Resumo
The chronic myeloid leukemia (CML) is characterized by presence of the Philadelphia chromosome (Ph), originated from the translocation between chromosomes 9 and 22. This chromosome generates an abnormal protein tyrosine kinase which is responsible for tumor cell proliferation. The emergence of tyrosine kinase inhibitors (TKIs) has transformed the treatment of CML and imatinib being the first representative of this class. Although treatment with imatinib has reached surprising results, approximately 30% of patients exhibited resistance, especially in later stages of the disease. This fact stimulated the development of novel BCR-ABL enzyme inhibitors drugs classified as tyrosine kinase inhibitors (TKIs) of second and third generations. The TKIs have different chemical functions in their structure, and the knowledge of synthetic methods for preparation of these compounds can be a powerful tool for the development of new derivatives. The five approved BCR-ABL Tyrosine Kinase inhibitors (TKI) used in Chronic Myeloid Leukemia (CML) are reviewed aiming the main synthetic routes, highlighting the advantages and disadvantages associated with them.
Keywords:
chronic myeloid leukemia; tyrosine kinase inhibitors; imatinib mesylate
Keywords:
chronic myeloid leukemia; tyrosine kinase inhibitors; imatinib mesylate
INTRODUÇÃO
Câncer ou neoplasia maligna é um termo genérico para denominar um grande grupo de doenças que tem em comum o crescimento desordenado das células. Sua principal característica é a multiplicação rápida de células anormais, que invadem os tecidos e órgãos, podendo espalhar-se para outras regiões do corpo, sendo esse processo conhecido como metástase.11
http://www2.inca.gov.br/wps/wcm/connect/cancer/site/oquee, acessada em Janeiro de 2017.
http://www2.inca.gov.br/wps/wcm/connect/...
As neoplasias estão incluídas em um grupo chamado de doenças crônicas não transmissíveis (DCNT) que são doenças multifatoriais que ocorrem ao longo da vida e apresentam longa duração. Os principais fatores de risco associados às DCNTs são tabagismo, consumo nocivo de álcool, inatividade física e alimentação não saudável. Com relação aos determinantes sociais para a ocorrência destas doenças podem ser citados: o sexo, a genética e a idade.22
http://portalsaude.saude.gov.br/index.php/o-ministerio/principal/leia-mais-o-ministerio/671-secretaria-svs/vigilancia-de-a-a-z/doencas-cronicas-nao-transmissiveis/14125-vigilancia-das-doencas-cronicas-nao-transmissiveis, acessada em Janeiro de 2017.
http://portalsaude.saude.gov.br/index.ph...
Os tipos de câncer com maior ocorrência de óbitos na população mundial estão descritos na Tabela 1.
O câncer é uma das principais causas de morte do mundo, e aproximadamente 30% das mortes são devido a quatro principais fatores de risco: alto índice de massa corporal, redução da ingestão de frutas e legumes, falta de atividade física, elevado consumo de tabaco e de álcool.33
http://www.inca.gov.br/estimativa/2016/, acessada em Janeiro de 2017.
http://www.inca.gov.br/estimativa/2016/...
No Brasil, as previsões para 2016 apontam para uma ocorrência de aproximadamente 600 mil novos casos.44
http://globocan.iarc.fr/Pages/bar_dev_sel.aspx, acessada em Janeiro de 2017.
http://globocan.iarc.fr/Pages/bar_dev_se...
Leucemia
O termo leucemia origina-se do grego e significa sangue branco.55 Contran, R.; Kumar, V.; Collins, T.; Patologia Estrutural e Funcional, 6ª ed., Guanabara: Koogan, 2000. É uma neoplasia que tem como característica o acúmulo de células jovens anormais na medula óssea que substituem as células sanguíneas normais.66
http://www2.inca.gov.br/wps/wcm/connect/tiposdecancer/site/home/leucemia, acessada em Janeiro de 2017.
http://www2.inca.gov.br/wps/wcm/connect/...
No Brasil, para 2016, estima-se um número de óbitos de 10.070, sendo 5.540 homens e 4.530 mulheres.33
http://www.inca.gov.br/estimativa/2016/, acessada em Janeiro de 2017.
http://www.inca.gov.br/estimativa/2016/...
A leucemia é classificada de acordo com o tipo de célula acometida e com a velocidade de progressão da doença.77 Fauci, A. S.; Braunwald, E.; Kasper, D. L.; Hauser, S. L.; Longo, D. L.; Jameson, J. L.; Loscalzo, J.; Harrison's Principles of Internal Medicine, 17th ed., McGraw-Hill: New York, 2008. Se as células afetadas forem linfoides, linfócitos ou tecido linfoide em formação é denominada leucemia linfoide. Quando o alvo são as células mieloides (células que mais tarde se transformam em eritrócitos, leucócitos e megacariócitos) é denominada leucemia mieloide.66
http://www2.inca.gov.br/wps/wcm/connect/tiposdecancer/site/home/leucemia, acessada em Janeiro de 2017.
http://www2.inca.gov.br/wps/wcm/connect/...
De acordo com a progressão da doença ela pode ser classificada em crônica ou aguda. Na fase crônica as células ainda conseguem realizar as funções dos glóbulos brancos, replicando-se mais lentamente, e podendo funcionar por um período mais longo. Algumas formas de leucemia crônica não apresentam sintomas iniciais, não podendo ser diagnosticadas durante anos. A fase aguda inicia-se depois que alguns glóbulos brancos perdem ou danificam a sua sequência de ácidos desoxirribonucleicos (DNA) permanecendo imaturos. Estes são conhecidos como células blásticas, que ainda mantém a sua capacidade para se multiplicar. Estas células não morrem como as normais e, ao se acumularem, interferem no funcionamento de órgãos vitais. Além disso, não realizam as suas funções, pois se multiplicam mais rápido que o normal, piorando a doença em curto espaço de tempo. As leucemias agudas requerem um tratamento rápido e agressivo.
Assim, as leucemias podem ser classificadas em quatro tipos mais comuns: Leucemia Linfoide Crônica (LLC), comum em adultos com mais de 55 anos; Leucemia Linfoide Aguda (LLA), prevalente em crianças (2-5 anos); Leucemia Mieloide Crônica (LMC), que acomete principalmente adultos do sexo masculino e Leucemia Mieloide Aguda (LMA), que é comum em adultos e crianças.66
http://www2.inca.gov.br/wps/wcm/connect/tiposdecancer/site/home/leucemia, acessada em Janeiro de 2017.
http://www2.inca.gov.br/wps/wcm/connect/...
Leucemia Mieloide Crônica (LMC)
A LCM foi descrita pela primeira vez em 1845, por John Hughes Bennet.88 Joske, D. J.; Med. J. Aust. 2008, 1, 277. No final de 1960, os pesquisadores Nowell e Hungerford, da Universidade da Pensilvânia, identificaram um cromossomo anormal presente nas células leucêmicas humanas.99 Geary, C. G.; British J. Haematol. 2000, 110, 2. Este cromossomo, chamado de Philadelphia (Ph), é resultado da translocação entre os braços do cromossomo 9 e 22, sendo encontrado em 95% dos pacientes com LMC.1010 Nowell, P. C.; Hungerford, D. A.; J. Natl. Cancer Inst. 1961, 27, 1013.,1111 Druker, B. J.; Sawyers, C. L.; Kantarjian, H.; Resta, D. J.; Reese, S. F.; Ford, J. M.; Capdeville, R.; Talpaz, M.; N. Engl. J. Med. 2001, 344, 1038.
O cromossomo Ph gera uma proteína híbrida BCR-ABL que apresenta atividade enzimática anormal da tirosina quinase, responsável pela patogênese da doença.1212 Daley, G. Q.; Van Etten, R. A.; Baltimore, D.; Science 1990, 247, 824. Em condições normais a tirosina quinase regula diversas atividades fundamentais como o ciclo celular, proliferação, diferenciação, mobilidade e a sobrevivência ou morte celular.1313 Baselga, J.; Science 2006, 312, 1175.
Na fisiopatologia, o oncogene BCR-ABL é responsável pela ativação da proteína tirosina quinase. Ao se ligar com o trifosfato de adenosina (ATP) esta proteína transfere fosfato para resíduos de tirosina em proteínas específicas. Estas, após fosforilações, realizam todas as atividades fundamentais de uma tirosina quinase de modo anormal levando à ocorrência de LMC.1111 Druker, B. J.; Sawyers, C. L.; Kantarjian, H.; Resta, D. J.; Reese, S. F.; Ford, J. M.; Capdeville, R.; Talpaz, M.; N. Engl. J. Med. 2001, 344, 1038. Desta forma, o bloqueio da ligação entre a proteína e o ATP é essencial para a interrupção de todas as etapas subsequentes. A descoberta deste mecanismo foi fundamental para o desenvolvimento de uma terapia alvo para o tratamento de pacientes com LMC.1414 Farderl, S.; Kantarjian, H. M.; Talpaz, M.; O'Brien, S.; Semin. Oncol. 2000, 27, 578.
Terapia da LMC e seu histórico
O histórico dos tratamentos empregados para a LMC mostra que os primeiros compostos testados foram a quinina (Figura 1) e o ferro, porém, o uso destas substâncias não apresentou nenhum resultado.
Em 1865, o médico alemão Lissauer administrou baixas doses de arsênico combinadas com iodo e cloreto de potássio (KCl) em uma paciente. Esse tratamento provocou a diminuição do tamanho do baço, do número de células brancas e a melhora da anemia, restaurando a sensação de bem-estar.99 Geary, C. G.; British J. Haematol. 2000, 110, 2.
Em 1878, Cutler e Bradford estudaram os efeitos do arsênio no hemograma e na doença. Eles realizaram a contagem total dos glóbulos brancos e vermelhos do sangue e descobriram que a administração desta associação, conhecida como solução de Fowler, causava a diminuição progressiva do número de glóbulos vermelhos e brancos do sangue.1515 Cutler, E. G.; Bradford, E. H.; Am. J. Med. Sci. 1878, 75, 74. Mesmo assim, esta solução, independentemente de sua toxicidade, continuou sendo utilizada para o tratamento da LMC até a introdução da radioterapia, em 1903.99 Geary, C. G.; British J. Haematol. 2000, 110, 2.
Em 1903, Nicholas Senn observou que o uso de irradiação por raios-X provocava rápida diminuição no baço e redução no número de leucócitos. Isto porque os raios-X impediam a replicação celular e causavam a morte das células, promovendo a melhora no hemograma e no bem-estar do paciente.1616 Senn, N.; Med. Records New York 1903, 64, 281. Os benefícios desta terapia foram tão evidentes que, pela primeira vez, o termo "remissão" passou a ser utilizado. Contudo, o uso de raios-X não aumentou a sobrevida dos pacientes que na época era de até três anos.1717 Hoffman, W. J.; Craver, L. F.; J. Am. Med. Assoc. 1931, 97, 836.
Outra medida terapêutica utilizada foi a esplenectomia, que consiste na remoção cirúrgica, completa ou parcial, do baço. As primeiras tentativas de esplenectomia datam de 1863, e a maior parte dos pacientes submetidos à esta intervenção encontrava-se em estágios avançados da doença.1818 Bryant, T.; Guy's Hosp. Rep. 1866, 12, 444. Em 1879, Gowers afirmou que a esplenectomia era invariavelmente fatal; contudo, a cirurgia era realizada apenas para dar maior conforto aos pacientes. Posteriormente, alguns médicos recomendaram a esplenectomia após o tratamento com radioterapia.99 Geary, C. G.; British J. Haematol. 2000, 110, 2.
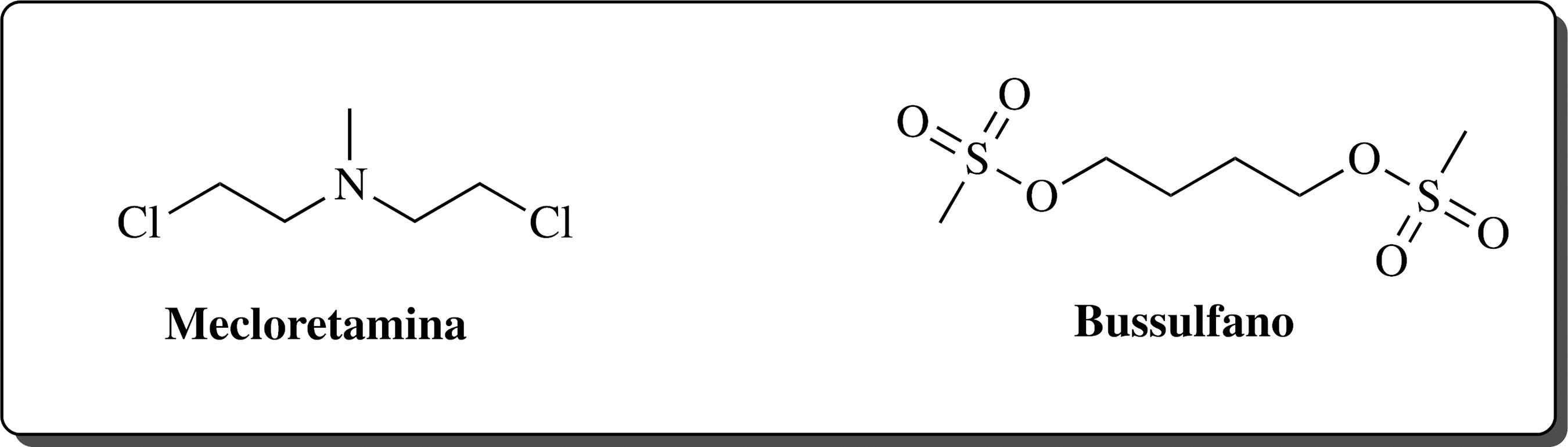
O primeiro agente antineoplásico utilizado no tratamento de LMC foi a mostarda nitrogenada (mecloretamina) (Figura 2). Desenvolvida em 1947, demonstrou que quando administrada por via intravenosa causava diminuição significativa no número de leucócitos. Embora a melhora clínica fosse evidente, não havia aumento da sobrevida dos pacientes.1919 Wintrobe, M. M.; Ann. Intern. Med. 1947, 27, 529.
