Abstract
Three simple, sensitive, economical and reproducible spectrophotometric methods (A, B and C) are described for determination of mesalamine in pure drug as well as in tablet dosage forms. Method A is based on the reduction of tungstate and/or molybdate in Folin Ciocalteu's reagent; method B describes the reaction between the diazotized drug and α-naphthol and method C is based on the reaction of the drug with vanillin, in acidic medium. Under optimum conditions, mesalamine could be quantified in the concentration ranges, 1-30, 1-15 and 2-30 µg mL-1 by method A, B and C, respectively. All the methods have been applied to the determination of mesalamine in tablet dosage forms. Results of analysis are validated statistically.
mesalamine; spectrophotometric analysis; validation
NOTA TÉCNICA
Simple and sensitive spectrophotometric methods for the analysis of mesalamine in bulk and tablet dosage forms
Bala Sekaran Chandra* * e-mail: balumphil@gmail.com ; Siva Santhosh Bhogela; Manjusha Shaik; Chaitanya Sravanthi Vadlamudi; Meghana Chappa; Naga Sirisha Maddirala
Department of Biotechnology, J.K.C. College, Guntur, Andhra Pradesh, India
ABSTRACT
Three simple, sensitive, economical and reproducible spectrophotometric methods (A, B and C) are described for determination of mesalamine in pure drug as well as in tablet dosage forms. Method A is based on the reduction of tungstate and/or molybdate in Folin Ciocalteu's reagent; method B describes the reaction between the diazotized drug and α-naphthol and method C is based on the reaction of the drug with vanillin, in acidic medium. Under optimum conditions, mesalamine could be quantified in the concentration ranges, 1-30, 1-15 and 2-30 µg mL-1 by method A, B and C, respectively. All the methods have been applied to the determination of mesalamine in tablet dosage forms. Results of analysis are validated statistically.
Keywords: mesalamine; spectrophotometric analysis; validation.
INTRODUCTION
Mesalamine (MSL), an anti-inflammatory drug, is used to treat and to maintain remission of mild to moderate ulcerative colitis or Crohn's disease.1-4 Mesalamine has the chemical name 5-amino-2-hydroxybenzoic acid; its structural formula is given in Figure 1. Mesalamine is thought to be the major therapeutically active part of the sulfasalazine molecule in the treatment of ulcerative colitis. Mesalamine is produced by bacterial action on sulfasalazine in the colon.5 The mechanism of action of mesalamine is unknown, but appears to be topical rather than systemic. Mucosal production of arachidonic acid metabolites (both through the cyclooxygenase pathways and through the lipoxygenase pathways) and hydroxyeicosatetraenoic acids is increased in patients with chronic inflammatory bowel disease, and it is possible that mesalamine diminishes inflammation by blocking cyclooxygenase and inhibiting prostaglandin production in the colon.6 Mesalamine acts as a scavenger of oxygen-derived free radicals,7 which are produced in greater numbers in patients with inflammatory bowel disease.
The therapeutic importance of MSL has necessitated the development of analytical methods for its determination in dosage forms in compliance with good manufacturing standards. The drug is officially listed in 2000 United States Pharmacopoeia and the Official method of its determination with high-performance liquid chromatography using mobile phase containing tetrabutylammonium hydrogen sulphate as an ion-pairing agent.8 Various other techniques such as UV-spectrophotometry,9 potentiometric titration,10 micellar electrokinetic chromatography,11 differential pulse voltammetry,12 HPLC and LC/MS/MS have been reported for assaying of MSL in pharmaceutical dosage forms and biological fluids.13-26 Visible spectrophotometry, because of simplicity and cost effectiveness, sensitivity and selectivity and fair accuracy and precision, has remained competitive in the era of chromatographic techniques for pharmaceutical analysis. Few visible spectrophotometric methods are found in the literature for the assay of MSL. In a method reported by Rafael et al.,10 hydrogen donating ability of MSL to 1, 1-diphenyl-2-picrylhydrazyl radicals is used as basis for its determination in pharmaceutical dosage forms. Patel et al.,27 reported three methods for the assay of MSL in tablet dosage forms with Bratton-Marshall reagent, p-dimethylaminobenzaldehyde and Gibb's reagent. Reaction of MSL with potassium iodate and potassium iodide was used by Navya Sloka et al.,28 for determining MSL. The reported visible spectrophotometric methods suffer from one or the other disadvantage such as poor accuracy and precision, use relatively expensive reagents, heating, control of temperature, narrow range of linear response and low molar absorptivity. Considering this drawback, there was a need to develop more advantageous spectrophotometric methods for the determination of MSL in pharmaceutical dosage forms.
