGF into the medium. The supernatant was then collected and used in the experiments shown in Figs 1E, 2A, C at the volume of 2 mL per monolayer of 1 x 107 A431 cells. Prior to adding the CM to the monolayers, the CM was incubated with the monoclonal antibody 7D11 (1:1000) at 37ºC for 1 h to eliminate contaminant virus particles and then titrated for any residual virus contamination on BSC-40 cells. The 7D11 antibody recognizes L1R, a specific antigen present on the surface of intracellular mature virions (IMV) (Wolffe et al. 1995) and blocks its penetration in the host cell. The titres obtained after 7D11 antibody treatment were very low [15 ±10 pfu/mL (n = 3)] indicating the success of the treatment. The African green monkey (Cercopithecus aethiops)-derived fibroblast cell line BSC-40 was cultured in modified Eagle's medium (MEM) supplemented with 7% (v/v) (FBS) and antibiotics in 5% CO2 at 37°C. The p21WAF1/Cip1 message was detected using a specific probe as described previously (Cereseto et al. 1996). The anti-phospho ERK1/2 antibody was purchased from Cell Signalling Technology (Beverly, MA, USA) and the anti-p21WAF1/Cip1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The pharmacological inhibitors were purchased either from Calbiochem (La Jolla, CA, USA) or Sigma (São Paulo, Brazil). Interferon-γ was purchased from R&D Systems (MN, USA).
The doses of drugs used throughout the experiments were established on the basis of experimental observations without causing any harm to the cells, given that no measurable effect on cell viability was verified by trypan blue dye exclusion.
Viral propagation and infection- WR, mutant VACV-VGF- (vSC20) (Buller et al. 1988) and the recombinant VACV vF13L-green fluorescent protein (GFP) chimera (Husain & Moss 2001) were propagated in BSC-40 cells and highly purified by sucrose gradient sedimentation as described previously (Joklik 1962). This method was appropriated for the purification of IMV, which correspond to the majority of the infectious progeny formed during VACV infection (Moss 2007).
For VACV infections, A431 cells were grown until they reach 80-90% confluence. After reaching the desired confluence, cells were FBS deprived for 12 h and then infected with VACV at the indicated MOI for the times shown. When appropriate, cells were incubated with the indicated pharmacological inhibitor for 30 min prior to and throughout VACV infection.
Virus infectivity assays- A431 cells were cultured in a 6-well culture dish at a density of 4.5 x 105 cells per well and infected with VACV. Infections were carried out at a MOI of 10 (4.5 x 106 pfu/well) for 3, 6, 12, 24, 36 and 48 h. Cultures were then washed with cold PBS, followed by three freeze/thaw cycles. Virus was collected from the supernatant of centrifuged cells and then assayed for infectivity as described previously (da Fonseca et al. 2002).
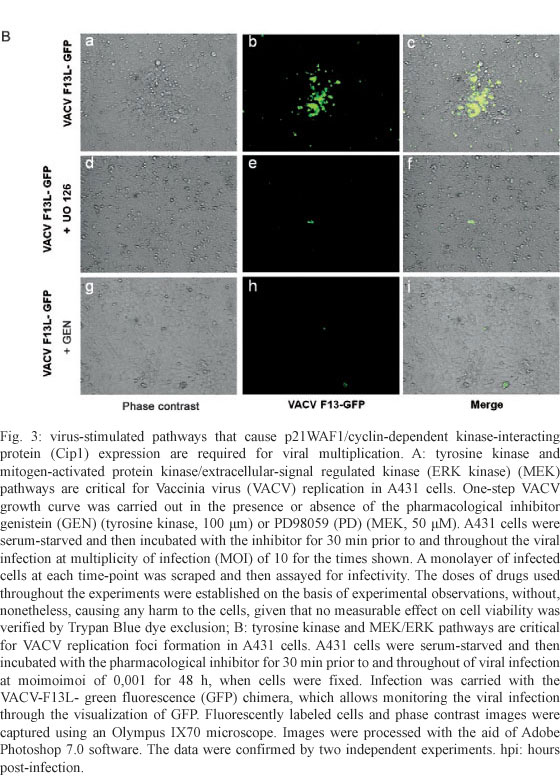
For infection foci imaging experiments, A431 cells were serum-starved and then incubated with the pharmacological inhibitor for 30 min prior to viral infection at a MOI of 0.001. Infection was allowed to proceed for 48 h in the continued presence of the inhibitor and then the cells were fixed. Infection was carried with the recombinant virus VACV-F13L-GFP, which allows for the monitoring of viral infection through the visualization of GFP. Fluorescently labelled cells and phase contrast images were captured using an Olympus IX70 microscope. Images were processed with the aid of Adobe Photoshop 7.0 software.
