Abstract
A LC-ESI-MS/MS method was developed and validated according to the European Union decision 2002/657/EC, for the determination of tetracyclines (TCs) in chicken-muscle since Europe is one of the main markets for Brazilian products. Linearity of r > 0.9979, limits of quantification in the range of 7.0-35.0 ng/g, average recoveries of 89.38 - 106.27%, within-day and between-day precision were adequate for all TCs. The decision limit and the detection capability were 93.00-106.46 ng/g and 95.84-114.38 ng/g, respectively. This method is suitable for application in surveillance programmes of residues of TCs in chicken-muscle samples.
tetracyclines; chicken-muscle; validation
ARTIGO
Development and validation of a method for the analysis of tetracyclines in chicken-muscle by liquid chromatography-electrospray-mass spectrometry in tandem (LC-ESI-MS/MS)
Thais Matsue Uekane* * e-mail: thais.uekane@gmail.com ; Francisco Radler Aquino Neto; Luiz Nelson F. Gomes
Instituto de Química, Universidade Federal do Rio de Janeiro, Cidade Universitária Ilha do Fundão, 21941-909 Rio de Janeiro - RJ, Brasil
ABSTRACT
A LC-ESI-MS/MS method was developed and validated according to the European Union decision 2002/657/EC, for the determination of tetracyclines (TCs) in chicken-muscle since Europe is one of the main markets for Brazilian products. Linearity of r > 0.9979, limits of quantification in the range of 7.0-35.0 ng/g, average recoveries of 89.38 - 106.27%, within-day and between-day precision were adequate for all TCs. The decision limit and the detection capability were 93.00-106.46 ng/g and 95.84-114.38 ng/g, respectively. This method is suitable for application in surveillance programmes of residues of TCs in chicken-muscle samples.
Keywords: tetracyclines; chicken-muscle; validation.
INTRODUCTION
Antibiotics are widely used in veterinary medicine for disease control. They are administed intravenous, intra mammary/ intrauterine and as feed additives. Tetracycline antibiotics are commonly used all over the world with broad antibacterial spectrum and bacteriostatic activity, as medicines for humans and animals, and as feed additives since they are well absorbed, exhibit low toxicity and are relatively inexpensive.1-8
The most common tetracyclines (TC) applied to food-producing animals are tetracycline (TC), chlortetracycline (CTC), oxytetracycline (OTC) and doxycycline (DC) (Figure 1).9-13
The high and in some cases improper use of TC antibiotics, may result in the presence of residues in edible animal tissues, which can be toxic and dangerous for human health and potencially cause allergic reactions in some hypersensitive individuals. Moreover, some studies suggest adverse effect in human intestinal flora from the long-term presence of low doses of TC antibiotics. Resistance to these antibiotics is common, and may generate the evolution of microorganisms, that becomes resistant to one tetracycline and usually exhibits resistance to other types of antibiotics, thus representing a risk for human health.2,10,12-15
The Ministry of Agriculture in Brazil established a residue control program for meat, witch stated that antibiotics used for human therapy must be avoided as feed additives. To ensure the safety of food for consumers the National Health Surveillance Agency (ANVISA) in 2003, established the maximum residue limit (MRL) of 100 ng/g for TC, OTC and CTC for meat products. This limit was also established by different organisms such as European Union, Food and Agriculture Organization of the United Nations (FAO/WHO) and Food and Drug Administration (FDA). Chicken-muscle is the second feedstock more consumed in Brazil and of great importance for Brazil and exporting countries economies. The noncompliance of the quality demanded by importing countries can result in economic lost, thus monitoring residues of TC antibiotics in chicken muscle is essencial.14,16-18
The European Union Decision 2002/657/EC defines the performance criteria for validation of analytical methods for residues analysis in animal products. Since Europe is one of the main markets for Brazilian products, validation of methods for quality control according with this decision is relevant.19