Em 1952, foi introduzido na terapia o primeiro fármaco - bussulfano (Figura 2) - que produziu remissão hematológica em 42%, gerando sobrevida na fase crônica de até 3,8 anos. Ele apresentou maior seletividade para o tecido hematopoiético, particularmente nas séries granulocíticas, embora os ensaios clínicos mostrassem que era tóxico para o epitélio germinal e para os pulmões. Após vários estudos utilizando diferentes dosagens do bussulfano, verificou-se que ele era capaz de controlar as manifestações da LMC de modo mais eficiente que a radioterapia, especialmente porque poderia ser administrado por via oral. No entanto, seu uso foi suspenso devido aos graves efeitos adversos, tais como mielossupressão prolongada, fibrose pulmonar, mielofibrose e endomiocardiofibrose.2020 Osarogiagbon, U. R.; McGlave, P. B.; Curr. Opin. Hematol. 1999, 6, 241.
A hidroxiuréia foi outro fármaco que se destacou devido aos excelentes resultados alcançados (Figura 3). Seu uso clínico como fármaco de primeira escolha teve início em 1972, devido à resposta hematológica completa de até 80%, e sobrevida média de 4,7 anos na fase crônica. Seu mecanismo consiste na interrupção da síntese de DNA, através da inibição da enzima ribonucleotídeo-redutase.2121 Guimarães, J. R. Q.; Manual de Oncologia, 2ª ed., BBS: São Paulo, 2006. Quando comparado com o bussulfano, a hidroxiuréria mostrou-se menos tóxica; no entanto, ambas as terapias não produziram remissão citogenética ou prevenção da progressão da doença para fase blástica.2222 Hehlmann, R.; Heimpel, H.; Hasford, J.; Kolb, H. J.; Pralle, H.; Hossfeld, D. K.; Queisser, W.; Löffler, H.; Hochhaus, A.; Heinze, B.; Blood 1993, 82, 398.
No início dos anos 80, o interferon alfa foi empregado e promoveu a remissão citogenética parcial, que consiste na redução do número de cromossomos Ph. Além disso, o uso do interferon alfa foi essencial para mostrar sua capacidade de aumentar a sobrevida do paciente.2323 Talpaz, M.; Semin. Hematol. 2001, 38, 22.,2424 Kantarjian, H.; O'Brien, S.; Cortes, J.; Shan, J.; Giles, F.; Garcia-Manero, G.; Verstovsek, S.; Faderl, S.; Rios, M. B.; Talpaz, M.; Cancer 2003, 98, 1430. Por não ser um tratamento curativo, os pacientes que apresentavam remissão citogenética eram mantidos na terapia, com doses de interferon por três vezes na semana, resultando em uma sobrevida de 5,5 anos.2222 Hehlmann, R.; Heimpel, H.; Hasford, J.; Kolb, H. J.; Pralle, H.; Hossfeld, D. K.; Queisser, W.; Löffler, H.; Hochhaus, A.; Heinze, B.; Blood 1993, 82, 398. Segundo Kantarjian, dos 274 pacientes tratados diariamente com interferon alfa, 80% alcançaram remissão hematológica completa (RHC) e 58% remissão citogenética completa (RCC), com sobrevida média de 7,4 anos.2424 Kantarjian, H.; O'Brien, S.; Cortes, J.; Shan, J.; Giles, F.; Garcia-Manero, G.; Verstovsek, S.; Faderl, S.; Rios, M. B.; Talpaz, M.; Cancer 2003, 98, 1430.,2525 Kantarjian, H. M.; J. Clin. Oncol. 1992, 10, 772. No entanto, as reações adversas levaram ao abandono do tratamento em cerca de 20% dos casos.2626 Hahn, E. A.; Glendenning, G. A.; Semin Hematol. 2003, 40, 31. Diversos fármacos foram testados em associação com o interferon alfa.2424 Kantarjian, H.; O'Brien, S.; Cortes, J.; Shan, J.; Giles, F.; Garcia-Manero, G.; Verstovsek, S.; Faderl, S.; Rios, M. B.; Talpaz, M.; Cancer 2003, 98, 1430.,2525 Kantarjian, H. M.; J. Clin. Oncol. 1992, 10, 772. Com a hidroxiuréia observou-se aumentou significativo da sobrevida dos pacientes, quando comparado ao uso da hidroxiuréia como monoterapia. Adicionalmente, foi alcançada remissão hematológica completa em 59% dos casos e 12% de remissão citogenética completa.2222 Hehlmann, R.; Heimpel, H.; Hasford, J.; Kolb, H. J.; Pralle, H.; Hossfeld, D. K.; Queisser, W.; Löffler, H.; Hochhaus, A.; Heinze, B.; Blood 1993, 82, 398. A associação com a citarabina (Figura 3) aumentou a sobrevida em 86%, quando comparada aos pacientes que mantiveram o uso de interferon alfa em monoterapia. Entretanto, a inclusão deste fármaco na politerapia aumentou a toxicidade do tratamento.2727 Guilhot, F.; Chastang, C.; Michallet, M.; N. Engl. J. Med. 1997, 337, 223.
O transplante de célula tronco hematopoiética (TCTH) - efetivo no tratamento da LMC por ser capaz de produzir resposta curativa - começou a ser utilizado em 1986; porém a idade dos pacientes - maior que 60 anos em 55% dos casos - e a falta de doadores compatíveis tornam essa modalidade terapêutica restrita.2828 Gratwohl, A.; Hermans, J.; Niederwieser, D.; Frassoni, F.; Arcese, W.; Gahrton, G.; Bandini, G.; Carreras, E.; Vernant, J. P.; Bosi, A.; Bone Marrow Transplant. 1993, 12, 509.
Nos últimos anos foi observada uma revolução no tratamento da LMC com o surgimento de uma nova classe de inibidores de tirosina quinase (ITQ).2929 Romeiro, N. C.; Avila, C. M.; Rev. Virtual Quim. 2010, 2, 59. Atualmente, estão disponíveis cinco ITQs: imatinibe, dasatinibe, nilotinibe, bosutinibe e ponatinibe. Uma ilustração do mecanismo de inibição da enzima tirosina quinase BCR-ABL pelos ITQs está descrita na Figura 4.
O primeiro inibidor específico da tirosina quinase, a tirfostina (1), foi descrito em 1988 por Yaish & colaboradores.3030 Yaish, P.; Gazit, A.; Gilon, C.; Levitzki, A.; Science 1988, 242, 933. Posteriormente, alguns estudos demonstraram que o esqueleto fenilaminopirimidina (FAP) (2) apresentava elevado potencial para inibir, de forma não específica, a quinase3131 Druker, B. J.; Lydon, N. B. Clin. Invest. 2000, 105, 3.,3232 Choi, H. G.; Sim, T.; Gray, N.; Zhou, W.; Chang, J. W.; Zhang, J.; Weisberg, E.; WO 144909 2010. (CA 154:64811) (Figura 5).
Em 1993, Druker e colaboradores sintetizaram e testaram uma série de moléculas contendo o esqueleto FAP,3333 Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A.; Nat. Rev. Drug Discovery 2002, 1, 493. dentre elas o imatinibe, que revelou ser capaz de eliminar de forma seletiva as células da LMC.3434 Goldman, J. M.; Lancet 2000, 355, 1031. Com o avanço dos testes clínicos, este fármaco passou a ser considerado como um "milagre". Estatísticas demonstraram que quase 100% dos pacientes tratados com o mesilato de imatinibe alcançavam a remissão total da doença.3535 Hunter, T.; J. Clin. Invest. 2007, 117, 2036.
O mesilato de imatinibe (Glivec®) marcou o início da era dos "tinibes",3636 Lima, L. M.; Quim. Nova 2007, 30, 1456. embora posteriormente, alguns pacientes apresentassem resistência a ele. Este fato mostrou a necessidade de se desenvolver novos fármacos desta classe, surgindo então os inibidores de segunda e terceira gerações, os quais atuam em mais de uma quinase sendo chamados inibidores "multi-quinases".
Inibidores de Tirosina Quinase
Considerando-se a relevância desta classe terapêutica e visando dar suporte aos futuros estudos de novos inibidores, este trabalho teve como objetivo revisar a literatura dos ITQs utilizados na terapêutica da LMC, bem como mostrar as principais rotas sintéticas para as suas obtenções.
A primeira geração de ITQs é representada pelo imatinibe, a segunda pelo dasatinibe e nilotinibe e a terceira pelo bosutinibe e ponatinibe3737 Quintás-Cardama, A.; Kantarjian, H.; Cortes, J.; Semin. Hematol. 2010, 47, 371. (Figura 6).
Imatinibe
O mesilato de imatinibe (STI571) - desenvolvido pela indústria suíça Novartis e aprovado pelo Food and Drug Administration (FDA) em 20013838 Pinto, A. C.; Barreiro, E. J.; Quim. Nova 2013, 36, 1557; Barreiro, E. J.; Pinto, A. C.; Rev. Virtual Quim. 2013, 5, 1059. - é um inibidor competitivo do sítio do ATP na enzima BCR-ABL, bloqueando sua atividade tirosina quinase ao impedir que o substrato seja fosforilado. Este bloqueio previne a transdução de sinais de energia necessários para a proliferação celular e apoptose. Além disso, o imatinibe inibe a proliferação das células de diferentes linhagens da LMC e das células progenitoras hematopoiéticas.3939 Savage, D. G.; Antman, K. H.; N. Engl. J. Med. 2002, 346, 683.
O imatinibe é administrado por via oral com doses entre 300 e 800 mg/dia. Pode ser utilizado na terapêutica de pacientes na fase aguda e na fase acelerada. No entanto, os melhores resultados são observados em pacientes recém-diagnosticados com a doença.4040 O'Brien, S. G.; Guilhot, F.; Larson, R. A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J. J.; Fischer, T.; Hochhaus, A.; Hughes, T.; N. Engl. J. Med. 2003, 348, 994.
Este fármaco tem uma rápida absorção (duas horas) quando administrado em altas doses, e sua biodisponibilidade é de 98%. Seu metabolismo é hepático, sendo realizado pela enzima citocromo P450 - isoforma CYP3A4 - e em menor extensão por outras isoformas. O objetivo do tratamento com imatinibe é encontrar uma resposta hematológica completa, alcançando como resultados uma contagem de leucócitos menor que 10x109/L, contagem de plaquetas menor de 450x109/L e ausência de células imaturas como mielócitos, promielócitos ou mieloblásticas no sangue periférico. Além disso, espera-se a eliminação dos sintomas da doença como, por exemplo a esplenomegalia.4141 Moen, M. D.; Mckeage, K.; Plosker, G. L.; Siddiqui, M. A.; Drugs 2007, 67, 299.
Embora o tratamento da LMC com imatinibe tenha alcançado resultados fantásticos, aproximadamente 20 a 30% dos pacientes têm apresentado resistência.4242 Apperley, J. F.; Lancet Oncol. 2007, 8, 1018. A resistência pode ser primária, quando desde o início o paciente não apresenta resposta alguma, ou pode ser resistência secundária, quando o paciente diminui ou não alcança mais resultados ao longo do tratamento.4343 Mahon, F. X.; Deininger, M. W. N.; Schultheis, B.; Chabrol, J.; Reiffers, J.; Goldman, J. M.; Melo, J. V.; Blood 2000, 96, 1070.
Os mecanismos de resistência podem ser explicados através das mutações no domínio quinase da proteína BCR-ABL, amplificação genética e super expressão do gene BCR-ABL, alteração da expressão de proteínas transmembranares de influxo e de efluxo e alterações na regulação de mecanismos de transdução de sinal.4444 Silva, V. P. N.; Dissertação de Mestrado, Universidade Nova de Lisboa, Portugal, 2011. A mutação no domínio quinase BCR-ABL - denominada de T315I - é a mais temida, visto que somente o ponatinibe pode ser utilizado para tratar esta resistência.4545 Gorre, M. E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P. N.; Sawyers, C. L.; Science 2001, 293, 876.,4646 Melo, J. V.; Chuah, C.; Cancer Lett. 2007, 249, 121.