RNA isolation and Northern blotting - A431 cells (5 x 106) were cultured in 75 cm2 tissue culture flasks and starved as indicated above. Viral infections were carried out at a MOI of 1 for the times shown. At the indicated times, total RNA was isolated using the phenol-chloroform protocol (de Magalhães 2001) and 15 μg RNA per sample were loaded, eletrophoresed on a 1.5% denaturing agarose-formaldehyde gel, transferred onto nylon membranes (GE Healthcare), UV cross-linked for 5 min and hybridized with the p21WAF1/Cip1 probe. Probes were labelled with alpha[32P] dCTP (GE Healthcare) to a specific activity 1-5 x 108 cpm/μg DNA by using a multiprime DNA labelling system from GE Healthcare. For the internal control of RNA loading, the membranes were stripped of the probe and re-probed with oligonucleotide for 18S rRNA labelled at the 5' end with gammaATP (32P) by using T4 phage PNK (GE Healthcare).
Electrophoretic mobility shift assay (EMSA)- A431 cells were cultured and starved as above and then VACV infected at a MOI of 1.0 or incubated with IFN-γ (500 IU/mL) for the indicated times (De Souza et al. 2005). Briefly, frozen cell pellets were thawed on ice and lysed with an equal volume of lysis buffer (100 M Tris/HCl, pH 8.0, 0.2 mM EDTA, 1% Triton X-100, 10% glycerol, 5 mM sodium pyrophosphate, 4 μg/mL leupeptin and 1 mM sodium orthovanadate). Lysates were scraped and collected into Eppendorf tubes and then centrifuged at 13,000 g for 20 min at 4°C. The protein concentration was determined using a Bio-Rad assay. Protein (10 μg) was pre-incubated with 1.2 μL of poly(dI-dC)·(dI-dC) (5.4 mg/mL) (Amersham Biosciences) at RT (25°C) for 10 min, followed by the addition of 1.25 μg of BSA, 0.125 μg of Escherichia coli DNA, 0.25 μg of yeast tRNA, 2% Ficoll 400 and 0.32 ng of labelled probe. The reaction mixtures were incubated at RT for 15 min and then analysed by 6% PAGE. The 5'-32P-end-labelled double-stranded probes corresponds to the p21WAF1/Cip1 cis-acting elements (only one strand is shown): p21GAS: 5'-ATCTCCTTCCCGGAAGCA - 3' (11) and the consensus SRE: 5' - GATGTCCATATTAGGACATC - 3' (13) were used in the assays. The supershift experiments were carried out by incubation of the cell extracts with anti-p21 antibody for 1 h before mixing it with the labelled probe for 15 min. Competition assays were carried out by incubating a 50-fold molar excess of unlabelled homologous or unrelated probe with the proteins for 10 min before adding the labelled probe.