TC antibiotics are biosynthetically produced, and as such, include a small percentage of impurities. The analysis of these residues in food can present several problems, once they have the tendency to bind irreversibly to the silanol groups on silicabased materials (C8, C18), due to the presence of the two ketone groups; form chelate complexes with divalent metal ions; combine with sample matrix proteins, can rapidly isomerize to form 4-epimers and anhydro tetracyclines. Epimers of TC antibiotics can be formed in aqueous conditions that are mildly acidic (pH 2-6), and it has been reported that this isomer is not formed in significant levels at pH 4 during the same day of extraction with buffer, nor when left in solution for up to 3 days at -20 ºC in the absence of light. Under strongly acidic conditions anhydroTC are formed by a loss of water (H on C-5a, HO on C-6) and proton transfer (O-11/O-12), thereby extending the aromatic nature of the D ring to include the C ring.9
Adequate treatment of samples is necessary to eliminate and/or reduce matrix interference, and is important to enrich the analyte of interest to obtain maximum measurement sensitivity. A deproteinizing agent is demanded as extracting solvent, such as buffers, acids or heat. Aqueous liquid extraction is a commonly technique employed for extraction of TC antibiotics in various matrices since provide greater solubility than most organic solvents, however has disadvantages of multiple extractions required, high consumption of time and reagents involved. Solid phase extraction (SPE) is commonly employed due to the TC antibiotics carbon backbone, aromatic region and varied functional groups for simultaneously cleanup and concentration of extracts, using several types of sorbents such as reversed phase, polymeric, phenyl, aminopropyl, cyane carbonyl, hydroxyl, and amino. Thus pretreatment of the extracs with aqueous liquid buffers before solid phase extractions techniques have been successfully applied to improve results of food based extraction.9,14,20-24
Several analytical procedures are available for the determination of TC antibiotics residues in animal tissues and chromatographic techniques are mainly used. High Performance Liquid Chromatography (HPLC) is a fast and reliable technique with high sensitivity for the analysis of TC antibiotics residues (identification, confirmation and quantification), varying its efficiency according to chromatographic conditions and type of detector used. Different detectors have been described for TC antibiotics analysis in chicken-muscle such as ultraviolet (UV), diode-array (DAD), fluorescence (FLD) and mass spectrometry (MS).9,20,23,25
The system of HPLC coupled with triple quadrupole mass analyzers in tandem (MS/MS) based on atmospheric pressure ionization (API) sources have been applied to the analysis of TC antibiotics in several foods, with a significant advantage for the absolute confirmation of these residues. The API techniques include electrospray (ESI), witch is suited to the introduction of polar and thermally labile compounds. ESI is a rapid, sensitive and selective analytical method for the determination of TC antibiotics in complex biological samples.5,14,26
Parameters that affect the sensitivity of ESI include mobile-phase additives, solution pH, flow rate, solvent composition and analyte concentration.27 Thus optimization of an extraction step and validation of a multi residue method, capable of quantifying TC antibiotics used worldwide in chicken-muscle is necessary.
The aim of this work was to develope and validate a method for identification, confirmation and quantification of TC antibiotics residues in chicken-muscle according to the European Union Decision 2002/657/EC using a sensitive and selective method such as LC-ESI-MS/MS.
EXPERIMENTAL
Chemicals and reagents
Acetonitrile, n-pentane, n-hexane, ethyl acetate and methanol HPLC grade and formic acid were Tedia Brazil (Rio de Janeiro, Brazil). Ethylenediaminetetraacetic acid disodium salt (EDTA Na2) was Reagen (Rio de Janeiro, Brazil), citric acid, hydrogen sodium phosphate and formic acid were Vetec (Rio de Janeiro, Brazil). High purity water was obtained by Milli-Q® purification system (Millipore, Bedford, MA, USA).
McIlvaine-Na2EDTA buffer 0.1 M was prepared by weighing 12.9 g of citric acid, 10.9 g hydrogen sodium phosphate and 37.18 g of EDTA Na2 to a 1 L of Milli-Q® water.28
Solid phase extraction (SPE) C18 cartridges (3 cc, 200 mg) were Varian (Walnut, Creek, CA, USA).