Síntese do imatinibe
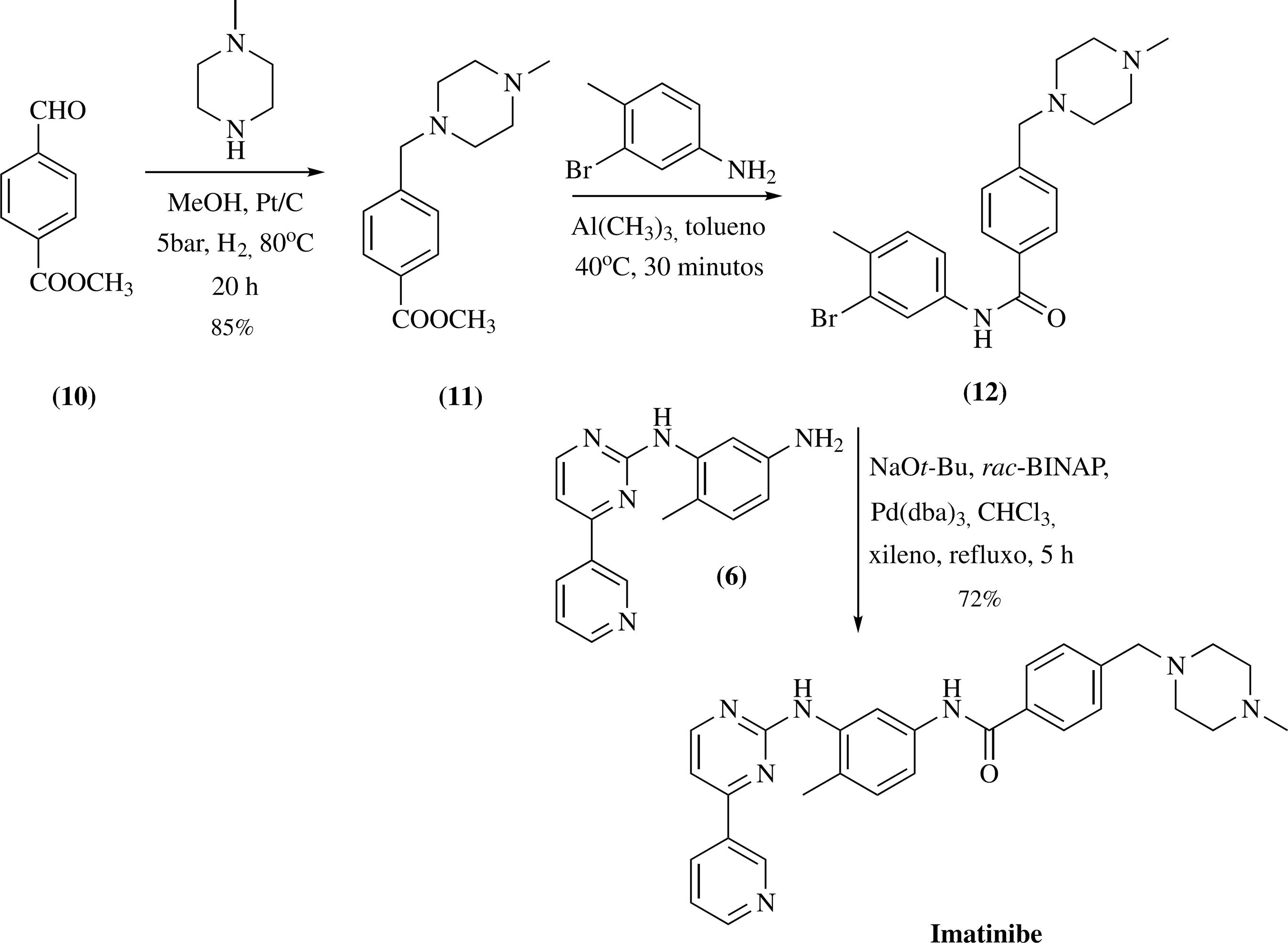
O primeiro processo para a obtenção do imatinibe é uma síntese convergente entre os intermediários 4 e 7, e foi descrito por Zimmermann e colaboradores.4747 a) Zimmermann, J.; EP0564409 1993. (CA120:107056); Zimmermann, J.; US5521184 1996. (CA125:114681) Neste trabalho, as principais limitações para a sua produção industrial são o uso da cianamida para a obtenção do derivado de guanidina 4, a necessidade de três etapas reacionais e o uso do sódio para a síntese do intermediário 7, que atualmente pode ser obtido em uma única etapa. Os rendimentos de todas as etapas desta rota sintética não foram descritos no documento original.
Recentemente, Boechat e colaboradores realizaram modificações na metodologia original.4848 Boechat, N.; Bastos, M. M.; Duarte, S. L.; Costa, J. C. S.; Mafra, J. C. M.; Daniel, L. C. C.; Rev. Virtual Quím. 2013, 5, 222. As principais vantagens deste método foram a obtenção do intermediário 7 em uma única etapa, e redução do grupo nitro do intermediário 5 para 6 através do sistema Fe/NH4Cl, em etanol e água, evitando assim o uso do Pd/C (Esquema 1). Após a avaliação experimental deste processo, verificou-se que a metodologia sintética é capaz de produzir o imatinibe em bom rendimento global e dentro das especificações farmacopéicas.
Metodologia de síntese original para a obtenção do imatinibe aperfeiçoada por Boechat e colaboradores
Posteriormente, os grupos de pesquisa de Kompella4949 Kompella, A.; Bhujanga, A. K. S. R.; Venkaiah, N. C.; Srinivas, R.; WO04108699 2004. (CA142:56339) e Szakacs5050 Szakács, Z.; Béni, S.; Varga, Z.; Orfi, L.; Kéri, G.; Noszál, B.; J. Med. Chem. 2005, 48, 249. propuseram melhorias na rota sintética descrita por Zimmermann e colaboradores, que permitiram a obtenção do imatinibe em bom rendimento global.
Em 2003, Loiseleur e colaboradores propuseram uma rota alternativa para a obtenção deste fármaco.5151 Loiseleur, O.; Kaufmann, D.; Abel, S.; Buerger, H. M.; Meisenbach, M.; Schmitz, B.; Sedelmeier, G.; WO03066613 2003. (CA139:180080) Os autores evitaram o uso da cianamida; no entanto, foi empregado o trimetilalumínio, que é altamente inflamável, e um catalisador à base de paládio (Pd), para a penúltima e última etapa sintética, respectivamente (Esquema 2). Estas condições reacionais não viabilizaram a aplicabilidade desta rota sintética em escala industrial. O rendimento da penúltima etapa não foi descrito.
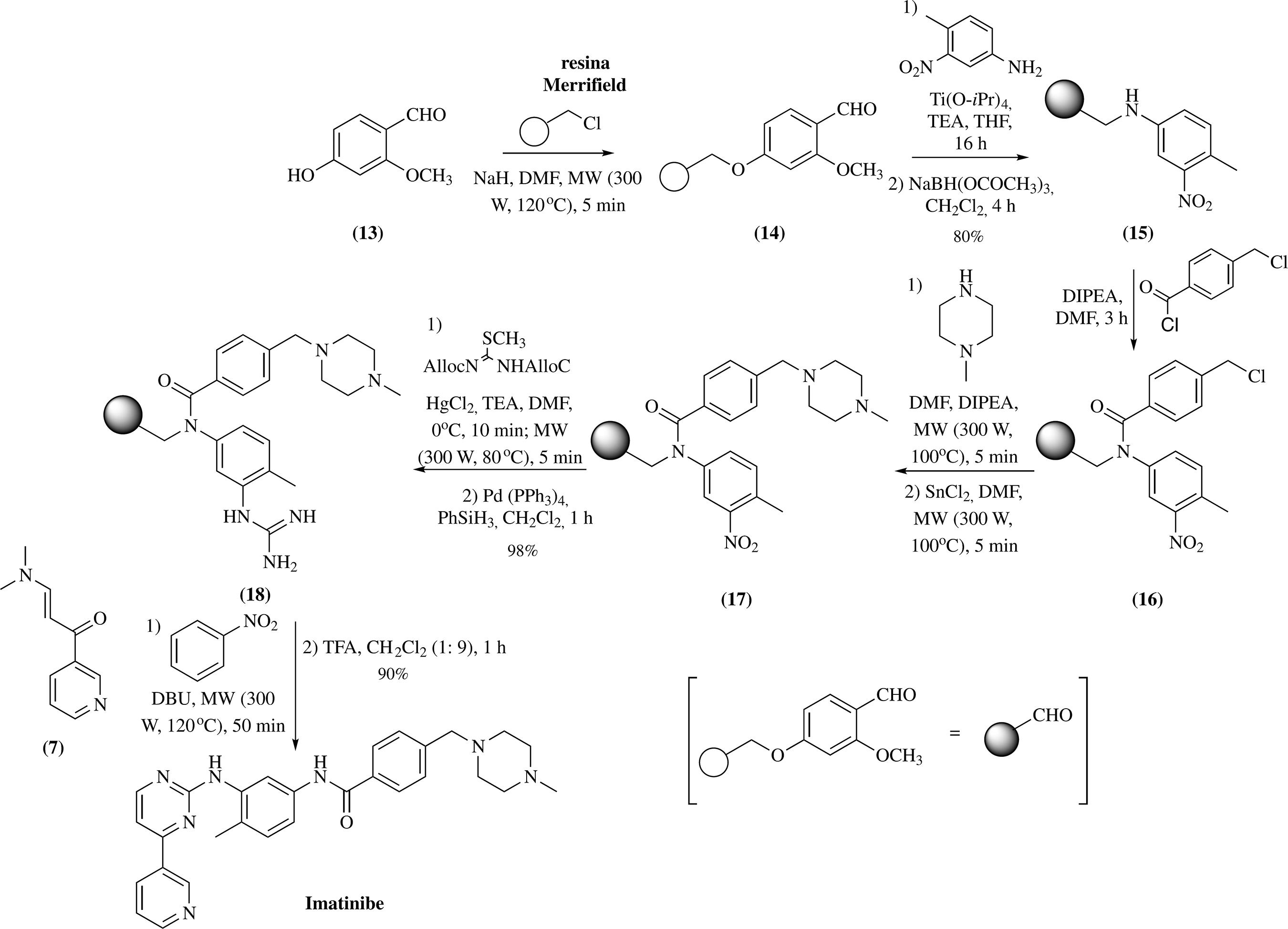
Em outra metodologia, descrita em 2007, o imatinibe pôde ser obtido a partir de uma síntese em fase sólida5252 Leonetti, F.; Capaldi, C.; Carotti, A.; Tetrahedron Lett. 2007, 48, 3455. (Esquema 3). A maior vantagem deste método foi o uso de micro-ondas, que permitiu a redução dos tempos reacionais e aumento dos rendimentos. Apesar dos benefícios do processo, é remota a possibilidade de um "scale-up", visto que o método tem baixa aplicabilidade industrial e não há rendimentos descritos para todas as etapas. Além disso, vários reagentes perigosos foram empregados, tais como cloreto de mercúrio (HgCl2) e fenilsilano (PhSiH3).
Em 2008, Liu e colaboradores5353 Liu, Y. F.; Wang, C. L.; Bai, Y. J.; Han, N.; Jiao, J. P.; Qi, X. L.; Org. Process Res. Dev. 2008, 12, 490. descreveram um processo com grande potencial para aumento de escala, visto que as condições reacionais eram mais simples para o uso industrial (Esquema 4).
A empresa Tianjin Weijie Technology patenteou um processo de obtenção do imatinibe em quatro etapas e com bom rendimento global (32%).5454 Han, P.; Lin, D.; Ma, C.; Peng, X.; Song, H.; Sun, Z.; Yang, M.; CN101735197 2010. (CA153:116266) Nesta metodologia, a amida 21 foi utilizada como material de partida (Esquema 5).