Immunoblotting- A431 cells (5 x 106) were cultured and starved as indicated above. Viral infections were carried out at a MOI of 1 for the times shown. Cells were then washed twice with cold PBS and lysed on ice with lysis buffer (100 mM Tris/HCl, pH 8.0, 0.2 mM
EDTA, 1% Triton X- 100, 10% glycerol, 5 mM sodium pyrophosphate, 4 μg/mL leupeptin and 1 mM sodium orthovanadate). Lysates were scraped and collected into Eppendorf tubes and then centrifuged at 13,000 g for 15 min at 4°C. Protein concentration was determined by using the Bio-Rad assay. For electrophoresis and immunoblotting, whole-cell lysates (25-30 μg) were separated by electrophoresis on an SDS/10% polyacrylamide gel and then transferred on to nitrocellulose membranes. Membranes were blocked overnight at 4°C with PBS containing 5% (w/v) non-fat dried milk and 0.1% (v/v) Tween 20. The membranes were washed three times with PBS containing 0.1% (v/v) Tween 20 and then incubated with the specific primary polyclonal antibody for p21/Cip1 (1:100) or anti total ERK1/2 (1:1500) in PBS containing 5% (w/v) BSA and 0.1% (v/v) Tween 20. After washing, the membranes were incubated with horseradish-peroxidase-conjugated secondary anti-rabbit antibody (1:3000 dilution). Immunoreactive bands were visualized by using an ECL® (enhanced chemiluminescence) detection system as described in the manufacturer's instructions (GE Healthcare).
RESULTS
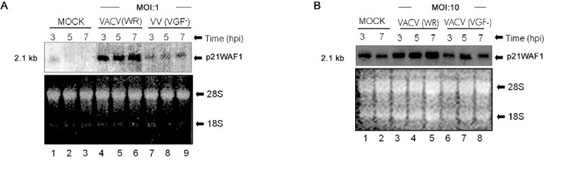
VACV stimulates the expression of p21WAF1/Cip1 in A431 cells - It has been shown that both VACV infection and purified VGF activate the EGFR on Y1173 in human A431 cells (King et al. 1986), which leads to the phosphorylation of phospholipase C-γ1 and an increase in phosphatidylinositol turnover (Kim et al. 1995). However, downstream targets of this pathway remain elusive. In order to evaluate the mRNA accumulation of the p21WAF1/Cip1 gene, Northern blot analysis of cells infected with either VACV (WR) or with the mutant virus VACV-VGF- at a MOI of 1 (+) or left uninfected (-)/MOCK for 3, 5 and 7 h was performed (Fig. 1A). Fig. 1A shows that VACV increases the transcription of p21WAF1/Cip1 to above basal levels between 3-7 h post-infection (hpi) (Fig. 1A, Lanes 4-6). Our results also demonstrate that the viral protein VGF is required for the transcription of p21WAF1/Cip1 because infection by a VGF- virus leads to only a minimal increase in the level of mRNA accumulation when compared with the WR infection (Fig. 1A, Lanes 7-9). A similar and slightly more intense pattern of p21/WAF1 mRNA accumulation was obtained in the experiments performed with a higher MOI (10), as shown in Fig. 1B. To extend these analyses to later times of infection, Northern blot assays of RNA samples obtained from MOCK or VACV-infected cells from 12-20 h were performed (Fig. 1C). As observed, after the initial increase in the p21WAF1/Cip1 message, infection with the wild-type virus was sufficient to maintain its elevated levels for at least 20 hpi (Fig. 1C, Lanes 1-10). Once again, similar results were obtained with a higher MOI (10), as shown in Fig. 1D.
Since VGF is secreted early during the VACV infection cycle (2 hpi), the next experiment was designed to investigate the role of this viral protein on VACV-stimulated p21WAF1/Cip1 expression. To this end, the CM was used to stimulate serum-starved A431 cells for different time periods. The CM corresponded to the supernatant collected from VACV-infected A431 monolayers for 4 h,
duration sufficient for the secretion of VGF into the medium. After being collected, the CM was submitted to anti-L1R antibody incubation for the neutralization of any residual virus. As observed, once again p21WAF1/Cip1 expression increased over basal levels at 3 h post-exposure to the CM (Fig. 1E, Lane 2) and was maintained for up to 5 h (Lane 4); however, it then declined to basal levels by 7 h (Lane 6). As a control, the same experiment was conducted with the CM obtained from uninfected cells and VGF-infected cells, and under these conditions, no increase in p21WAF1/Cip1 transcript was observed (data not shown).