Analytical standard of Tetracycline, Chlortetracycline, Oxytetracycline, Doxycycline and Demeclocycline (DMCTC), used as internal standard (IS), were purchased from Sigma-Aldrich (St Louis, MO, USA). DMCTC was used as IS because it is an obsolete antibiotic (Figure 1).
Standard solutions
Stock standard solutions of TC, CTC, OTC, DC e DMCTC were prepared by dissolving each compound separetly in methanol to obtain a final concentration of 1.0 mg/mL. Stock standard solutions were stored at - 20 ºC in the dark, wrapped in aluminum foil. These standard solutions were diluted in methanol to give a series of working standard solutions that were storage at 4 ºC in the dark, wrapped in aluminum foil.
Experimental procedure
Chicken-muscle samples were obtained at local market, processed in a blender and analyzed for the presence of the five TC antibiotics. The blank chicken-muscles samples were homogenized to form a pool of chicken-muscle that was used for the development and validation experiments.
Sample preparation procedure
For sample preparation, a procedure was adapted and optmized from an article published by Oka and co-workers.12
An aliquot of 1.0 gram of chicken-muscle pool was placed in a centrifuge tube, fortified with standard solution, shaken with vortex for 30 s and left for 30 min at room temperature to ensure the appropriate distribution in the matrix. A volume of 4.0 mL of McIlvaine-Na2EDTA 0.1 M buffer (pH 4) was added, the mixture was shaken with vortex for 15 s at maximum speed and centrifuged at 2600 rpm for 5 min at 10 ºC. The supernatant was transferred to a clean centrifuge tube, extraction was repeated by adding a volume of 4.0 mL of McIlvaine-Na2EDTA 0.1 M buffer to the remaining chicken-muscle and centrifuged.
To the combined supernatants, a degreasing step was tested with n-pentane and n-hexane, a 4.0 mL of n-pentane or n-hexane was added to the combined supernatants and centrifuged at 2600 rpm for 15 min at 10 ºC. Both n-pentane and n-hexane showed similar recoveries gave clean extracts and are easy to use. n-Pentane was chosen for its high volatility, low cost, miscibility and relative safety.
The aqueous layer was applied to SPE C18, previously activated with methanol and Milli-Q water. After sample loading, the cartridge was washed with Milli-Q water and TC antibiotics were eluted with a mixture of ethyl acetate and methanol (95:5). The solvent was removed under a stream of nitrogen at 40 ºC, and the residue was dissolved in 100 µL of mobile phase. An aliquot (10 µL) was injected into the LC-ESI-MS/MS system.
LC-ESI-MS/MS
Analyses were performed with a 1200 L Varian LC-MS/MS triple quadrupole (Walnut, Creek, CA, USA). The mass spectrometer was equipped with an electrospray (ESI) interface operating in the positive mode. The LC was equipped with two mobile phase pumps (ProStar 210), a degassit on line, an auto sampler (ProStar 410) and a column thermostat. The ESI interface was calibrated using a polypropyleneglycol solution (PPG) provided by manufacturer, and ESI parameters were optimized for each TC by direct infusion of individually standard solution into the mass spectrometer.
The parameters of the mass spectrometer were: needle 5000 V, shield 600 V, housing 50 ºC, capillary voltage 50 V and detector voltage 1500 V. High purity nitrogen at 25 psi was used as nebulizer gas and as drying gas at 19 psi at 360 ºC; high purity argon was used as the collision gas at 2.0 mTorr. The mass spectrometer was operated in selective reaction monitoring (SRM) mode to confirm the identity of TCs in the samples by selecting specific precursor-to-product ion for each TC and to quantify using the most abundant transition.
Separations were conducted using a Pursuit C18 (100 x 2.0 mm I.D., 5 µm) column with a Polaris C18 (2.0 mm, 3 µm) guard column both Varian (Walnut, Creek, CA, USA) at 25 ºC. Mobile phase solvent A was water 0.1% formic acid and solvent B was acetonitrile 0.1% formic acid, at a flow rate of 0.3 mL/min, in a gradient that begins with 10% of solvent B, increases linearly in 3.0 min to 75% of solvent B maintaining this proportion for 2.5 min, then return to initial conditions in 9.0 min.