Metodologia sintética para a obtenção do imatinibe descrita pela Tianjin Weijie Technology
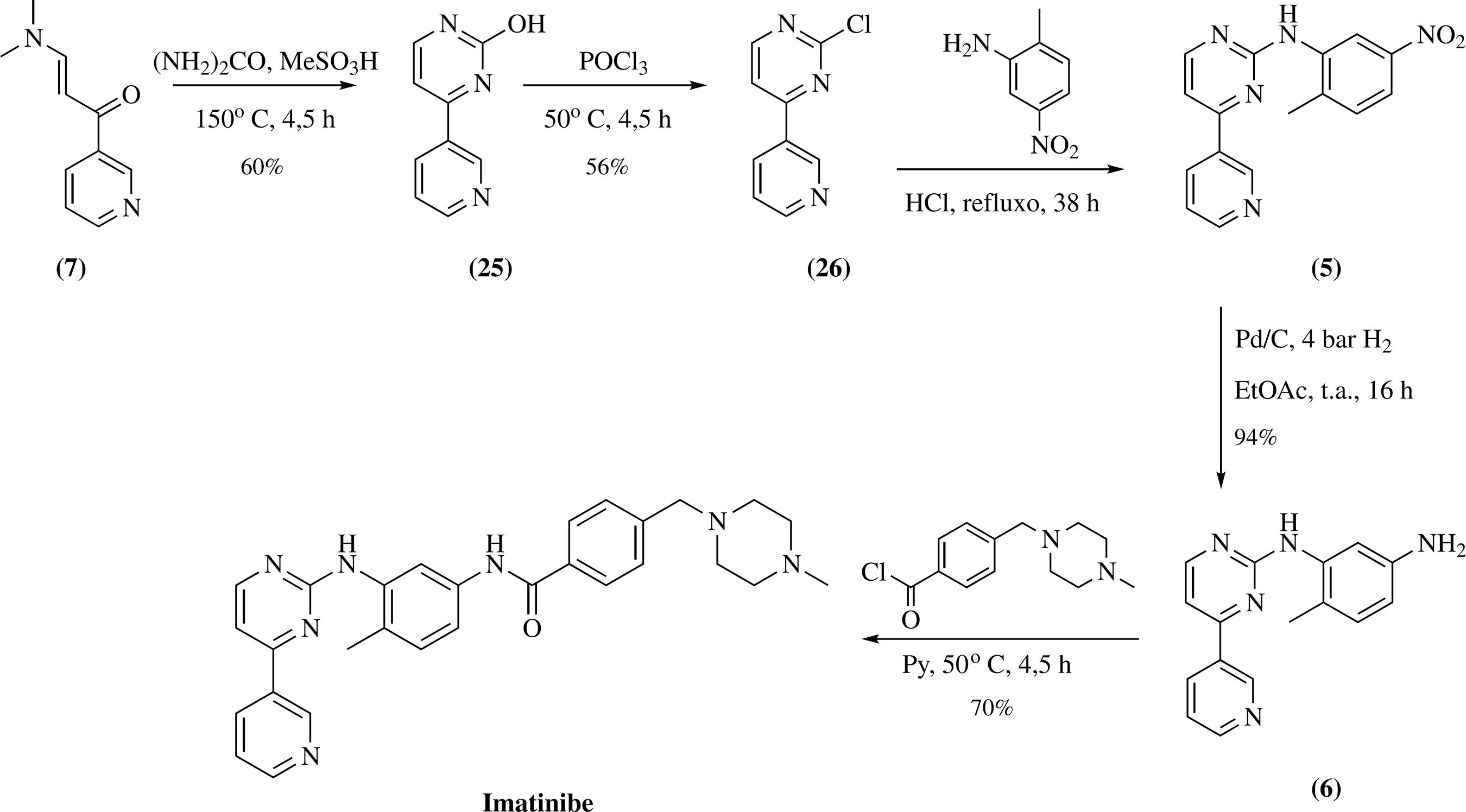
O uso da cianamida foi evitado na metodologia proposta por Wehrstedt e colaboradores. Contudo, como não há descrição do rendimento da terceira etapa, e foi necessário o uso de purificação por coluna cromatográfica para a obtenção do intermediário 5 em alto grau de pureza, este método tornou-se menos suscetível à aplicação na escala industrial5555 Wehrstedt, K. D.; Wildner, W.; Güthner, T.; Holzrichter, K.; Mertschenk, B.; Ulrich, A.; J. Hazard. Mater. 2009, 170, 829. (Esquema 6).
A conversão do imatinibe para mesilato é descrita através da reação entre o imatinibe e o ácido metanossulfônico, na presença de carvão ativo e metanol, à temperatura ambiente.5656 Pathi, S. L.; Kankan, R. N.; Puppala, R.; IN20110617
2011. (CA162:365447)
57 Lee, J. J.; Kang, S. G.; Jang, S. J.; KR2015010142
2015. (CA162:272978)
58 Xu, H.; Zhang, Z.; Wang, W.; CN104230885
2014. (CA162:137895)
59 Xu, X.; Min, T.; Shi, W.; Yaoxue Shijian Zazhi
2014, 32, 290.
60 Gruza, H.; Mirek, S.; Jezewski, A.; Wrzosek, A.; PL215042 B1
2013. (CA161:628850)
61 Liu, Z.; Yang, K.; Yu, L.; Wu, L.; Guo, X.; Chen, Z.; Huaxue Shiji
2014, 36, 574.
62 Xu, M.; He, L.; Guo, F.; CN103664874
2014. (CA160:515640)
63 Zhao, C.; Li, X.; Wang, X.; CN103588754
2014. (CA160:356375)-6464 Fors, B. P.; Watson, D. A.; Biscoe, M. R.; Buchwald, S. L.; J. Am. Chem. Soc.
2008, 130, 13552.
Devido à importância do imatinibe no cenário mundial e por ele não estar mais sob patente, nos últimos anos inúmeras metodologias têm sido descritas para a sua obtenção.5656 Pathi, S. L.; Kankan, R. N.; Puppala, R.; IN20110617
2011. (CA162:365447)
57 Lee, J. J.; Kang, S. G.; Jang, S. J.; KR2015010142
2015. (CA162:272978)
58 Xu, H.; Zhang, Z.; Wang, W.; CN104230885
2014. (CA162:137895)
59 Xu, X.; Min, T.; Shi, W.; Yaoxue Shijian Zazhi
2014, 32, 290.
60 Gruza, H.; Mirek, S.; Jezewski, A.; Wrzosek, A.; PL215042 B1
2013. (CA161:628850)
61 Liu, Z.; Yang, K.; Yu, L.; Wu, L.; Guo, X.; Chen, Z.; Huaxue Shiji
2014, 36, 574.
62 Xu, M.; He, L.; Guo, F.; CN103664874
2014. (CA160:515640)
63 Zhao, C.; Li, X.; Wang, X.; CN103588754
2014. (CA160:356375)-6464 Fors, B. P.; Watson, D. A.; Biscoe, M. R.; Buchwald, S. L.; J. Am. Chem. Soc.
2008, 130, 13552. Todas visam melhorar o rendimento global do processo e baseiam-se nos métodos já descritos anteriormente.
Dasatinibe
O dasatinibe (BMS-354825 - Sprycel®) - comercializado pela Bristol-Myers Squibb (BMS) - e aprovado pelo FDA em 20066565 Cortes, J.; Rousselot, P.; Kim, D.; Ritchie, E.; Hamerschlak, N.; Coutre, S.; Hochhaus, A.; Guilhot, F.; Saglio, G.; Apperley, J.; Ottmann, O.; Shah, N.; Erben, P.; Branford, S.; Agarwal, P.; Gollerkeri, A.; Baccarani, M.; Blood 2007, 109, 3207. - é um inibidor da forma ativa e inativa da enzima BCR-ABL, além de inibir outras quinases, tais como a família SRC, que é uma tirosina quinase intracelular, que está envolvida em processos como o crescimento celular.6666 O'Hare, T.; Walters, D. K.; Stoffregen, E. P.; Jia, T.; Manley, P. W.; Mestan, J.; Cowan-Jacob, S. W.; Lee, F. Y.; Heinrich, M. C.; Deininger, M. W. N.; Druker, B. J.; Cancer Res. 2005, 65, 4500; O'Hare, T.; Cancer Cell 2009, 16, 401.
O dasatinibe é utilizado na terapia de pacientes em fase crônica com dose inicial de 100 mg/dia, inferior ao imatinibe, que necessita de doses entre 300 e 800 mg/dia.4040 O'Brien, S. G.; Guilhot, F.; Larson, R. A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J. J.; Fischer, T.; Hochhaus, A.; Hughes, T.; N. Engl. J. Med.
2003, 348, 994. Pacientes em fase aguda ou crise blástica utilizam doses de 70 mg duas vezes ao dia.6767 Quintas-Cardama, A.; Kantarjian, H.; O'brien, S.; Borthakur, G.; Bruzzi, J.; Munden, R.; Cortes, J.; J. Clin. Oncol.
2007, 25, 3908. Seu tempo de meia vida é de 3 a 5 horas, sendo metabolizado pela enzima 3A4 do citocromo P450 e eliminado principalmente pelas fezes (85%).6868
http://www.medicinanet.com.br/bula/4806/sprycel.htm, acessada em Janeiro de 2017.
http://www.medicinanet.com.br/bula/4806/...
Devido à habilidade em inibir a proliferação da maioria das células mutantes resistentes ao imatinibe, o dasatinibe mostrou ser uma boa alternativa para o tratamento de pacientes que não apresentam boa resposta ao imatinibe. No entanto, uma alta toxicidade deste fármaco foi observada em pacientes nas fases mais avançadas da doença devido às elevadas doses utilizadas na terapia.6969 Tokarski, J. S.; Newitt, J. A.; Chang, C. Y. J.; Cheng, J. D.; Wittekind, M.; Kiefer, S. E.; Kish, K.; Lee, F. Y. F.; Borzillerri, R.; Lombardo, L. J.; Xie, D.; Zhang, Y.; Klei, H. E.; Cancer Res. 2006, 66, 5790.,7070 Delamain, M. T.; Conchon, M.; Rev. Bras. Hematol. Hemoter. 2008, 30, 37.
Síntese do dasatinibe
A rota sintética original para a obtenção do dasatinibe, descrita pela Bristol-Myers Squibb (BMS), consiste de três etapas reacionais a partir da reação entre o aminotiazol 27 e a 2-cloro-6-metilanilina (28), formando a amida 29. Posteriormente, 29 foi tratada com excesso de hidreto de sódio e o ânion gerado foi usado na reação com a pirimidina 30. A última etapa foi realizada através da reação de substituição nucleofílica aromática entre o derivado 31 e a piperazina 32, e o rendimento reacional não foi descrito7171 Das, J.; Padmanabha, R.; Chen, P.; Norris, D. J.; Doweyko, A. M. P.; Barrish, J.; Wityak, J.; Louis, L. J.; Lee, F. Y. F.; WO04085388
2004. (CA140:253556)
72 Das, J.; Padmanabha, R.; Chen, P.; Norris, D. J.; Doweyko, A. M. P.; Barrish, J. C.; Wityak, J.; US pat. 04007302
2004. (CA140:253556)
73 Das, J.; Padmanabha, R.; Chen, P.; Norris, D. J.; Doweyko, A. M. P.; Barrish, J. C.; Wityak, J.; Lombardo, J. J.; Lee, F. Y. F.; US pat. 040054186
2004. (CA140:253556)-7474 Barrish, J. C.; Wityak, J.; Das, J.; Chen, P.; Norris, D. J.; Shieven, G.; US pat. 050288303
2005. (CA133:321878) (Esquema 7).
A metodologia para a obtenção do amino tiazol 27 (Esquema 8) foi descrita em 2006 por cientistas da BMS envolvidos na síntese do dasatinibe e, portanto, não está relatada na patente original.7575 Lombardo, L. J.; Lee, F. Y.; Chen, P.; Norris, D.; Barrish, J. C.; Behnia, K.; Castaneda, S.; Cornelius, L. A. M.; Das, J.; Doweyko, A. M.; Fairchild, C.; Hunt, J. T.; Inigo, I.; Johnston, K.; Kamath, A.; Kan, D.; Klei, H.; Marathe, P.; Pang, S.; Peterson, R.; Pitt, S.; Schieven, G. L.; Schmidt, R. J.; Tokarski, J.; Wen, M. L.; Wityak, J.; Borzilleri, R. M.; J. Med. Chem. 2004, 47, 6658.,7676 Das, J.; Chen, P.; Norris, D.; Padmanabha, R.; Lin, J.; Moquin, R. V.; Shen, Cook, L. S.; Doweyko, A. M.; Pitt, S.; Pang, S.; Shen, D. R.; Fang, Q.; de Fex, H. F.; McIntyre, K. W.; Shuster, D. J.; Gillooly, K. M.; Behnia, K.; Schieven, G. L.; Wityak, J.; Barrish, J. C.; J. Med. Chem. 2006, 49, 6819.
Preparação do carboxilato 27 utilizado como material de partida na rota sintética original para a síntese do dasatinibe
Em 2006, a BMS descreveu uma outra rota sintética para a síntese do dasatinibe, que foi preparado em escala de multigramas com rendimento global de 61% (Esquema 9). A metodologia teve início com a desprotonação do 2-cloro tiazol (35), seguida da adição do ânion ao isocianato 36. Posteriormente, o intermediário 37 foi tratado com o 1-(clorometil)-4-metoxi-benzeno (38) para a formação do derivado 39 em 95% de rendimento. Este foi submetido à uma reação de substituição nucleofílica aromática para a formação do intermediário 41, que após desproteção levou à formação do composto 42 em 99% de rendimento. A etapa final desta rota consistiu na reação entre a piperazina 32 e o intermediário 42 para a formação do dasatinibe. Apesar de ser um método eficiente, a maior limitação do processo é a dificuldade na síntese do material de partida 35, que é obtido através da cloração direta do tiazol, com a formação de regioisômeros, necessitando de purificação para uso.7575 Lombardo, L. J.; Lee, F. Y.; Chen, P.; Norris, D.; Barrish, J. C.; Behnia, K.; Castaneda, S.; Cornelius, L. A. M.; Das, J.; Doweyko, A. M.; Fairchild, C.; Hunt, J. T.; Inigo, I.; Johnston, K.; Kamath, A.; Kan, D.; Klei, H.; Marathe, P.; Pang, S.; Peterson, R.; Pitt, S.; Schieven, G. L.; Schmidt, R. J.; Tokarski, J.; Wen, M. L.; Wityak, J.; Borzilleri, R. M.; J. Med. Chem. 2004, 47, 6658.,7676 Das, J.; Chen, P.; Norris, D.; Padmanabha, R.; Lin, J.; Moquin, R. V.; Shen, Cook, L. S.; Doweyko, A. M.; Pitt, S.; Pang, S.; Shen, D. R.; Fang, Q.; de Fex, H. F.; McIntyre, K. W.; Shuster, D. J.; Gillooly, K. M.; Behnia, K.; Schieven, G. L.; Wityak, J.; Barrish, J. C.; J. Med. Chem. 2006, 49, 6819.