This paper describes three sensitive, accurate, precise, simple and economical methods for the determination of MSL in dosage forms. The proposed methods are validated as per the guidelines of the International Conference on Harmonisation.29
EXPERIMENTAL
Instrumentation
Elico Double beam UV visible spectrophotometer model SL-159 with 1 cm matched quartz cells was used for all spectral and absorbance measurements.
Materials and reagents
All the chemicals and reagents used were of analytical grade and solutions were prepared in double distilled water. 1:3 diluted Folin-Ciocalteu reagent (FCR) was prepared by dissolving accurately measured 25 mL of commercially available FCR (Fisher Scientific, Mumbai) in 75 mL of water. 3% Vanillin (Merck, Mumbai) in methanol (Merck, Mumbai) and aqueous solutions of 0.5 M NaOH (Merck, Mumbai), 4 M NaOH, 1% sodium nitrite (Sdfine-Chem limited, Mumbai), 1 N HCl (Fisher Scientific, Mumbai), 5 N HCl, 2% sulphamic acid (Merck, Mumbai), and 0.5% α-Naphthol (Molychem, Mumbai) were prepared for analysis.
MSL was kindly provided by Matrix laboratories, Hyderabad, India, and was used as received. Commercial samples of tablets namely Asacol (Win medicare, New Delhi), Meseacol (Sun Max pharma limited, Ahmedabad) and Salofalk GR (Germen remedies, Mumbai) containing 400, 400 and 250 mg of mesalamine, respectively were purchased from local pharmacy market and employed in the study.
Preparation of drug solutions
Stock solution of MSL was prepared by dissolving 100 mg of MSL in 50 mL of distilled water in a 100 mL volumetric flask and then make up to the mark with distilled water (1 mg mL-1). Working standard solutions containing 100 µg mL-1 (methods A and C) and 50 µg mL-1 (method B) of MSL were prepared by further dilution of stock solution.
Recommended procedures
Method A
Into a series of 10 mL volumetric flasks, volumes of MSL working standard solution equivalent to 1-30 µg mL-1 were transferred. To each flask, 1 mL of 1:3 diluted FCR and 2 mL of 0.5 M NaOH was added and brought up to the volume with water and mixed well. The absorbance of the colored product formed after 15 min at room temperature (25 ± 1 °C) was measured at 655 nm against the reagent blank prepared similarly omitting the drug.
Method B
Aliquots of the working standard solution containing 1-15 µg mL-1 of MSL were transferred into a series of 10 mL volumetric flasks. A volume of 1.0 mL of 1% sodium nitrite was added to each flask followed by 1.0 mL of 1 N HCl and cooled in an ice bath. After 5 min, 1.0 mL of 2% sulphamic acid was added to each flask. Then volumes of 0.5 mL of 0.5% α-naphthol and 1 mL of 4 M NaOH solutions were added. The contents were diluted to the mark with distilled water and mixed well. The absorbance of the colored azo-dye was measured at 510 nm against the reagent blank prepared similarly omitting the drug.
Method C
Into a series of 10 mL volumetric flasks, volumes of MSL working standard solution equivalent to 2-30 µg mL-1 were transferred. To each flask, 1 mL of 3% vanillin and 2 mL of 5 N HCl was added and brought up to the volume with water and mixed well. The absorbance of the chromogenic Schiff's base formed at room temperature (25 ± 1 °C) was measured at 395 nm against the reagent blank prepared similarly omitting the drug.
In all the above procedures, the analytical curves were constructed by plotting the absorbance versus final concentration of MSL. The amount of the drug was computed either from analytical curve or from regression equation.
Procedure for tablet dosage forms
Five tablets were weighed accurately and ground into a fine powder. An accurately weighed amount of the finely powdered tablets equivalent to 100 mg of MSL was transferred into a 100 mL calibrated flask, shaken for about 10 min with 50 mL water and diluted to volume with water, mixed well and filtered using a Whatman No. 1 filter paper. The filtrate was further diluted to working concentrations for use in the spectrophotometric methods A (100 µg mL-1), B (50 µg mL-1) and C (100 µg mL-1). The assay was completed following the recommended procedures for the determination of MSL.
Procedure for reference method
Aliquots of standard solution of MSL ranging from 0.5 to 5 mL (1000 µg mL-1) were transferred into a series of 10 mL volumetric flasks. To each flask, 2 mL of ethanolic para dimethyl amino benzaldehyde and 1 mL of nitric acid (4 N) were added. The solutions were warmed on a water bath for 10 min at 60-70 °C. The resulting solutions were cooled to room temperature and the volume was brought up to the mark with water. The absorbance of the light yellow colored chromogen was measured at 440 nm against the reagent blank. The amount of MSL present in the sample solution was computed from the respective analytical curve or regression equation.