Validation
The European Union Decision 2002/657/EC defines the performance criteria for validation of analytical methods for residues analysis in animal products and was used for validation experiments of this study.
For specificity, ten blank chicken-muscles samples of different origins were separated, each tissue was divided in two aliquots, one aliquot was fortified at MRL for all TCs (100 ng/g) and internal standard at 200 ng/g and the other fortified only with internal standard at 200 ng/g, both aliquots were extracted and analyzed in parallel as described in above.
Linearity for TCs was assessed by matrix samples fortified with five different levels of concentration including zero, covering a range of 0-200 ng/g as recommended by 2002/657/EC decision; the concentration of the internal standard was constant through the tested range. Calibration curves were constructed with these samples, each one of them injected 3 times.
Limit of quantification (LQ) was assessed by calculating signal-to-noise of matrix samples fortified with different levels of concentration of standard solution. The concentration where the signal-to-noise was equal to 10 was considered the LQ.
Accuracy was expressed in terms of recovery of TCs from tissue pool at three levels of concentration (½ MRL, 1.0 MRL and 1.5 MRL) in triplicate. The concentration found in the chicken muscle was compared with the theoretical concentration of the fortified tissues.
Within-day precision was expressed by the relative standard deviation (%) of the analysis conducted in the same day in the same concentration as for recovery. Between-day precision was assessed by ANOVA test of the analysis conducted in two different days in the same concentration as for recovery.
The Decision 2002/657/EC included the calculation of decision limit (CCα) and detection capability (CCβ). The first is defined as the limit above which it can be concluded, with an error probability of α, that a sample is noncompliant (higher than the MRL), and the second is defined as the smallest content of the substance that may be detected, identified, and/or quantified in a sample with an error probability of β. For these calculations, blank samples were fortified at MRL, extracted and analyzed; CCα and CCβ were calculated using the Equations 1 and 2:19, 24

Stability of the analyte during storage is important since it may give rise to deviations in the outcome of the results of analysis. TC stability was determined in stock standard solution of 100 ng/mL prepared in methanol for each analyte. Aliquots of solution were storage at 4 ºC and -20 ºC in the dark, after analyzing one fresh aliquot. Aliquots from the solution were analyzed each week for two months and stability was determined when variation of 10% of the inital area was observed.29
RESULTS AND DISCUSSION
Through the direct infusion of TCs standard solutions, breakdown curves and TC precursor-to-product ions in the electrospray mass spectra (ESI MS) were obtained and collision energies were optimized to maximize the relative abundance for each transition ion, these were used for selective monitoring reaction (SMR) experiment. TCs in the positive ESI MS gave the molecular ion species [M + H - NH3]+ and [M + H - NH3 - H2O]+, except for DC. The loss of ammonia occurs from the carboxy-amide moiety in the A ring and the loss of ammonia and water depends on the presence of a hydroxy group in the C ring.12,21
According to the Decision 2002/657/EC, confirmatory methods for residues should provide information on the chemical structure of the analyte. LC with mass spectrometry detection methods requires four identification points, one precursor ion and two products ions meet these requirements.19 For confirmation of TCs it was monitored one precursor ion and three products ions, except for DC, from these transitions it was seleted the precursor-to-product ion that presented higher intensity for quantification purposes (Table 1).
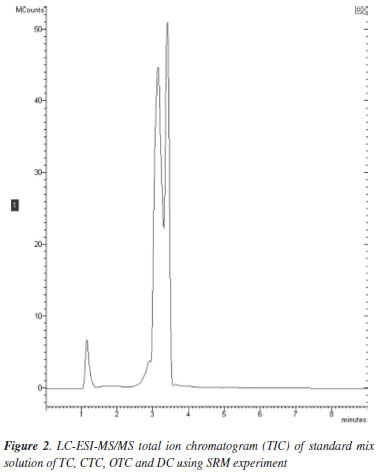
A TC mixture standard solution chromatogram did not show a full separation profile (Figure 2). TCs have retention time coincident. However, the use of SRM experiment allows unequivocal identification of these substances since it monitors precursor-to-product ion specific for each TC (Figure 3).