To investigate whether the increased levels of p21WAF1/Cip1 transcript observed upon VACV infection were accompanied by an elevated production of its gene product, immunoblotting of A431 cells infected with either VACV (WR) or with the mutant virus VACV-VGF-, or were left uninfected (-) for the times shown (Fig. 1F). As shown in Fig. 1D (Lanes 2, 5, 8), the wild-type virus was sufficient to stimulate the accumulation of the gene product. Consistent with the requirement of VGF for VACV-stimulated expression of the p21WAF1/Cip1 message, VGF was equally necessary for the accumulation of the gene product, since p21WAF1/Cip1 was not detected upon infection with the mutant VACV-VGF- (Lanes 3, 6, 9).
A tyrosine kinase and MEK-dependent pathway is required for VACV-stimulated p21WAF1/Cip1 expression in A431 cells - Next, we investigated the signal transduction pathway(s) associated with the increase in p21WAF1/Cip1 transcription upon viral infection. To do so, cells were incubated with the pharmacological inhibitors genistein (GEN) (phosphotyrosine, 100 μM), bisindolylmaleimide (Bis) (PKC, 0.25 μM) or PD98059 (MEK, 50 μM) for 30 min prior to exposure to the CM for additional 4 h. After incubation, total RNA was extracted, electrophoresed and hybridized with labelled p21WAF1/Cip1 probe (Fig. 2A). As shown in Fig. 2A, VGF requires a tyrosine kinase-dependent pathway(s) to stimulate gene transcription, since incubation with GEN prior to exposure to the supernatant of VACV-infected cells (CM) caused the p21WAF1/Cip1 message to return to basal levels (Lane 4). In addition, the signal transduction pathway is partially dependent on MEK/ERK because pre-treatment with PD98059 resulted in an accentuated reduction in transcript accumulation (compare Lanes 2 and 8), whereas pharmacological inhibition of PKC Bis did not seem to play a relevant role in gene induction (Lanes 5, 6). In line with these observations, p21WAF1/Cip1 expression, upon VACV infection, did not seem to rely on PKC because treatment with the PKC inhibitor Bis caused an increase over basal levels in transcript accumulation (Fig. 2B, Lanes 5, 6). Pre-treatment with GEN and PD98059, on the other hand, reduced virus-stimulated gene transcription, as evidenced in Fig. 2B (compare Lanes 2, 3 and 7). In order to evaluate the effect of pharmacological inhibitors in p21WAF1/Cip1 protein expression, immunoblot analysis was performed (Fig. 2C). As observed, the signalling pathway(s) leading to the production of the gene product upon VGF exposure (CM) were also dependent on tyrosine kinase(s) (data not shown) and partially relied on MEK, as shown in Fig. 2C (compare Lanes 7 and 9, upper and middle panels).
VACV-stimulated p21WAF1/Cip1 expression is STAT-1-independent - Because both EGF and IFN-γ-stimulated p21WAF1/Cip1 expression levels are mediated via a STAT-1-dependent pathway (Chin et al. 1996, Johannessen et al. 1999), we asked whether viral infection could lead to STAT-1 activation, which, in turn, could act as a p21WAF1 gene activator. To address this question, cells were left uninfected (-), infected with VACV-WR (+) at an MOI of 1 for 12 h or incubated with IFN-γ (500 IU/mL) for 6 h (Fig. 2D). As observed through EMSA, shown in Fig. 2D, the exposure of A431 cells to IFN-γ caused STAT-1 (GAF) to associate with the GAS sequence present in the 5' regulatory region of p21WAF1/Cip1 (Lane 3), but VACV infection did not stimulate the formation of a GAS/GAF DNA/protein in complex (Lanes 1, 2). The identity and specificity of the complex formed upon IFN-γ stimulation was confirmed through competition (Lanes 5, 6) and super shift assays (Lane 7). Together these results show that STAT-1 is not the transcriptional activator present and is, consequently, not responsible for inducing p21WAF1 gene expression in VACV infected A431 cells.