Em outra metodologia, a preparação do dasatinibe foi realizada através de uma síntese convergente onde a formação do anel tiazólico ocorre nas últimas etapas da rota (Esquema 10). A reação inicia-se com a obtenção da enona 48, a partir de uma adição à carbonila no substrato 47. Em seguida, ocorre a formação do intermediário tiazólico 49 em 71% de rendimento. Este foi tratado com a piperazina 32 para a formação do fármaco com bom rendimento global.7777 Chen, B. C.; Zhao, R.; Wang, B.; WO05076990 2005. (CA143:211902),7878 Chen, B. C.; Droghini, R.; Lajeunesse, J.; Dimarco, J. D.; Galella, M.; Chidambaram, R.; WO05077945 2005. (CA143:248376)
Uma outra rota sintética com poucas etapas reacionais foi proposta para a preparação deste fármaco. Este processo inicia-se com uma reação de substituição nucleofílica aromática na 4,6-dicloro-2-metil-pirimidina (30) formando 50, que após acoplamento de Buchwald-Hartwig com 51 levou à formação do dasatinibe em 80% de rendimento global7878 Chen, B. C.; Droghini, R.; Lajeunesse, J.; Dimarco, J. D.; Galella, M.; Chidambaram, R.; WO05077945 2005. (CA143:248376) (Esquema 11).
Preparação do dasatinibe através do acoplamento de Buchwald-Hartwig de um aminotiazole 51 e uma pirimidina 50
Em 2010, a empresa de Bio-engenharia Nanjing Cavendish descreveu duas metodologias para a preparação do dasatinibe.7979 Hou, W.; Xu, Y.; Yan, R.; Yang, H.; CN101812060 2010. (CA153:406452) Na primeira, o dasatinibe foi preparado em quatro etapas, partindo da 4,6-dicloro-2-metil-pirimidina (30). O segundo método utilizou como materiais de partida a 4-amino-6-bromo-2-metil-pirimidina (56) e o 2-clorotiazol-5-carboxilato de metila (57) (Esquema 12). Neste processo, a primeira etapa consistiu de uma reação de substituição nucleofílica aromática no intermediário 57. Posteriormente, o derivado 58 foi submetido à uma hidrólise do éster fornecendo o composto 59 em 78% de rendimento. O rendimento global do método foi de 42%.
Rotas sintéticas descritas pela empresa de Bio-engenharia Nanjing Cavendish para a obtenção do dasatinibe
Nilotinibe
O nilotinibe (AMN107 - Tasigna®) - desenvolvido pela Novartis e aprovado em 2007 pelo FDA - é utilizado em pacientes adultos com LMC, em fase crônica ou acelerada, resistentes ou intolerantes ao imatinibe. É estruturalmente semelhante ao imatinibe e foi descoberto a partir de um planejamento racional que objetivava melhor afinidade e especificidade de ligação contra BCR-ABL em relação ao imatinibe.8080 Weisberg, E.; Manley, P. W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S. W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; Kung, A. L.; Mestan, J.; Daley, G. Q.; Callahan, L.; Catley, L.; Cavazza, C.; Azam, M.; Neuberg, D.; Wright, R. D.; Gilliland, D. G.; Griffin, J. D.; Cancer Cell.
2005, 7, 129.,8181
http://www.cancer.gov/cancertopics/druginfo/fda-nilotinib, acessada em Janeiro de 2017.
http://www.cancer.gov/cancertopics/drugi...
Apresenta atividade contra a maioria das mutações resistentes ao imatinibe, com exceção domínio de mutação T315I do gene BCR-ABL, que permaneceu insensível. Além disso, inibe o receptor de tirosina quinase e marcador tumoral (c-Kit) e o receptor do fator de crescimento derivado das plaquetas (PDGFR).8080 Weisberg, E.; Manley, P. W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S. W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; Kung, A. L.; Mestan, J.; Daley, G. Q.; Callahan, L.; Catley, L.; Cavazza, C.; Azam, M.; Neuberg, D.; Wright, R. D.; Gilliland, D. G.; Griffin, J. D.; Cancer Cell. 2005, 7, 129.,8282 Deininger, M. W.; Clin. Cancer Res. 2008, 14, 4027.
A dose usual do tratamento é de 200 mg duas vezes ao dia. O pico de concentração plasmática é alcançado três horas após a ingestão oral. Os principais metabólitos encontrados são produtos de oxidação e hidroxilação, que ocorrem na fase I do metabolismo hepático. Seus metabólitos são inativos e a principal via de eliminação é pelas fezes. As reações adversas mais relatadas pelos pacientes foram erupção cutânea, prurido, náusea, fadiga, dor de cabeça, dor abdominal, constipação e diarreia.8383 Lipton, J. H.; Pasic, I.; Leuk. Res. 2017, 55, 65.
Pacientes em fase crônica apresentaram uma resposta hematológica de 62%, sendo 41% completa; enquanto na fase aguda a resposta hematológica foi de 44% e 22% a completa. Com relação à resposta citogenética, observou-se um índice de 42% e 12%, respectivamente, nos dois grupos.8484 Le Coutre, P.; Ottmann, O. G.; Giles, F.; Kim, D. W.; Cortes, J.; Gattermann, N.; Apperley, J. F.; Larson, R. A.; Abruzzese, E.; O'Brien, S. G.; Kuliczkowski, K.; Hochhaus, A.; Mahon, F. X.; Saglio, G.; Gobbi, M.; Kwong, Y. L.; Baccarani, M.; Hughes, T.; Martinelli, G.; Radich, J. P.; Zheng, M.; Shou, Y.; Kantarjian, H.; Blood 2008, 111, 1834.
Recentemente, Hochhaus e colaboradores publicaram um estudo comparativo entre a incidência de mutações em pacientes que fizeram uso do nilotinibe ou imatinibe. Os resultados mostraram que pacientes tratados com nilotinibe apresentaram menor número de mutações BCR-ABL do que pacientes tratados com imatinibe.8585 Hochhaus, A.; Saglio, G.; Larson, R. A.; Kim, D. W.; Etienne, G.; Rosti, G.; De Souza, C.; Kurokawa, M.; Kalaycio, M. E.; Hoenekopp, A.; Fan, X.; Shou, Y.; Kantarjian, H. M.; Hughes, T. P.; Blood 2013, 121, 3703.
Síntese do nilotinibe
Em 2004, foi depositada pela Novartis a primeira patente para a obtenção do nilotinibe sem conter nenhum dado de rendimento reacional. O método consiste em uma síntese convergente entre os intermediários 64 e 65. A obtenção de 64 consiste na formação do sal da guanidina 62, seguida de uma reação de condensação com a enaminona 07 levando à formação da pirimidina 63. Posteriormente, o grupo éster de 63 foi hidrolisado para o ácido carboxílico correspondente 64 e acoplado com anilina 65, utilizando fosforocianidato de dietila (66) como reagente de acoplamento (Esquema 13). Nesta patente também foi descrita a metodologia de preparação da anilina 65 através de uma rota com quatro etapas reacionais.8686 Breitenstein, W.; Furet, P.; Jacob, S.; Manley, P. W.; WO04005281 2004. (CA140:77161) Contudo, em 2006, a Novartis descreveu quatro diferentes rotas sintéticas para a obtenção do derivado 65 com rendimentos globais variando entre 18 e 53%.8787 Abel, S.; Acemoglu, M.; Erb, B.; Krell, C.; Sclafani, J.; Meisenbach, M.; Prashad, M.; Sheih, W. C.; Xue, S.; WO06135640 2006. (CA146:81866)
Síntese convergente do nilotinibe descrita pela Novartis, através da reação entre o intermediário 64 e a anilina 65
Estudos posteriores mostraram que o nilotinibe poderia ser sintetizado através da condensação direta da anilina 65 com éster metílico 63 (Esquema 14). A reação foi realizada à temperatura ambiente, na presença de terc-butóxido de potássio, levando à formação do produto final em 67% de rendimento isolado. Este método oferece vantagens, tais como a eliminação da etapa de hidrólise e a necessidade de ativação do ácido 64 para a formação do nilotinibe. Estas mudanças favorecem a preparação do fármaco em escala industrial e diminuem o custo, além de eliminar o uso de reagentes de acoplamento, que são relativamente caros e dificultam a purificação.8888 McKenna, J.; Shieh, W. C.; WO06135641 2006. (CA146:81883)
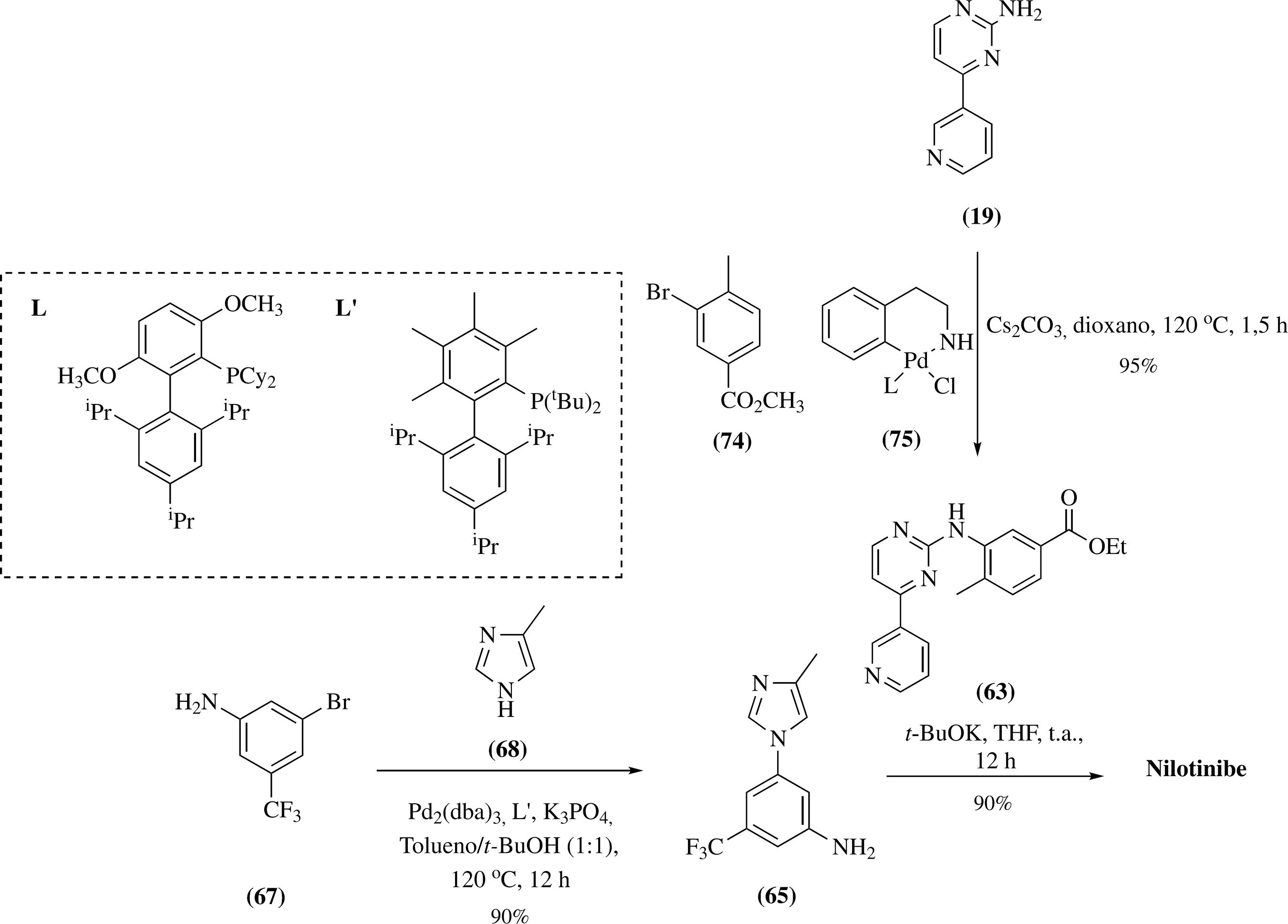
Uma rota sintética mais simples foi desenvolvida por pesquisadores da Ariad Pharmaceuticals (Esquema 15). Esta metodologia inicia-se com o acoplamento do 4(5)-metilimidazole (68) com a 3-bromo-5-trifluorometilanilina (67), levando à formação da mistura de anilinas 65 como regioisômeros na proporção de 85: 15. Neste processo, a combinação do quelante 8-hidroxiquinolina (69) e iodeto de cobre facilitou a reação de acoplamento, ocorrendo a formação do produto com rendimentos razoáveis. A separação e purificação dos isômeros 65 pôde ser obtida através de cromatografia em coluna e recristalização, respectivamente. Na etapa posterior, foi realizada a reação entre a anilina 65 com o cloreto de ácido 70, ocorrendo a formação do derivado 71 em 95% de rendimento. A etapa final consiste em um acoplamento de Buchwald que permite a obtenção do nilotinibe em 89% de rendimento.8989 Huang, W. S.; Shakespeare, W.; Synthesis 2007, 2121.