Method validation
The described method was fully validated according to 2002/657/EC decision. Since the creation of Codex Alimentarius several food guidelines were produced in order to protect consumer health, some of its validation parameters were also taken into consideration.30 Experimental results were checked for the presence of outliers through Grubbs test, none of the results were considered an outlier and all results were used for validation process.
Specificity
Specificity means the ability of a method to distinguish between the analytes being measured and other substances, this can vary according to class of compound or matrix. The presence of interfering peaks that could interfere on the identification and quantification of TCs was verified with the application of the whole procedure as described in section above.
A peak that appeared at retention time of approximately 1.0 min on transitions (precursor-to-product ion) of TC, DC and OTC (Figure 3) could not be identified due to the lack of standards, this peak could be due to impurities or degradation products of these standards, such epimers, however no interference from matrix around the retention time of the five TCs was observed, thus the method was considered specific for this analysis.
Linearity
Calibration curves were obtained by least-square linear regression analysis of the peak area ratio of the substance of interest to internal standard versus analyte concentration for each tetracycline in chicken-muscle samples. The analytical procedure was linear up to 200 ng/g for all TCs, since the determination coefficient (r) values were 0.99795, 0.99695; 0.99835 and 0.99859 for TC, CTC, OTC and DC respectively and were higher than recommended by National Institute of Metrology, Standardization and Industrial Quality (Inmetro, The Brazilian NMI).31 The MRL established by the National Health Surveillance Agency for TCs was in the center of this concentration range. LQ was below ½ MRL for all TCs and are shown in Table 2.
Accuracy and precision (within-day and between-day)
The average recovery and the relative standard deviation were calculated for each concentration and compared according to the European Union decision, this states that recovery must be in the range of 80-110% and according to Codex Alimentarius in the range of 70-110% for mass fraction between 10 and 100 ng/g and 80-110% for mass fraction > 100 ng/g.
Table 3 presents the mean recovery of this method for each TC at 50 ng/g (½ MRL), 100 ng/g (1.0 MRL) and 150 ng/g (1.5 MRL). Recovery values were considered satisfactory for chicken-muscle samples in all fortification levels and were in the range of 89.38 and 106.27%, similar to obtained from other matrices such as bovine, swine, shrimp, fish and milk.1,32-34
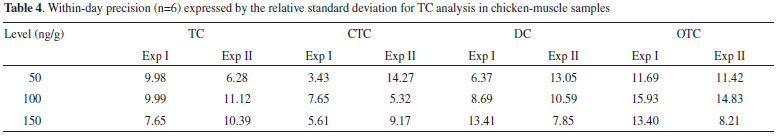
A relative standard deviation (RSD) up to 20% for within-day precision is acceptable according to European Union decision and Codex Alimentarius. Table 4 presents the within-day precision of the method at the 3 different levels of interest; these were considered satisfactory for all TCs.
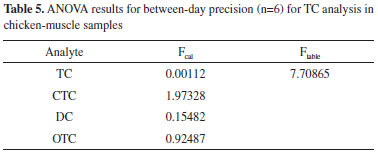
According to ANOVA test, between-day precision was equivalent since the Fcal values were lower than the Ftable values for all TCs on the two days of analysis for the three levels of concentration (½, 1.0, 1.5 MRL), thus this method has adequate between-day precision for all TCs (Table 5).
Decision limit (CCα) and detection capability (CCβ)
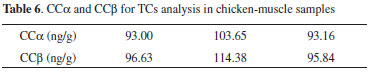
CCα and CCβ were calculated by spiking a pool of tissue samples at MRL using Equation 1 and 2 and are presented in Table 6. The values for CCα and CCβ were in accordance to other studies of TCs in other food matrices.21,24,34
Stability
The stability of the standard solutions in methanol was 2 weeks at 4 ºC in the dark for all TCs and 4 weeks at -20 ºC in the dark for all TCs, working standard solutions were renewed after 2 weeks of use and stock standard solutions were renewed after one month of use. Through this period it was observed on the chromatograms an emerging peak/an increase of the peak with retention time of approximately 1.0 min on transitions (precursor-to-product ion) of TCs, this could be due to the formation of epimers, however this was not confirmed due to the lack of standards.