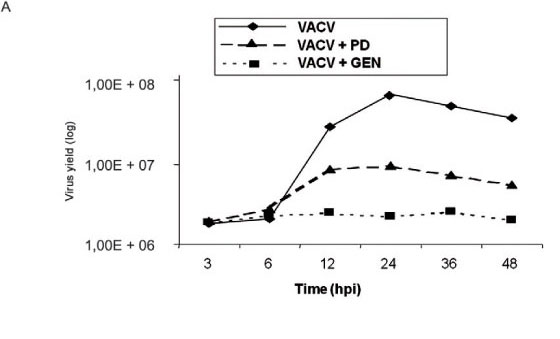
Virus-stimulated pathways leading to p21WAF1/Cip expression are required for viral replication - To investigate whether virus-stimulated pathways that cause p21WAF1/Cip expression are biologically relevant, a one-step viral growth curve was designed. A431 cells were serum-starved and then incubated either in the presence or absence of GEN (tyrosine kinase inhibitor, 100 μm) or PD98059 (MEK inhibitor, 50 μM) for 30 min prior to and throughout the viral infection at a MOI of 10 for the times shown. A monolayer of infected cells at each time-point was scraped and then assayed for infectivity. Fig. 3A shows that, in accordance with the requirement for a tyrosine kinase-dependent pathway(s), VACV multiplication was drastically affected. In line with the partial requirement of MEK/ERK for virus-stimulation of p21WAF1/Cip expression, PD98059 was also detrimental for VACV, although this effect was not as dramatic as that observed with GEN (Fig. 3A). Also, viral multiplication was not altered by the addition of the PKC inhibitor Bis (data not shown). In order to corroborate the requirement of tyrosine kinase and MEK/ERK pathways for VACV biology, the size of the foci of infection was monitored in A431 cells treated with GEN or PD98059 (Fig. 3B), as VACV plaques are not visualized in this cell line (King et al. 1986). In this assay we used the VACV-F13L-GFP chimera, which allows for the visualization of infected cells through the emission of GFP. As expected, treatment with both inhibitors greatly reduced the size of foci of viral infection, as demonstrated in Fig. 3B.
DISCUSSION
VACV infection appears to mitogenically sensitize the host cells and thus create the most adequate intracellular environment for the virus to propagate (Twardzik et al. 1985, King et al. 1986, de Magalhães et al. 2001). We have previously shown, using the nontransformed mouse fibroblast cell line A31, that both VACV and VGF are required to stimulate the MAPKs, which in turn lead to the transcriptional activation of growth-related genes, such as c-fos and early growth response gene (egr-1) (de Magalhães et al. 2001, Andrade et al. 2004). Moreover, activation of MEK/ERK and further expression of EGR-1 during the viral cycle have been proved to be beneficial for viral multiplication, as functional inactivation of EGR-1 by siRNA resulted not only in the reduction of viral yield but also in diminished viral plaque formation (Silva et al. 2006). Our data also showed that a tyrosine kinase-dependent pathway(s) is required to transmit the signals to MEK/ERK, which is a downstream target (Andrade et al. 2004).
The intracellular environment has also been shown to play a pivotal role in Orthopoxvirus biology (McFadden 2005). This assumption is based on the following observations: activation of MEK/ERK signalling pathway in mouse fibroblasts renders the cells favourable for viral multiplication, as occurs with VACV infection (Andrade et al. 2004) and it also restrains viral growth, as reported for the myxoma virus (Wang et al. 2004). In addition, since the availability of cellular resources, such as host factors required for the expression of intermediate and late-viral genes (Broyles 2003, Katsafanas & Moss 2004, Knutson et al. 2009), can be restrictive to viral multiplication, viral stimulation of cell cycle progression from a quiescent stage (Go/G1) to the S phase (DNA synthesis) seems to be the way that viral multiplication succeeds (Katz et al. 2005, Mo et al. 2009). In fact, VACV-VGF- replicates better in cycling cells than in quiescent ones (Buller et al. 1985) and VACV infection affects cell cycle progression by increasing the percentage of cells in the S phase (Wali & Strayer 1999).
In the present study, we investigated whether the EGF-regulated expression of p21WAF1/Cip1 in A431 cells (Chin et al. 1996, Toyoda et al. 1998) was also paralleled by VACV infection. Our data show that VACV regulated p21WAF1/Cip1 expression in a time and VGF-dependent manner (Fig. 1).