Em 2009, Chen e colaboradores descreveram um processo de obtenção do nilotinibe que utiliza condições semelhantes à síntese original.8686 Breitenstein, W.; Furet, P.; Jacob, S.; Manley, P. W.; WO04005281 2004. (CA140:77161) No entanto, a amina em 63 foi protegida com carbamato de terc-butil (Boc) antes da hidrólise do éster. Isto permitiu o acoplamento do intermediário 64 com a anilina 65 em excelente rendimento9090 Chen, Y.; Wang, L.; Zhou, H.; Wang, C.; Zhongguo Yiyao Gongye Zazhi 2009, 40, 401. (Esquema 16).
Preparação do nilotinibe utilizando a proteção de amina em 63 com Boc para a melhoria do rendimento do processo
Posteriormente, a Teva Pharmaceuticals patenteou uma reação de acoplamento catalisada por cobre, para gerar o intermediário chave anilina 65 (Esquema 17). A metodologia desenvolvida melhorou a regiosseletividade e o isômero-1,5 foi detectado como uma impureza com 0,13% de rendimento. O processo utilizou a 8-hidroxiquinolina (69) como quelante, iodeto de cobre e uma mistura de óxido de cálcio e hidróxido de sódio. A reação foi aquecida à 120 °C por 69 horas, e o produto final foi purificado por precipitação utilizando uma mistura de acetato de etila/éter de petróleo. A segunda etapa consistiu na reação entre o intermediário 65 com o cloreto de ácido 72, obtido in situ, levando à formação do nilotinibe em 81% de rendimento.9191 Wang, Y.; Li, J.; Kansal, V. K.; Zhu, J.; Lifshitz-Liron, R.; Mistry, D.; Vasoya, S. L.; Ariyamuthu, S.; Piraski, G.; He, X.; WO10009402 2010. (CA152:192145) Rendimentos mais elevados (94%) foram observados quando o cloreto de ácido 72 foi isolado e, em seguida, acoplado com a anilina 65 (Esquema 17).
Um método alternativo também descrito pela Teva Pharmaceuticals inicia-se com a preparação do cloreto de ácido 72, seguido de uma reação de acoplamento utilizando THF como solvente, N,N-Diisopropiletilamina (DIPEA) como base, na presença de 4-dimetilaminopiridina (DMAP) como um co-catalisador. A etapa final para a formação do nilotinibe consiste em outro acoplamento entre os derivados 73 e 68, utilizando iodeto de cobre, 69 e 1,8-diazabiciclo-[5.4.0]-undec-7-eno (DBU), à 120 oC por 48 horas (Esquema 18). O baixo rendimento reacional desta etapa (44%) foi devido às perdas durante a filtração sob celite, utilizada para a purificação do produto.9191 Wang, Y.; Li, J.; Kansal, V. K.; Zhu, J.; Lifshitz-Liron, R.; Mistry, D.; Vasoya, S. L.; Ariyamuthu, S.; Piraski, G.; He, X.; WO10009402 2010. (CA152:192145)
Metodologia de síntese do nilotinibe em três etapas reacionais, desenvolvida pela Teva Pharmaceuticals
Em 2012, Buchwald e colaboradores descreveram um outro processo que consiste no acoplamento do 4(5)-metilimidazole (68) com o 3-(trifluorometil)-5-bromoanilina (67), levando à formação regiosseletiva do intermediário 65 em 90% de rendimento. Este excelente resultado foi atribuído ao uso do Pd como catalisador. Este tipo de catálise também foi empregada com sucesso para a preparação do derivado 63, utilizado na última etapa da síntese do fármaco. Contudo, uma das principais limitações desta metodologia é a necessidade de remoção dos resíduos do metal no produto final9292 Buchwald, S. L.; Ueda, S.; Su, M.; J. Am. Chem. Soc. 2012, 134, 700. (Esquema 19).
Metodologia descrita por Buchwald e colaboradores para a síntese do intermediário 63 e do nilotinibe
Bosutinibe
O bosutinibe (SKI606 - Bosulif®) - comercializado pela Pfizer e aprovado pelo FDA, em setembro de 2012 - é utilizado em pacientes em fase crônica, acelerada ou blástica com cromossomo Ph positivo, resistentes ou intolerantes ao imatinibe.9393
http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm318203.htm, acessada em Janeiro de 2017.
http://www.fda.gov/Drugs/InformationOnDr...
Assim como o dasatinibe, o bosutinibe é um duplo inibidor ativo contra SCR e BCR-ABL (inibe a maioria das células resistentes ao imatinibe, exceto a T315I), que não apresenta atividade inibitória contra o c-Kit ou PDGFR.9494 Puttini, M.; Coluccia, A. M.; Boschelli, F.; Cleris, L.; Marchesi, E.; Donella-Deana, A.; Ahmed, S.; Redaelli, S.; Piazza, R.; Magistroni, V.; Andreoni, F.; Scapozza, L.; Formelli, F.; Gambacorti-Passerini, C.; Cancer Res. 2006, 66, 11314. Possui um padrão de inibição semelhante ao imatinibe, contudo utiliza menores doses para atingir seu efeito. Acredita-se que devido à similaridade do padrão de inibição, ambos os fármacos compartilham de um mecanismo comum de inibição.9595 Remsing Rix, L. L.; Rix, U.; Colinge, J.; Hantschel, O.; Bennett, K. L.; Stranzl, T.; Mueller, A.; Baumgartner, C.; Valent, P.; Augustin, M.; Till, J. H.; Superti-Furga, G.; Leukemia 2009, 23, 477.
A dosagem indicada é de 500 mg ao dia, sendo necessária a ingestão acompanhada de um alimento. Entre 4 a 6 horas, o medicamento atinge o seu pico plasmático, sendo metabolizado principalmente pelo CYP3A4. Os seus metabólitos não são ativos e a sua eliminação é pelas fezes.9696
http://www.pfizer.com/products/#bosulif, acessada em Janeiro de 2017.
http://www.pfizer.com/products/#bosulif...
Síntese do bosutinibe
Os métodos descritos na literatura para a síntese do bosutinibe apresentam algumas desvantagens, tais como baixos rendimentos, alto custo dos reagentes e elevados tempos reacionais. A metodologia proposta por Boschelli e colaboradores consiste num processo de seis etapas cuja aplicabilidade foi limitada devido ao rendimento global de 8,3%.9797 Boschelli, D. H.; Ye, F.; Wang, Y. D.; Dutia, M.; Johnson, S. L.; Wu, B.; Miller, K.; Powell, D. W.; Yaczko, D.; Young, M.; Tischler, M.; Arndt, K.; Discafani, C.; Etienne, C.; Gibbons, J.; Grod, J.; Lucas, J.; Weber, J. M.; Boschelli, F.; J. Med. Chem. 2001, 44, 3965. A primeira etapa consiste em uma reação de alquilação da hidroxila, seguida da nitração e posterior redução do grupo nitro em 77 para formar 78. Para a obtenção de 79, inicialmente ocorreu a formação da amidina, seguido da ciclização do anel. Na quarta etapa, o oxicloreto de fosfóro (POCl3) foi utilizado como reagente numa reação de cloração. Após uma substituição nucleofílica aromática em 80, seguida de uma substituição nucleofílica bimolecular em 81, o bosutinibe foi obtido com elevado grau de pureza9797 Boschelli, D. H.; Ye, F.; Wang, Y. D.; Dutia, M.; Johnson, S. L.; Wu, B.; Miller, K.; Powell, D. W.; Yaczko, D.; Young, M.; Tischler, M.; Arndt, K.; Discafani, C.; Etienne, C.; Gibbons, J.; Grod, J.; Lucas, J.; Weber, J. M.; Boschelli, F.; J. Med. Chem. 2001, 44, 3965. (Esquema 20).
Metodologia com seis etapas reacionais para a síntese do bosutinibe descrita por Boschelli e colaboradores
O método proposto por Sutherland e colaboradores é composto por cinco etapas e apresenta rendimento global de 32%. É considerado um processo eficiente, mesmo que alguns reagentes possuam elevado custo e algumas etapas apresentem longos tempos reacionais, configurando limitações importantes9898 Sutherland, K. W.; Feigelson, G. B.; Boschelli, D. H.; Blum, D. M.; Strong, H. L.; US pat. 0043537 2005. (CA142:261413) (Esquema 21).
Processo de síntese do bosutinibe com cinco etapas e rendimento global de 32%, descrito por Sutherland e colaboradores
A síntese do bosutinibe com maior potencial para aplicação em escala industrial foi descrita em 2010 por Yin e colaboradores9999 Yin, X. J.; Xu, G. H.; Sun, X.; Peng, Y.; Ji, X.; Jiang K.; Li, F.; Molecules 2010, 15, 4261. (Esquema 22). Esta metodologia inicia-se com o ácido 3-metoxi-4-hidroxibenzóico (88), que foi esterificado e alquilado com 1-bromo-3-cloropropano, levando à formação do intermediário 89. A nitração seguida da redução do grupo nitro no derivado 89 permitiu a formação do composto 78, o qual foi condensado com 3,3-dietoxiproprionitrila, ciclizado e clorado para a obtenção do derivado 4-cloroquinolina 80. Este, após reação de substituição nucleofílica aromática com a 2,4-dicloro-5-metoxianilina formou o derivado 81, que finalmente reagiu com a N-metilpiperazina para obter o bosutinibe.
Síntese descrita em 2010 para obtenção do bosutinibe em escala industrial com cinco etapas
Ponatinibe
Em 14 de dezembro de 2012 o FDA concedeu aprovação acelerada para ponatinibe (AP24534 - Iclusig®). Desenvolvido pela Ariad Pharmaceuticals, é indicado para o tratamento de pacientes adultos com fase crônica, fase acelerada ou blástica de LMC, que são resistentes ou intolerantes às terapias anteriores com quaisquer inibidores de tirosina quinase.100100
http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203469lbl.pdf, acessada em Janeiro de 2017.
http://www.accessdata.fda.gov/drugsatfda...
A aprovação acelerada foi baseada nos resultados de um estudo multicêntrico internacional. Os ensaios clínicos com 449 pacientes resistentes ou intolerantes ao tratamento com inibidores da tirosina quinase mostraram que ele inibe a mutação T315I.101101 Leonetti, F.; Stefanachi, A.; Nicolotti, O.; Catto, M.; Pisani, L.; Cellamare, S.; Carotti, A.; Curr. Med. Chem. 2011, 18, 2943.
A dose recomendada e a posologia para ponatinibe é de 45 mg uma vez ao dia. Os efeitos colaterais mais comuns relatados no ensaio clínico incluem hipertensão, erupção cutânea, dor abdominal, fadiga, dor de cabeça, pele seca, constipação, febre, dor nas articulações e náuseas. No entanto, também foram observados casos de trombose arterial e hepatotoxicidade.100100
http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203469lbl.pdf, acessada em Janeiro de 2017.
http://www.accessdata.fda.gov/drugsatfda...