CONCLUSIONS
The simple, fast, reliable and multiresidue method developed and validated for chicken-muscle samples showed good linearity (r >0.9979), LQ below the ½ MRL for all TCs, mean recovery over 80%, with adequate within-day and between-day precision, CCα and CCβ were 93.00-106.46 and 95.84-114.38 ng/g, respectively for all TCs according to European Union and Codex Alimentarius guidelines. This method is suitable for application in surveillance programmes of residues of tetracyclines in chicken-muscle samples and will be applied in the future on real samples.
ACKNOWLEDGEMENTS
The authors thank CNPq, FAPERJ and FUJB for the financial support for the development of this work.
REFERENCES
1. Bogialli, S.; Curini, R.; Di Corcia; A.; Lagana, A.; Rizzuti, G.; J. Agric. Food Chem. 2006, 54, 1564.
2. Denobile, M.; Nascimento, E. S.; Rev. Bras. Ciênc. Farm. 2004, 40, 209.
3. Michalova, E.; Novotna, P.; Schlegelova, J.; Vet. Med. 2004, 49, 79.
4. Cherlet, M.; Schelkens, M.; Croubels, S.; De Backer, P.; Anal. Chim. Acta 2003, 492, 199.
5. Nakazawa, H.; Ino, S.; Kato, K.; Watanabe, T.; Ito, Y.; Oka, H.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 1999, 732, 55.
6. Pena, A.; Carmona, A.; Barbosa, A.; Lino, C.; Silveira, I.; Castillo, B.; J. Pharm. Biomed. Anal. 1998, 18, 839.
7. Cooper, A. D.; Stubbings, G. W. F.; Kelly, M.; Tarbin, J. A.; Farrington, W. H. H.; Shearer, G.; J. Chromatogr., A 1998, 812, 321.
8. Oka, H.; Yoshimoto, I.; Ito, Y.; Hayakawa, J.; Harada, K.; Suzuki, M.; Odani, H.; Maeda, K.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 1997, 693, 337.
9. Anderson, C. R.; Rupp, H. S.; Wu, W. H.; J. Chromatogr., A 2005, 1075, 23.
10. Al-Ghamdi, M. S.; Al-Mustafa, Z. H.; El-Morsy, F.; Al-Faky, A.; Haider, I.; Essa, H.; Publ. Health 2000, 114, 300.
11. Anadón, A.; Martinez-Larrañaga, M. R.; Livest. Prod. Sci. 1999, 59, 183.
12. Oka, H.; Ito, Y.; Ikai, Y.; Kagami, T.; Harada, K.; J. Chromatogr., A 1998, 812, 309.
13. De Wasch, K.; Okerman, L.; Croubels, S.; De Brabander, H.; van Hoof, J.; De Backer, P.; Analyst 1998, 123, 2737.
14. Blasco, C.; Di Corcia, A.; Picó, Y.; J. Food Chem. 2009, 116, 1005.
15. Toldrá, F.; Reig, M.; Trends Food Sci. Technol. 2006, 17, 482.
16. Ministério da Agricultura, Pecuária e Abastecimento (MAPA); Lei nº 193 de maio de 1998 - Aprovar o regulamento técnico para o licenciamento e a renovação de licença de antimicrobianos de uso veterinário, anexo, elaborado pela Secretaria de Defesa Agropecuária.
17. Agência Nacional de Vigilância Sanitária (ANVISA); Resolução 899 de 29 de maio de 2003 - Guia para Validação de Métodos Analíticos e Bioanalíticos.
18. http://www.sindan.org.br/sd/sindan/index.html, acessada em Junho 2007 e Outubro 2010.
19. European Commission; Official Journal of the European Union L221, 8, 2002.
20. Schneider, M. J.; Braden, S. E.; Reyes-Herrera, I.; Donoghue, D. J.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2007, 846, 8.