Our data also support the conclusion that virus-stimulated p21WAF1/Cip1 expression in A431 cells is mediated through a tyrosine kinase-dependent pathway(s) (Fig. 2A), in accordance with the requirement of Y1173 to activate EGFR, as reported by others (Downward et al. 1984). Evidence also showed that the expression of p21WAF1/Cip1 after VACV infection relied partially on the MEK/ERK pathway (Fig. 2 A-C), which is in line with our previous observation (Andrade et al. 2004) and those reported by others (Akashi et al. 1999). This observation diverges from EGF stimulation of p21WAF1/Cip1 in A431 cells, which is independent of MEK/ERK (Toyoda et al. 1998).
We also provide evidence that VACV-stimulated gene expression in A431 cells is independent of STAT-1 (Fig. 2D) because no GAS-GAF complex formed after viral infection. This observation is divergent from the EGF and IFN-γ-requirement of STAT-1 for p21WAF1/Cip1 expression via the GAS-GAF interaction (Chin et al. 1996). Moreover, VACV down-regulation of the IFN-γ-stimulated GAS-GAF complex formation (Fig. 2D, Lane 4) is consistent with viral inactivation of activated STAT-1 (Najarro et al. 2001) and viral secretion of the viral analogue of the IFN-γ receptor (Smith et al. 1997). VACV infection and IFN-γ stimulation of A431 cells appeared to simultaneously increase the mRNA level of CKI, suggesting that cooperative and yet independent pathways might be associated with the gene regulation (data not shown).
Thus, VACV-stimulated gene expression appears to be dependent on a tyrosine kinase-dependent, STAT-1-independent pathway, which partially recruits MEK/ERK to deliver downstream signals for gene expression. Because other cis-acting elements are found in the 5' regulatory region of p21WAF1/Cip1, such as AP-1, SIE and ISRE, among others (Chin et al. 1996), it is reasonable to assume that the signals are delivered to a regulatory sequence other than the GAS element. Our previous data investigating the association of c-fos expression with tyrosine kinase and MEK/ERK pathways upon VACV infection (de Magalhães et al. 2001, Andrade et al. 2004) provide support for this assumption. Moreover, VACV infection also activates c-Jun with kinetics that parallel those verified for c-fos expression (our unpublished observations) and both proteins associate to form the AP-1 complex, which in turn binds to the AP-1 regulatory sequence found in the promoter region of genes regulated via tyrosine kinase and MEK/ERK-dependent pathways. Thus, the expression of p21WAF1/Cip1 in A431 cells could be achieved via the recruitment of the AP-1 regulatory sequence. However, the detailed VACV-stimulated pathway(s) associated with p21WAF1/Cip1 expression, in this particular cell line, remains to be further characterized.
Irrespective of a positive or negative role of p21WAF1/Cip1 in cell cycle progression, the signalling pathways associated with CKI expression are similar to those that regulate viral replication. Thus, even if p21WAF1/Cip1 negatively regulates cell cycle progression, a circumstance that is unfavourable for viral replication, the signals conveyed by these pathways seem to activate downstream molecules that, collectively, could counteract p21WAF1/Cip1. This hypothesis is highly suggestive because pharmacological inhibition of MEK/ERK and protein tyrosine kinase(s) was found to be detrimental for viral replication and dissemination (Fig. 3) and is consistent with the reduced growth of VGF- VACV observed in quiescent cells (Buller et al. 1988 and data not shown). The results emphasize a biological role for these signalling pathways in ultimately creating the favourable conditions required for the generation of viral progeny.
Received 15 December 2009
Accepted 1 April 2010
Financial support: FAPEMIG, CAPES, CNPq (AAA, ACTCP and BSAFB were recipients of pre-doctoral fellowships; CAB, EGK and PCPF are recipients of research fellowships) AAA and BSAFB contributed equally to this work.
- Akashi M, Osawa Y, Koeffler HP, Hachiya M 1999. p21WAF1 expression by an activator of protein kinase C is regulated mainly at the post-transcriptional level in cells lacking p53: important role of RNA stabilization. Biochem J 337: 607-616.