RESULTS AND DISCUSSION
Reaction mechanism
Method A
In the alkaline medium, MSL react instantaneously with the FCR resulting in blue colored product. The colored product is due to a common reaction mechanism i.e., the oxidation of the drug and the reduction of FCR (blue color). Folin-Ciocalteu reagent is a mixture of acids and involves the following chemical species: 3 H2O.P2O5.13 WO3. 5MoO3.10 H2O and 3H2O.P2O5.14WO3.4MoO 3.10 H2O
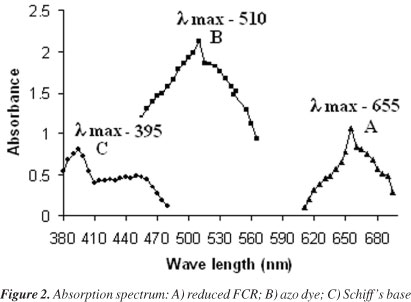
The blue color formation by FCR with drug seems analogous to Folin phenol protein reaction.30 MSL probably reduce 1, 2 or 3 oxygen atoms of tungstate and/or molybdate in the FCR producing reduced species which have a characteristic intense blue colour with Λmax at 655 nm (Figure 2). The colored product was stable for about 5 h.
Method B
Primary aromatic amine upon treatment with nitrous acid in an ice-cooled solution forms diazonium salt

Under proper conditions diazonium salts react with certain aromatic compounds to yield products of general formula Ar-N=N-Ar1, called as azo compounds and the reaction is called as diazo coupling reaction.31

Method B involves the diazo-coupling reaction of diazonium salt of MSL with α-naphthol in alkaline medium. MSL was treated with sodium nitrite solution at 0-5 ºC in acidic medium, resulting in the formation of diazonium chloride ion. The excess nitrite is removed with sulphamic acid. The formed diazonium chloride ion is coupled with α-naphthol to form a red colored azo dye in an alkaline medium having Λmax at 510 nm (Figure 2). The red color azo dye is stable for about 3 h. The possible reaction mechanism had been shown in Scheme 1.
Method C
Schiff bases are formed by a condensation reaction between an aromatic amine and an aldehyde or ketone.32

Aromatic aldehydes have lead to numerous applications as analytical reagents. Aromatic aldehydes like vanillin, cinnamaldehyde and p-dimethylaminobenzaldehyde have been applied to the colorimetric determination of drugs with aromatic amines in acidic medium.33-35 Method C is based on the formation of chromogenic schiff's base between the primary amino group of MSL and aldehyde group of vanillin. The produced chromogen exhibits Λmax at 395 nm (Figure 2) and is stable for 2 h. The probable reaction mechanism is given in Scheme 2.
Optimization of reagent concentration
Preliminary experiments were performed to determine the optimum concentration of the reagents used in the estimation of MSL. The influence of the reagent concentration on the rate of reaction was studied and optimized. The optimum concentration of the reagents was maintained throughout the experiment.
Method A
Effect of concentration of Folin-Ciocalteu reagent
The influence of the volume of 1:3 diluted FCR on the intensity of the color developed at constant MSL concentration (20 µg mL-1) was examined in the range 0.2-2.0 mL of FCR. The maximum absorbance was obtained with 1 mL of FCR; above this volume the absorbance remained unchanged. Therefore, 1 mL of FCR was used in all further measurements.
Effect of NaOH
The effect of the volume of 0.5 M NaOH on the color development was investigated by adding different volumes (0.5-3.0 mL) of 0.5 M NaOH to 1 mL of MSL (20 µg). It was found that the maximum absorbance of the blue color was reached with 2 mL of the 0.5 M NaOH, and remained constant with higher volumes. Therefore, 2 mL of the 0.5 M NaOH was used throughout the experimental investigations.
Effect of time
To optimize the reaction time for color development, 1 mL of FCR and 2 mL of 0.5 M NaOH and 1 mL of MSL (20 µg) were added and kept at room temperature for varied time. The maximum intensity of color was obtained at 15 min and remained constant up to 5 h. Therefore, 15 min of reaction time was used throughout the determination process.
Method B
Effect of sodium nitrite and sulphamic acid
The effect of adding various amounts of sodium nitrite solution on absorbance of 10 µg mL-1 MSL was examined. The concentration of sodium nitrite was varied between 0.2-2.0 mL of 1% sodium nitrite in water. The results showed that 1 mL of 1% sodium nitrite gave maximum absorbance. Thus, 1 mL of 1% sodium nitrite was chosen for the procedure. The excess of nitrite could be removed by the addition of 1 mL of 2% sulfamic acid. An excess of sulphamic acid has no effect on color.