21. Chico, J.; Rúbies, A.; Centrich, F.; Companyó, R.; Prat, M. D.; Granados, M.; J. Chromatogr., A 2008, 1213, 189.
22. Queiroz, S. C. N.; Collins, C. H.; Jardim I. C. S. F.; Quim. Nova 2001, 24, 68.
23. Oka, H.; Ito, Y.; Ikai, Y.; Kagami, T.; Harada, K.; J. Chromatogr., A 2000, 812, 309.
24. Samanidou, V. F.; Nikolaidou, K. I.; Papadoyannis, I. N.; J. Sep. Sci. 2005, 28, 2247.
25. Goto, T.; Ito, Y.; Yamada, S.; Matsumoto, H.; Oka, H.; J. Chromatogr., A 2005, 1100, 193.
26. Cooper, A. D.; Stubbings, G. W. F.; Kelly, M.; Tarbin, J. A.; Farrington, W. H. H.; Shearer, G.; J. Chromatogr., A 1998, 812, 321.
27. Kamel, A. M.; Brown, P. R.; Munson, B.; Anal. Chem. 1999, 71, 968.
28. Ruela, I. C. A.; Lima, J. A.; Souza, S. V. C.; Junqueira, R. G.; Ciênc. Tecnol. Aliment. 2005, 25, 139.
29. Cherlet, M.; Croubels, S.; De Backer, P.; J. Chromatogr., A 2006, 1102, 116.
30. Codex Alimentarius-Resíduos de Medicamentos Veterinários em los Alimentos, 2ª ed., Roma, 1993, vol 3.
31. Instituto Nacional de Metrologia. Normatização e Qualidade Industrial (INMETRO); Orientações sobre Validação de Métodos de Ensaios Químicos; DOQ-CGCRE-008, Revisão: 01 de março de 2003.
32. Andersen, W. C.; Roybal, J. E.; Gonzales, S. A.; Turnipseed, S. B.; Pfenning, A. P.; Kuck, L. R.; Anal. Chim. Acta 2005, 529, 145.
33. Schneider, M. J.; Darwish, A. M.; Freeman, D. W.; Anal. Chim. Acta 2007, 586, 269.
34. Cinquina, A. L.; Longo, F.; Anastasi, G.; Giannetti, L.; Cozzani, R.; J. Chromatogr., A 2003, 987, 227.
Recebido em 20/12/09; aceito em 8/7/10; publicado na web em 9/11/10
- 1. Bogialli, S.; Curini, R.; Di Corcia; A.; Lagana, A.; Rizzuti, G.; J. Agric. Food Chem. 2006, 54, 1564.
- 2. Denobile, M.; Nascimento, E. S.; Rev. Bras. Ciênc. Farm. 2004, 40, 209.
- 3. Michalova, E.; Novotna, P.; Schlegelova, J.; Vet. Med. 2004, 49, 79.
- 4. Cherlet, M.; Schelkens, M.; Croubels, S.; De Backer, P.; Anal. Chim. Acta 2003, 492, 199.
- 5. Nakazawa, H.; Ino, S.; Kato, K.; Watanabe, T.; Ito, Y.; Oka, H.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci 1999, 732, 55.
- 6. Pena, A.; Carmona, A.; Barbosa, A.; Lino, C.; Silveira, I.; Castillo, B.; J. Pharm. Biomed. Anal. 1998, 18, 839.
- 7. Cooper, A. D.; Stubbings, G. W. F.; Kelly, M.; Tarbin, J. A.; Farrington, W. H. H.; Shearer, G.; J. Chromatogr., A 1998, 812, 321.
- 8. Oka, H.; Yoshimoto, I.; Ito, Y.; Hayakawa, J.; Harada, K.; Suzuki, M.; Odani, H.; Maeda, K.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 1997, 693, 337.
- 9. Anderson, C. R.; Rupp, H. S.; Wu, W. H.; J. Chromatogr., A 2005, 1075, 23.
- 10. Al-Ghamdi, M. S.; Al-Mustafa, Z. H.; El-Morsy, F.; Al-Faky, A.; Haider, I.; Essa, H.; Publ. Health 2000, 114, 300.