Síntese do ponatinibe
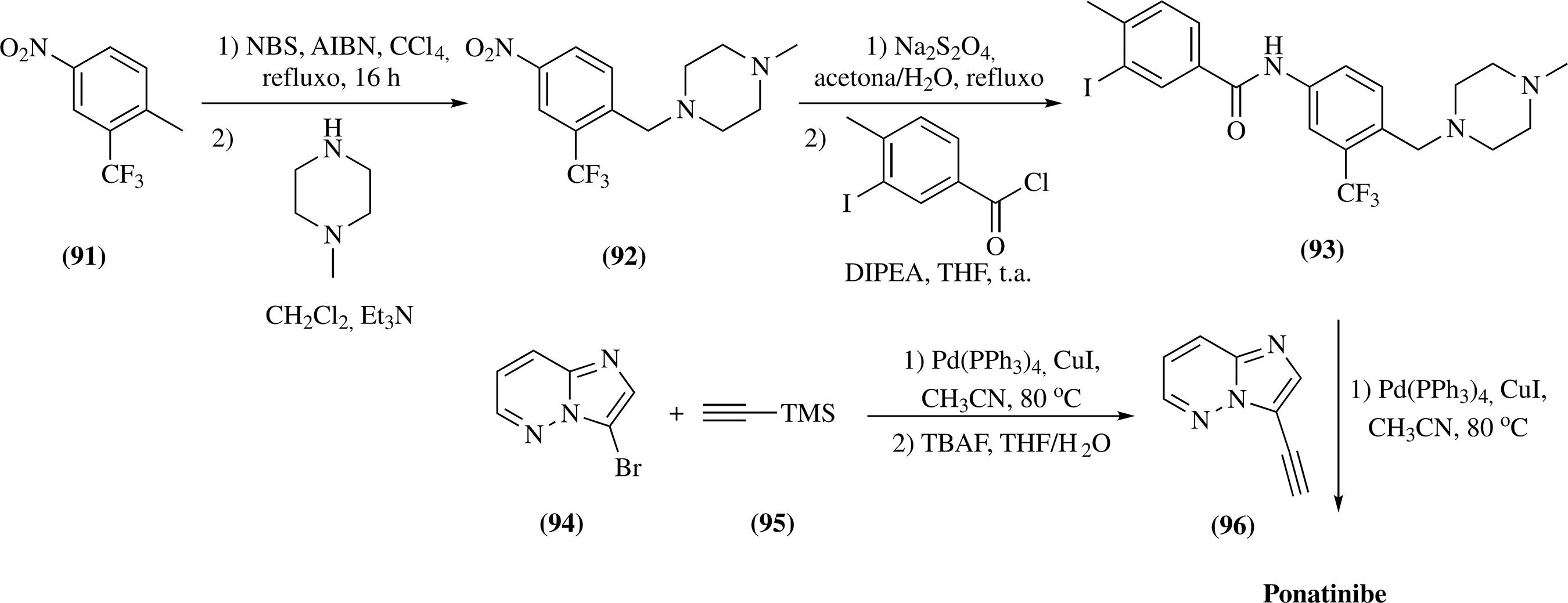
A primeira síntese do ponatinibe, descrita por Zou e colaboradores, consiste num processo convergente e simples que realiza dois acoplamentos de Sonogashira para a introdução de uma ligação tripla entre dois ligantes.102102 Zou, D.; Huang, W. S.; Thomas, R. M.; Romero, J. A. C.; Qi, J.; Wang, Y.; Zhu, X.; Shakespeare, W.C.; Sundaramoorthi, R.; Metcalf, C.A. III; Dalgarno, D. C.; Sawyer, T. K. WO 075869 2007. (CA147:143466) Para a preparação do primeiro ligante 93 foi realizada a reação entre o 2-trifluorometil-4-nitrotolueno (91), o N-bromosuccinimida (NBS) e a N-metilpiperazina para a formação do intermediário 92. A redução do grupo nitro em 92, seguido do acoplamento com cloreto de 3-iodo-4-metilbenzoila, permitiu a síntese do intermediário 93. A segunda porção molecular 96 foi obtida a partir da reação com PdCl2(PPh3)2 ou Pd(PPh3)4, CuI em acetonitrila, a 80 °C; e este, após o acoplamento de Sonogashira com 93, levou à formação do ponatinibe102102 Zou, D.; Huang, W. S.; Thomas, R. M.; Romero, J. A. C.; Qi, J.; Wang, Y.; Zhu, X.; Shakespeare, W.C.; Sundaramoorthi, R.; Metcalf, C.A. III; Dalgarno, D. C.; Sawyer, T. K. WO 075869 2007. (CA147:143466) (Esquema 23). Neste trabalho, os rendimentos reacionais não foram descritos.
Em outra abordagem, os autores propuseram realizar o acoplamento de Sonogashira no início da síntese (Esquema 24). Esta metodologia inicia-se com a preparação do intermediário 98 através da reação entre 96 e o ácido 3-iodo-4-metil benzóico (97).102102 Zou, D.; Huang, W. S.; Thomas, R. M.; Romero, J. A. C.; Qi, J.; Wang, Y.; Zhu, X.; Shakespeare, W.C.; Sundaramoorthi, R.; Metcalf, C.A. III; Dalgarno, D. C.; Sawyer, T. K. WO 075869 2007. (CA147:143466) Os rendimentos das etapas não foram descritos.
Rota sintética para a preparação do ponatinibe, que realiza o acoplamento de Sonogashira no início da síntese
Em 2014, Kovi e colaboradores descreveram uma metodologia sintética para a preparação do ponatinibe em sete etapas103103 Kovi, R.; Kannapan, J.; Thakor, S. F.; Patel, R. A.; US pat. 0343282 2014. (CA161:769806) (Esquema 25). Segundo os autores, este método foi capaz de reduzir drasticamente a complexidade do processo quando comparado aos anteriores. Inicialmente, o 1-metil-2-trifluorometil-4-nitrobenzeno (100) reagiu com NBS, para a formação do intermediário 101. Este, após reação com ftalimida de potássio seguida de redução catalítica, levou à formação do composto 103. Posteriormente, foram realizadas mais quatro etapas reacionais para a formação do ponatinibe. Os rendimentos reacionais de algumas das etapas não foram descritos.
Metodologia descrita por Kovi e colaboradores para a preparação do ponatinibe em sete etapas reacionais
Outra metodologia para a preparação do ponatinibe foi descrita por Ding e colaboradores. Neste trabalho, o composto pôde ser sintetizado através de uma rota sintética em duas etapas. A primeira consiste em uma reação de adição à carbonila levando à formação do intermediário 93 em 91% de rendimento. Este, após ser submetido a uma reação de acoplamento de Sonogashira, permitiu a formação do ponatinibe104104 Ren, X.; Pan, P.; Zhang, Z.; Wang, D.; Lu, X.; Li, Y.; Wen, D.; Long, H.; Luo, J.; Feng, Y.; Zhuang, X.; Zhang, F.; Liu, J.; Leng, F.; Lang, X.; Bai, Y.; She, M.; Tu, Z.; Pan, J.; Ding, K.; J. Med. Chem. 2012, 56, 879. (Esquema 26).
CONCLUSÕES
O imatinibe foi o primeiro representante da classe dos inibidores de tirosina quinase BCR-ABL utilizado no tratamento da LMC. Atualmente, não há mais nenhuma patente que impeça a produção desta substância, e com isso, inúmeras metodologias de síntese encontram-se descritas. Todos os métodos visam diminuir o custo de produção com objetivo de reduzir o valor final do medicamento.
Para o dasatinibe e nilotinibe, ITQs de segunda geração, há um menor número de rotas sintéticas. Contudo, pode-se observar que nos últimos anos muitos grupos investiram na pesquisa de novos processos de preparação destas substâncias devido à proximidade de expiração de suas patentes. Um dos grandes benefícios dos ITQs de segunda geração é a redução das doses necessárias para tratar o paciente.
Com relação à terceira geração, bosutinibe e ponatinibe, o número de metodologias sintéticas é muito inferior, pois eles foram recentemente aprovados pelo FDA (ambos em 2012).
Finalmente, pode-se dizer que o emprego dos ITQs e a descoberta do mesilato de imatinibe revolucionaram o tratamento da LMC. No entanto, ainda existe a necessidade de desenvolver novos análogos que tenham a capacidade de inibir as formas mutantes resistentes aos fármacos de primeira, segunda e terceira gerações. Outra importante necessidade é o uso de fármacos que apresentem baixa ou nenhuma toxicidade, visto que eles são de uso contínuo. Para isto, é importante o conhecimento dos métodos de obtenção dos intermediários e ITQs visando a síntese de novos derivados.
-
#
Programa de Pós-Graduação em Farmacologia e Química Medicinal - P-GFQM
-
‡
Programa de Bolsas Institucionais - PROBIN
AGRADECIMENTOS
Os autores agradecem à CAPES, FAPERJ e ao CNPq pelas bolsas e auxílios financeiros aos projetos desenvolvidos no Laboratório de Síntese de Fármacos de Farmanguinhos.
REFERÊNCIAS
-
1http://www2.inca.gov.br/wps/wcm/connect/cancer/site/oquee, acessada em Janeiro de 2017.
» http://www2.inca.gov.br/wps/wcm/connect/cancer/site/oquee -
2http://portalsaude.saude.gov.br/index.php/o-ministerio/principal/leia-mais-o-ministerio/671-secretaria-svs/vigilancia-de-a-a-z/doencas-cronicas-nao-transmissiveis/14125-vigilancia-das-doencas-cronicas-nao-transmissiveis, acessada em Janeiro de 2017.
» http://portalsaude.saude.gov.br/index.php/o-ministerio/principal/leia-mais-o-ministerio/671-secretaria-svs/vigilancia-de-a-a-z/doencas-cronicas-nao-transmissiveis/14125-vigilancia-das-doencas-cronicas-nao-transmissiveis -
3http://www.inca.gov.br/estimativa/2016/, acessada em Janeiro de 2017.
» http://www.inca.gov.br/estimativa/2016/ -
4http://globocan.iarc.fr/Pages/bar_dev_sel.aspx, acessada em Janeiro de 2017.
» http://globocan.iarc.fr/Pages/bar_dev_sel.aspx -
5Contran, R.; Kumar, V.; Collins, T.; Patologia Estrutural e Funcional, 6ª ed., Guanabara: Koogan, 2000.
-
6http://www2.inca.gov.br/wps/wcm/connect/tiposdecancer/site/home/leucemia, acessada em Janeiro de 2017.
» http://www2.inca.gov.br/wps/wcm/connect/tiposdecancer/site/home/leucemia -
7Fauci, A. S.; Braunwald, E.; Kasper, D. L.; Hauser, S. L.; Longo, D. L.; Jameson, J. L.; Loscalzo, J.; Harrison's Principles of Internal Medicine, 17th ed., McGraw-Hill: New York, 2008.
-
8Joske, D. J.; Med. J. Aust. 2008, 1, 277.
-
9Geary, C. G.; British J. Haematol. 2000, 110, 2.
-
10Nowell, P. C.; Hungerford, D. A.; J. Natl. Cancer Inst. 1961, 27, 1013.
-
11Druker, B. J.; Sawyers, C. L.; Kantarjian, H.; Resta, D. J.; Reese, S. F.; Ford, J. M.; Capdeville, R.; Talpaz, M.; N. Engl. J. Med. 2001, 344, 1038.
-
12Daley, G. Q.; Van Etten, R. A.; Baltimore, D.; Science 1990, 247, 824.
-
13Baselga, J.; Science 2006, 312, 1175.
-
14Farderl, S.; Kantarjian, H. M.; Talpaz, M.; O'Brien, S.; Semin. Oncol. 2000, 27, 578.
-
15Cutler, E. G.; Bradford, E. H.; Am. J. Med. Sci. 1878, 75, 74.
-
16Senn, N.; Med. Records New York 1903, 64, 281.
-
17Hoffman, W. J.; Craver, L. F.; J. Am. Med. Assoc. 1931, 97, 836.
-
18Bryant, T.; Guy's Hosp. Rep. 1866, 12, 444.
-
19Wintrobe, M. M.; Ann. Intern. Med. 1947, 27, 529.
-
20Osarogiagbon, U. R.; McGlave, P. B.; Curr. Opin. Hematol. 1999, 6, 241.
-
21Guimarães, J. R. Q.; Manual de Oncologia, 2ª ed., BBS: São Paulo, 2006.
-
22Hehlmann, R.; Heimpel, H.; Hasford, J.; Kolb, H. J.; Pralle, H.; Hossfeld, D. K.; Queisser, W.; Löffler, H.; Hochhaus, A.; Heinze, B.; Blood 1993, 82, 398.
-
23Talpaz, M.; Semin. Hematol. 2001, 38, 22.
-
24Kantarjian, H.; O'Brien, S.; Cortes, J.; Shan, J.; Giles, F.; Garcia-Manero, G.; Verstovsek, S.; Faderl, S.; Rios, M. B.; Talpaz, M.; Cancer 2003, 98, 1430.
-
25Kantarjian, H. M.; J. Clin. Oncol. 1992, 10, 772.
-
26Hahn, E. A.; Glendenning, G. A.; Semin Hematol. 2003, 40, 31.
-
27Guilhot, F.; Chastang, C.; Michallet, M.; N. Engl. J. Med. 1997, 337, 223.
-
28Gratwohl, A.; Hermans, J.; Niederwieser, D.; Frassoni, F.; Arcese, W.; Gahrton, G.; Bandini, G.; Carreras, E.; Vernant, J. P.; Bosi, A.; Bone Marrow Transplant. 1993, 12, 509.
-
29Romeiro, N. C.; Avila, C. M.; Rev. Virtual Quim. 2010, 2, 59.
-
30Yaish, P.; Gazit, A.; Gilon, C.; Levitzki, A.; Science 1988, 242, 933.
-
31Druker, B. J.; Lydon, N. B. Clin. Invest. 2000, 105, 3.
-
32Choi, H. G.; Sim, T.; Gray, N.; Zhou, W.; Chang, J. W.; Zhang, J.; Weisberg, E.; WO 144909 2010 (CA 154:64811)
-
33Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A.; Nat. Rev. Drug Discovery 2002, 1, 493.
-
34Goldman, J. M.; Lancet 2000, 355, 1031.
-
35Hunter, T.; J. Clin. Invest. 2007, 117, 2036.
-
36Lima, L. M.; Quim. Nova 2007, 30, 1456.
-
37Quintás-Cardama, A.; Kantarjian, H.; Cortes, J.; Semin. Hematol. 2010, 47, 371.
-
38Pinto, A. C.; Barreiro, E. J.; Quim. Nova 2013, 36, 1557; Barreiro, E. J.; Pinto, A. C.; Rev. Virtual Quim. 2013, 5, 1059.
-
39Savage, D. G.; Antman, K. H.; N. Engl. J. Med. 2002, 346, 683.
-
40O'Brien, S. G.; Guilhot, F.; Larson, R. A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J. J.; Fischer, T.; Hochhaus, A.; Hughes, T.; N. Engl. J. Med. 2003, 348, 994.
-
41Moen, M. D.; Mckeage, K.; Plosker, G. L.; Siddiqui, M. A.; Drugs 2007, 67, 299.
-
42Apperley, J. F.; Lancet Oncol. 2007, 8, 1018.
-
43Mahon, F. X.; Deininger, M. W. N.; Schultheis, B.; Chabrol, J.; Reiffers, J.; Goldman, J. M.; Melo, J. V.; Blood 2000, 96, 1070.