- Andrade AA, Silva PN, Pereira AC, De Sousa LP, Ferreira PC, Gazzinelli RT, Kroon EG, Ropert C, Bonjardim CA 2004. The Vaccinia virus accinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem J 381: 437-446.
- Barnes DW 1982. Epidermal growth factor inhibits growth of A431 human epidermoid carcinoma in serum-free cell culture. J Cell Biol 93: 1-4.
- Brown JP, Twardzik DR, Marquardt H, Todaro GJ 1985. Vaccinia virus encodes a polypeptide homologous to epidermal growth factor and transforming growth factor. Nature 313: 491-492.
- Broyles SS 2003. Vaccinia virus transcription. J Gen Virol 84: 2293-2303.
- Buller RM, Chakrabarti S, Moss B, Fredrickson T 1988. Cell proliferative response to Vaccinia virus is mediated by VGF. Virology 164: 182-192.
- Buller RM, Smith GL, Cremer K, Notkins AL, Moss B 1985. Decreased virulence of recombinant Vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature 317: 813-815.
- Burnet FM 1936. The uses of the developing egg in virus research. Spec Rep Ser Med Res Counc 220: 555-556.
- Cereseto A, Diella F, Mulloy JC, Cara A, Michieli P, Grassmann R, Franchini G, Klotman ME 1996. p53 functional impairment and high p21waf1/cip1 expression in human T-cell lymphotropic/leukemia virus type I-transformed T cells. Blood 88: 1551-1560.
- Chin YE, Kitagawa M, Su WC, You ZH, Iwamoto Y, Fu XY 1996. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science 272: 719-722.
- da Fonseca FG, Trindade GS, Silva RL, Bonjardim CA, Ferreira PC, Kroon EG 2002. Characterization of a Vaccinia-like virus isolated in a Brazilian forest. J Gen Virol 83: 223-228.
- de Magalhaes JC, Andrade AA, Silva PN, Sousa LP, Ropert C, Ferreira PC, Kroon EG, Gazzinelli RT, Bonjardim CA 2001. A mitogenic signal triggered at an early stage of Vaccinia virus infection: implication of MEK/ERK and protein kinase A in virus multiplication. J Biol Chem 276: 38353-38360.
- De Sousa LP, Brasil BS, Silva BM, Freitas MH, Nogueira SV, Ferreira PC, Kroon EG, Bonjardim CA 2005. Plasminogen/plasmin regulates c-fos and egr-1 expression via the MEK/ERK pathway. Biochem Biophys Res Commun 329: 237-245.
- Downward J, Parker P, Waterfield MD 1984. Autophosphorylation sites on the epidermal growth factor receptor. Nature 311: 483-485.
- el-Deiry WS, Tokino T, Velculescu VE, Levy DV DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B 1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817-825.
- Gill GN, Lazar CS 1981. Increased phosphotyrosine content and inhibition of proliferation in EGF-treated A431 cells. Nature 293: 305-307.
- Hunter T, Ling N, Cooper JA 1984. Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature 311: 480-483.
- Husain M, Moss B 2001. Vaccinia virus F13L protein with a conserved phospholipase catalytic motif induces colocalization of the B5R envelope glycoprotein in post-Golgi vesicles. J Virol 75: 7528-7542.
- Johannessen LE, Knardal SL, Madshus IH 1999. Epidermal growth factor increases the level of the cyclin-dependent kinase (CDK) inhibitor p21/CIP1 (CDK-interacting protein 1) in A431 cells by increasing the half-lives of the p21/CIP1 transcript and the p21/CIP1 protein. Biochem J 337: 599-606.
- Joklik WK 1962. The purification of four strains of poxvirus. Virology 18: 9-18.
- Katsafanas GC, Moss B 2004. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J Biol Chem 279: 52210-52217.
- Katz RA, Greger JG, Skalka AM 2005. Effects of cell cycle status on early events in retroviral replication. J Cell Biochem 94: 880-889.