Effect of HCl
The influence of acidity on the development of color was studied using different volumes (0.2-2.0 mL) of 1 N HCl. The maximum color intensity was observed with 1 mL of 1 N HCl and therefore 1 mL of 1 N HCl solution was used throughout the experiment.
Effect of concentration of coupling agent
To optimize the concentration of coupling agent, different volumes (0.1-1.5 mL) of 0.5% α-naphthol were added to the mixture under study. It was found that 0.5 mL of α-naphthol solution was sufficient for maximum and stable color development. There was a decrease in absorbance at lower concentration of 0.5% α-naphthol, whereas no change in absorbance was observed at higher concentration.
Effect of NaOH
The effect of NaOH concentration on the absorbance was studied; volumes from 0.2-2.0 mL of 4 M NaOH solutions were examined. The investigations showed that 1 mL of NaOH gave maximum absorbance, therefore 1 mL of 4 M NaOH was chosen for the procedure. Other alkaline solutions (1% aqueous sodium carbonate and 4 M potassium hydroxide) were tried, but best results were obtained by using NaOH.
Method C
Effect of concentration of vanillin
The effect of the concentration of 3% vanillin on the colored product was studied using different volumes in the range 0.2-2.0 mL. The absorbance increased with increase in concentration of 3% vanillin. The maximum absorbance was obtained with 1 mL; above this concentration absorbance remained constant. Thus, 1 mL was used for subsequent studies.
Effect of HCl
To achieve the maximum absorbance value for an analyte by the formation of stable chromogenic Schiff's base, the selection of optimum acidity is essential. The influence of acidity on the development of color was studied using different volumes (0.5-4.0 mL) of 5 N HCl. The maximum color intensity was observed with 2 mL of
5 N HCl and therefore 2 mL of 5 N HCl solution was used throughout the experiment.
Method validation
Analytical data
The optical characteristics such as Beer's law limits, Sandell's sensitivity and molar absorptivity were calculated for the proposed methods and the results are summarized in Table 1. Regression analysis of the Beer's law plot at their Λmax revealed a good correlation. Graphs of absorbance versus concentration are described by regression equation Y = mx + c (where Y is the absorbance, m is the slope, x is the concentration of drug in µg mL-1 and c is the intercept) obtained by least squares method.
Limit of detection and limit of quantification
Limit of detection (LOD) and limit of quantification (LOQ) decide about the sensitivity of the method. LOD is the lowest detectable concentration of the analyte by the method while LOQ is the minimum quantifiable concentration. LOD and LOQ were calculated by equations:36 LOD = δ3.3/s and LOQ = δ10/s, respectively, where δ is the standard deviation of five replicate determination values of blank and s is slope of calibration. The results (Table 1) indicating proposed methods are highly sensitive.
Precision and accuracy
The precision and accuracy of the proposed method was determined by carrying out 6 replicate determinations of fixed concentration of MSL, within Beer's limit, by the proposed methods. The calculated standard deviation and relative standard deviation for the proposed methods showed that the precision was good (Table 1). The accuracy of an analytical method expresses the closeness between the reference value and the found value. Accuracy was evaluated as percentage relative error between the measured concentrations and concentrations taken for MSL. The results obtai-ned are compiled in Table 1 and shown that the accuracy is good.
Recovery studies
The validity and reliability of the proposed methods was assessed by the recovery studies via standard addition method. The recovery studies were carried out by adding a fixed concentration of bulk sample of MSL to the pre-analyzed formulation and the total concentration was once again determined using the proposed methods. The % recovery of the added pure drug was calculated as:37

where Ct is the total drug concentration measured after standard addition; Cs, drug concentration in the formulation sample; Ca, drug concentration added to formulation. The results (Table 2) revealed that any small change in the drug concentration in the solutions could be accurately determined by the proposed analytical methods. The closeness of the recoveries suggests a lack of interference from tablet excipients and thereby establishes some degree of selectivity.
Robustness
A measure of the capacity of the analytical procedure to remain unaffected by small but deliberate variations in method performance parameters, which provides an indication of its reliability during normal usage. The robustness of the proposed methods A, B and C relative to each operational parameter was challenged. The operational parameters investigated were as follows:
Method A
Volume of 1:3 diluted FCR: 1 ± 0.2 mL; volume of 0.5 M NaOH:
2 ± 0.2 mL; reaction time: 15 ± 2 min.