-
44Silva, V. P. N.; Dissertação de Mestrado, Universidade Nova de Lisboa, Portugal, 2011.
-
45Gorre, M. E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P. N.; Sawyers, C. L.; Science 2001, 293, 876.
-
46Melo, J. V.; Chuah, C.; Cancer Lett. 2007, 249, 121.
-
47a) Zimmermann, J.; EP0564409 1993 (CA120:107056); Zimmermann, J.; US5521184 1996 (CA125:114681)
-
48Boechat, N.; Bastos, M. M.; Duarte, S. L.; Costa, J. C. S.; Mafra, J. C. M.; Daniel, L. C. C.; Rev. Virtual Quím. 2013, 5, 222.
-
49Kompella, A.; Bhujanga, A. K. S. R.; Venkaiah, N. C.; Srinivas, R.; WO04108699 2004 (CA142:56339)
-
50Szakács, Z.; Béni, S.; Varga, Z.; Orfi, L.; Kéri, G.; Noszál, B.; J. Med. Chem. 2005, 48, 249.
-
51Loiseleur, O.; Kaufmann, D.; Abel, S.; Buerger, H. M.; Meisenbach, M.; Schmitz, B.; Sedelmeier, G.; WO03066613 2003 (CA139:180080)
-
52Leonetti, F.; Capaldi, C.; Carotti, A.; Tetrahedron Lett. 2007, 48, 3455.
-
53Liu, Y. F.; Wang, C. L.; Bai, Y. J.; Han, N.; Jiao, J. P.; Qi, X. L.; Org. Process Res. Dev. 2008, 12, 490.
-
54Han, P.; Lin, D.; Ma, C.; Peng, X.; Song, H.; Sun, Z.; Yang, M.; CN101735197 2010 (CA153:116266)
-
55Wehrstedt, K. D.; Wildner, W.; Güthner, T.; Holzrichter, K.; Mertschenk, B.; Ulrich, A.; J. Hazard. Mater. 2009, 170, 829.
-
56Pathi, S. L.; Kankan, R. N.; Puppala, R.; IN20110617 2011 (CA162:365447)
-
57Lee, J. J.; Kang, S. G.; Jang, S. J.; KR2015010142 2015. (CA162:272978)
-
58Xu, H.; Zhang, Z.; Wang, W.; CN104230885 2014 (CA162:137895)
-
59Xu, X.; Min, T.; Shi, W.; Yaoxue Shijian Zazhi 2014, 32, 290.
-
60Gruza, H.; Mirek, S.; Jezewski, A.; Wrzosek, A.; PL215042 B1 2013 (CA161:628850)
-
61Liu, Z.; Yang, K.; Yu, L.; Wu, L.; Guo, X.; Chen, Z.; Huaxue Shiji 2014, 36, 574.
-
62Xu, M.; He, L.; Guo, F.; CN103664874 2014 (CA160:515640)
-
63Zhao, C.; Li, X.; Wang, X.; CN103588754 2014 (CA160:356375)
-
64Fors, B. P.; Watson, D. A.; Biscoe, M. R.; Buchwald, S. L.; J. Am. Chem. Soc. 2008, 130, 13552.
-
65Cortes, J.; Rousselot, P.; Kim, D.; Ritchie, E.; Hamerschlak, N.; Coutre, S.; Hochhaus, A.; Guilhot, F.; Saglio, G.; Apperley, J.; Ottmann, O.; Shah, N.; Erben, P.; Branford, S.; Agarwal, P.; Gollerkeri, A.; Baccarani, M.; Blood 2007, 109, 3207.
-
66O'Hare, T.; Walters, D. K.; Stoffregen, E. P.; Jia, T.; Manley, P. W.; Mestan, J.; Cowan-Jacob, S. W.; Lee, F. Y.; Heinrich, M. C.; Deininger, M. W. N.; Druker, B. J.; Cancer Res. 2005, 65, 4500; O'Hare, T.; Cancer Cell 2009, 16, 401.
-
67Quintas-Cardama, A.; Kantarjian, H.; O'brien, S.; Borthakur, G.; Bruzzi, J.; Munden, R.; Cortes, J.; J. Clin. Oncol. 2007, 25, 3908.
-
68http://www.medicinanet.com.br/bula/4806/sprycel.htm, acessada em Janeiro de 2017.
» http://www.medicinanet.com.br/bula/4806/sprycel.htm -
69Tokarski, J. S.; Newitt, J. A.; Chang, C. Y. J.; Cheng, J. D.; Wittekind, M.; Kiefer, S. E.; Kish, K.; Lee, F. Y. F.; Borzillerri, R.; Lombardo, L. J.; Xie, D.; Zhang, Y.; Klei, H. E.; Cancer Res. 2006, 66, 5790.
-
70Delamain, M. T.; Conchon, M.; Rev. Bras. Hematol. Hemoter. 2008, 30, 37.
-
71Das, J.; Padmanabha, R.; Chen, P.; Norris, D. J.; Doweyko, A. M. P.; Barrish, J.; Wityak, J.; Louis, L. J.; Lee, F. Y. F.; WO04085388 2004 (CA140:253556)
-
72Das, J.; Padmanabha, R.; Chen, P.; Norris, D. J.; Doweyko, A. M. P.; Barrish, J. C.; Wityak, J.; US pat. 04007302 2004 (CA140:253556)
-
73Das, J.; Padmanabha, R.; Chen, P.; Norris, D. J.; Doweyko, A. M. P.; Barrish, J. C.; Wityak, J.; Lombardo, J. J.; Lee, F. Y. F.; US pat. 040054186 2004 (CA140:253556)
-
74Barrish, J. C.; Wityak, J.; Das, J.; Chen, P.; Norris, D. J.; Shieven, G.; US pat. 050288303 2005 (CA133:321878)
-
75Lombardo, L. J.; Lee, F. Y.; Chen, P.; Norris, D.; Barrish, J. C.; Behnia, K.; Castaneda, S.; Cornelius, L. A. M.; Das, J.; Doweyko, A. M.; Fairchild, C.; Hunt, J. T.; Inigo, I.; Johnston, K.; Kamath, A.; Kan, D.; Klei, H.; Marathe, P.; Pang, S.; Peterson, R.; Pitt, S.; Schieven, G. L.; Schmidt, R. J.; Tokarski, J.; Wen, M. L.; Wityak, J.; Borzilleri, R. M.; J. Med. Chem. 2004, 47, 6658.
-
76Das, J.; Chen, P.; Norris, D.; Padmanabha, R.; Lin, J.; Moquin, R. V.; Shen, Cook, L. S.; Doweyko, A. M.; Pitt, S.; Pang, S.; Shen, D. R.; Fang, Q.; de Fex, H. F.; McIntyre, K. W.; Shuster, D. J.; Gillooly, K. M.; Behnia, K.; Schieven, G. L.; Wityak, J.; Barrish, J. C.; J. Med. Chem. 2006, 49, 6819.
-
77Chen, B. C.; Zhao, R.; Wang, B.; WO05076990 2005 (CA143:211902)
-
78Chen, B. C.; Droghini, R.; Lajeunesse, J.; Dimarco, J. D.; Galella, M.; Chidambaram, R.; WO05077945 2005 (CA143:248376)
-
79Hou, W.; Xu, Y.; Yan, R.; Yang, H.; CN101812060 2010 (CA153:406452)
-
80Weisberg, E.; Manley, P. W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S. W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; Kung, A. L.; Mestan, J.; Daley, G. Q.; Callahan, L.; Catley, L.; Cavazza, C.; Azam, M.; Neuberg, D.; Wright, R. D.; Gilliland, D. G.; Griffin, J. D.; Cancer Cell. 2005, 7, 129.
-
81http://www.cancer.gov/cancertopics/druginfo/fda-nilotinib, acessada em Janeiro de 2017.
» http://www.cancer.gov/cancertopics/druginfo/fda-nilotinib -
82Deininger, M. W.; Clin. Cancer Res. 2008, 14, 4027.
-
83Lipton, J. H.; Pasic, I.; Leuk. Res. 2017, 55, 65.
-
84Le Coutre, P.; Ottmann, O. G.; Giles, F.; Kim, D. W.; Cortes, J.; Gattermann, N.; Apperley, J. F.; Larson, R. A.; Abruzzese, E.; O'Brien, S. G.; Kuliczkowski, K.; Hochhaus, A.; Mahon, F. X.; Saglio, G.; Gobbi, M.; Kwong, Y. L.; Baccarani, M.; Hughes, T.; Martinelli, G.; Radich, J. P.; Zheng, M.; Shou, Y.; Kantarjian, H.; Blood 2008, 111, 1834.
-
85Hochhaus, A.; Saglio, G.; Larson, R. A.; Kim, D. W.; Etienne, G.; Rosti, G.; De Souza, C.; Kurokawa, M.; Kalaycio, M. E.; Hoenekopp, A.; Fan, X.; Shou, Y.; Kantarjian, H. M.; Hughes, T. P.; Blood 2013, 121, 3703.
-
86Breitenstein, W.; Furet, P.; Jacob, S.; Manley, P. W.; WO04005281 2004. (CA140:77161)
-
87Abel, S.; Acemoglu, M.; Erb, B.; Krell, C.; Sclafani, J.; Meisenbach, M.; Prashad, M.; Sheih, W. C.; Xue, S.; WO06135640 2006 (CA146:81866)
-
88McKenna, J.; Shieh, W. C.; WO06135641 2006 (CA146:81883)
-
89Huang, W. S.; Shakespeare, W.; Synthesis 2007, 2121.
-
90Chen, Y.; Wang, L.; Zhou, H.; Wang, C.; Zhongguo Yiyao Gongye Zazhi 2009, 40, 401.
-
91Wang, Y.; Li, J.; Kansal, V. K.; Zhu, J.; Lifshitz-Liron, R.; Mistry, D.; Vasoya, S. L.; Ariyamuthu, S.; Piraski, G.; He, X.; WO10009402 2010 (CA152:192145)
-
92Buchwald, S. L.; Ueda, S.; Su, M.; J. Am. Chem. Soc. 2012, 134, 700.
-
93http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm318203.htm, acessada em Janeiro de 2017.
» http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm318203.htm -
94Puttini, M.; Coluccia, A. M.; Boschelli, F.; Cleris, L.; Marchesi, E.; Donella-Deana, A.; Ahmed, S.; Redaelli, S.; Piazza, R.; Magistroni, V.; Andreoni, F.; Scapozza, L.; Formelli, F.; Gambacorti-Passerini, C.; Cancer Res. 2006, 66, 11314.
-
95Remsing Rix, L. L.; Rix, U.; Colinge, J.; Hantschel, O.; Bennett, K. L.; Stranzl, T.; Mueller, A.; Baumgartner, C.; Valent, P.; Augustin, M.; Till, J. H.; Superti-Furga, G.; Leukemia 2009, 23, 477.
-
96http://www.pfizer.com/products/#bosulif, acessada em Janeiro de 2017.
» http://www.pfizer.com/products/#bosulif -
97Boschelli, D. H.; Ye, F.; Wang, Y. D.; Dutia, M.; Johnson, S. L.; Wu, B.; Miller, K.; Powell, D. W.; Yaczko, D.; Young, M.; Tischler, M.; Arndt, K.; Discafani, C.; Etienne, C.; Gibbons, J.; Grod, J.; Lucas, J.; Weber, J. M.; Boschelli, F.; J. Med. Chem. 2001, 44, 3965.
-
98Sutherland, K. W.; Feigelson, G. B.; Boschelli, D. H.; Blum, D. M.; Strong, H. L.; US pat. 0043537 2005 (CA142:261413)
-
99Yin, X. J.; Xu, G. H.; Sun, X.; Peng, Y.; Ji, X.; Jiang K.; Li, F.; Molecules 2010, 15, 4261.
-
100http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203469lbl.pdf, acessada em Janeiro de 2017.
» http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203469lbl.pdf -
101Leonetti, F.; Stefanachi, A.; Nicolotti, O.; Catto, M.; Pisani, L.; Cellamare, S.; Carotti, A.; Curr. Med. Chem. 2011, 18, 2943.
-
102Zou, D.; Huang, W. S.; Thomas, R. M.; Romero, J. A. C.; Qi, J.; Wang, Y.; Zhu, X.; Shakespeare, W.C.; Sundaramoorthi, R.; Metcalf, C.A. III; Dalgarno, D. C.; Sawyer, T. K. WO 075869 2007 (CA147:143466)
-
103Kovi, R.; Kannapan, J.; Thakor, S. F.; Patel, R. A.; US pat. 0343282 2014 (CA161:769806)
-
104Ren, X.; Pan, P.; Zhang, Z.; Wang, D.; Lu, X.; Li, Y.; Wen, D.; Long, H.; Luo, J.; Feng, Y.; Zhuang, X.; Zhang, F.; Liu, J.; Leng, F.; Lang, X.; Bai, Y.; She, M.; Tu, Z.; Pan, J.; Ding, K.; J. Med. Chem. 2012, 56, 879.
Datas de Publicação
-
Publicação nesta coleção
Ago 2017
Histórico
-
Recebido
05 Out 2016 -
Aceito
10 Jan 2017