- Kim HS, Lee YH, Min DS, Chang JS, Ryu SH, Ahn BY, Suh PG 1995. Tyrosine phosphorylation of phospholipase C-gamma 1 by Vaccinia virus growth factor. Virology 214: 21-28.
- King CS, Cooper JA, Moss B, Twardzik DR 1986. Vaccinia virus growth factor stimulates tyrosine protein kinase activity of A431 cell epidermal growth factor receptors. Mol Cell Biol 6: 332-336.
- Knutson BA, Oh J, Broyles SS 2009. Downregulation of Vaccinia virus intermediate and late promoters by host transcription factor YY1. J Gen Virol 90: 1592-1599.
- McFadden G 2005. Poxvirus tropism. Nat Rev Microbiol 3: 201-213.
- Mo M, Fleming SB, Mercer AA 2009. Cell cycle deregulation by a poxvirus partial mimic of anaphase-promoting complex subunit 11. Proc Natl Acad Sci USA 106: 19527-19532.
- Moss B 2007. Poxviridae: the viruses and their replication. In DM Knipe, PM Howley (eds.), Fields virology, vol.2, 5th ed., Lippincott Williams & Wilkins, Philadelphia, p. 2905-2945.
- Najarro P, Traktman P, Lewis JA 2001. Vaccinia virus blocks gamma interferon signal transduction: viral VH1 phosphatase reverses Stat1 activation. J Virol 75: 3185-3196.
- Seet BT, Johnston JB, Brunetti CR, Barrett JW, Everett H, Cameron C, Sypula J, Nazarian SH, Lucas A, McFadden G 2003. Poxviruses and immune evasion. Annu Rev Immunol 21:377-423.
- Sherr CJ, Roberts JM 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13: 1501-1512.
- Silva PN, Soares JA, Brasil BS, Nogueira SV, Andrade AA, de Magalhaes JC, Bonjardim MB, Ferreira PC, Kroon EG, Bruna-Romero O, Bonjardim CA 2006. Differential role played by the MEK/ERK/EGR-1 pathway in orthopoxviruses Vaccinia and cowpox biology, Biochem J 398: 83-95.
- Smith GL, Symons JA, Khanna A, Vanderplasschen A, Alcami A 1997. Vaccinia virus immune evasion. Immunol Rev 159: 137-154.
- Toyoda M, Gotoh N, Handa H, Shibuya M 1998. Involvement of MAP kinase-independent protein kinase C signaling pathway in the EGF-induced p21(WAF1/Cip1) expression and growth inhibition of A431 cells. Biochem Biophys Res Commun 250: 430-435.
- Twardzik DR, Brown JP, Ranchalis JE, Todaro GJ, Moss B 1985. Vaccinia virus-infected cells release a novel polypeptide functionally related to transforming and epidermal growth factors. Proc Natl Acad Sci USA 82: 5300-5304.
- Tzahar E, Moyer JD, Waterman H, Barbacci EG, Bao J, Levkowitz G, Shelly M, Strano S, Pinkas-Kramarski R, Pierce JH, Andrews GC, Yarden Y 1998. Pathogenic poxviruses reveal viral strategies to exploit the ErbB signaling network. EMBO J 17: 5948-5963.
- Wali A, Strayer DS 1999. Infection with Vaccinia virus alters regulation of cell cycle progression. DNA Cell Biol 18: 837-843.
- Wang F, Ma Y, Barrett JW, Gao X, Loh J, Barton E, Virgin HW, McFadden G 2004. Disruption of Erk-dependent type I interferon induction breaks the Myxoma virus species barrier. Nat Immunol 5: 1266-1274.
- Wolffe EJ, Vijaya S, Moss B 1995. A myristylated membrane protein encoded by the Vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 211: 53-63.
- Zhou Y, Wang S, Yue BG, Gobl A, Oberg K 2002. Effects of interferon alpha on the expression of p21cip1/waf1 and cell cycle distribution in carcinoid tumors. Cancer Invest 20: 348-356.
Publication Dates
-
Publication in this collection
21 May 2010 -
Date of issue
May 2010
History
-
Accepted
01 Apr 2010 -
Received
15 Sept 2009