- 11. Anadón, A.; Martinez-Larrañaga, M. R.; Livest. Prod. Sci. 1999, 59, 183.
- 12. Oka, H.; Ito, Y.; Ikai, Y.; Kagami, T.; Harada, K.; J. Chromatogr., A 1998, 812, 309.
- 13. De Wasch, K.; Okerman, L.; Croubels, S.; De Brabander, H.; van Hoof, J.; De Backer, P.; Analyst 1998, 123, 2737.
- 14. Blasco, C.; Di Corcia, A.; Picó, Y.; J. Food Chem 2009, 116, 1005.
- 15. Toldrá, F.; Reig, M.; Trends Food Sci. Technol 2006, 17, 482.
-
16Ministério da Agricultura, Pecuária e Abastecimento (MAPA); Lei nº 193 de maio de 1998 - Aprovar o regulamento técnico para o licenciamento e a renovação de licença de antimicrobianos de uso veterinário, anexo, elaborado pela Secretaria de Defesa Agropecuária.
-
17Agência Nacional de Vigilância Sanitária (ANVISA); Resolução 899 de 29 de maio de 2003 - Guia para Validação de Métodos Analíticos e Bioanalíticos.
-
18http://www.sindan.org.br/sd/sindan/index.html, acessada em Junho 2007 e Outubro 2010.
» link - 19. European Commission; Official Journal of the European Union L221, 8, 2002.
- 20. Schneider, M. J.; Braden, S. E.; Reyes-Herrera, I.; Donoghue, D. J.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2007, 846, 8.
- 21. Chico, J.; Rúbies, A.; Centrich, F.; Companyó, R.; Prat, M. D.; Granados, M.; J. Chromatogr., A 2008, 1213, 189.
- 22. Queiroz, S. C. N.; Collins, C. H.; Jardim I. C. S. F.; Quim. Nova 2001, 24, 68.
- 23. Oka, H.; Ito, Y.; Ikai, Y.; Kagami, T.; Harada, K.; J. Chromatogr., A 2000, 812, 309.
- 24. Samanidou, V. F.; Nikolaidou, K. I.; Papadoyannis, I. N.; J. Sep. Sci. 2005, 28, 2247.
- 25. Goto, T.; Ito, Y.; Yamada, S.; Matsumoto, H.; Oka, H.; J. Chromatogr., A 2005, 1100, 193.
- 26. Cooper, A. D.; Stubbings, G. W. F.; Kelly, M.; Tarbin, J. A.; Farrington, W. H. H.; Shearer, G.; J. Chromatogr., A 1998, 812, 321.
- 27. Kamel, A. M.; Brown, P. R.; Munson, B.; Anal. Chem. 1999, 71, 968.
- 28. Ruela, I. C. A.; Lima, J. A.; Souza, S. V. C.; Junqueira, R. G.; Ciênc. Tecnol. Aliment. 2005, 25, 139.
- 29. Cherlet, M.; Croubels, S.; De Backer, P.; J. Chromatogr., A 2006, 1102, 116.
- 30. Codex Alimentarius-Resíduos de Medicamentos Veterinários em los Alimentos, 2Ş ed., Roma, 1993, vol 3.
- 31. Instituto Nacional de Metrologia. Normatização e Qualidade Industrial (INMETRO); Orientações sobre Validação de Métodos de Ensaios Químicos; DOQ-CGCRE-008, Revisão: 01 de março de 2003.
- 32. Andersen, W. C.; Roybal, J. E.; Gonzales, S. A.; Turnipseed, S. B.; Pfenning, A. P.; Kuck, L. R.; Anal. Chim. Acta 2005, 529, 145.
- 33. Schneider, M. J.; Darwish, A. M.; Freeman, D. W.; Anal. Chim. Acta 2007, 586, 269.
- 34. Cinquina, A. L.; Longo, F.; Anastasi, G.; Giannetti, L.; Cozzani, R.; J. Chromatogr., A 2003, 987, 227.
Publication Dates
-
Publication in this collection
10 Feb 2011 -
Date of issue
2011
History
-
Received
20 Dec 2009 -
Accepted
08 July 2010