Method B
Volume of 1% sodium nitrite: 1 ± 0.2 mL; volume of 1 N HCl:
1 ± 0.2 mL; volume of 0.5% α-naphthol: 0.5 ± 0.1 mL; volume of 4 M NaOH: 1 ± 0.2 mL.
Method C
Volume of 3% vanillin: 1 ± 0.2 mL; volume of 5 N HCl: 2 mL ± 0.2 mL.
The robustness of the proposed methods was assessed by analyzing the content of mesalamine in tablet dosage form (Asacol, containing 400 mg of active component) under variable experimental conditions. A sample solution containing 30 µg mL-1 (methods A and C) and 15 µg mL-1 (method B) of the drug was prepared and assayed 5 times by the proposed methods. The mean percent recovery and relative standard deviation were calculated. The mean percent recovery was in the range of 99.54-100.46, 100.15-101.78 and 99.28-100.55, with relative standard deviation of 0.91-1.12%, 0.73-1.05% and 0.97-1.49% for methods A, B and C, respectively. The results of mean recovery and relative standard deviation indicate good sensitivity and appreciable recovery. Thus, the operational conditions of the proposed methods are robust.
Application of the proposed methods to tablet dosage forms
The proposed methods were applied to the determination of MSL in commercially available tablets. The same samples were analyzed, simultaneously by the proposed methods and the reference method.27 The results were compared statistically by applying Student's t-test for accuracy and the variance ratio F-test for precision with results from the reference method at a 95% confidence level. The calculated t-test and F-values (Table 3) did not exceed the tabulated values of 2.77 and 6.39, respectively, indicating no significant difference between the proposed methods and the reference method in terms of accuracy and precision.
CONCLUSIONS
The proposed methods were found to be simple, economical, selective and sensitive. The methods do not require heat treatment, expensive reagents and sophisticated instruments. The statistical parameters and recovery study data clearly indicate the reproducibility and accuracy of the methods. Analysis of the authentic samples containing MSL showed no interference from the common excipients. Hence, these methods could be considered for the determination of MSL in the quality control laboratories.
ACKNOWLEDGEMENTS
The authors express their gratitude to the management J.K.C College, Guntur, Andhra Pradesh for providing research facilities.
REFERENCES
1. Hanauer, S.; Schwartz, J.; Robinson, M.; Roufail, W.; Arora, S.; Cello, J.; Safdi, M.; Am. J. Gastroenterol. 1993, 88, 1188.
2. Sninsky, C. A.; Cort, D. H.; Shanahan, F.; Powers, B. J.; Sessions, J. T.; Pruitt, R. E.; Jacobs, W. H.; Lo, S. K.; Targan, S. R.; Cerda, J. J.; Ann. Int. Med. 1991, 115, 350.
3. Bondesen, S.; Hegnhoj, J.; Larsen, F.; Hansen, S. H.; Hansen, C. P.; Rasmussen, S. N.; Dig. Dis. Sci. 1991, 36, 1735.
4. Thomson, A. B. R.; Aliment. Pharmacol. Ther. 1991, 5, 449.
5. Scott, H.; Noah Wilson, R.; Athos, B.; Anna, Z.; Rie, M.; Peter, B.; Frederick, M.; Sarathchandra, R.; Robert, B.; Sonia, F.; Inflamm. Bowel. Dis. 2006, 12, 251.
6. Small, R. E.; Schraa, C. C.; Pharmacotherapy1994, 14, 385.
7. Sutherland, L. R.; May, G. R.; Shaffer, E. A.; Ann. Int. Med. 1993, 118, 540.
8. United States Pharmacopoeia, 24th ed., United States Pharmacopoeial Convention: Rockville, 2000 [CD-ROOM] .
9. Rakesh Kumar, S.; Pankaj Singh, P.; Pragya, G.; Int. J. Pharm. Sci. Res. 2010, 1, 44.
10. Rafael, J. A.; Jabor, J. R.; Casagrande, R.; Georgetti, S. R.; Borin, M. F.; Fonseca, M. J. V.; Rev. Bras. Cienc. Farm. 2007, 43, 97.
11. Gotti, R.; Pomponio, R.; Bertucci, C.; Cavrini, V.; J. Chromatogr., A 2001, 916, 175.
12. Biljana, N.; Branimir, I.; J. Pharm. Biomed. Anal. 2003, 31, 169.
13. Haney, P. W.; Dash, A. K.; J. Chromatogr., A 1997, 765, 233.
14. Cendrowska, I.; Drewnowska, M.; Grzezliewicz, A.; Butkiewicz, K.; J. Chromatogr., A 1990, 509, 195.
15. Kersten, B. S.; Catalano, T.; Rozenman, Y.; J. Chromatogr., A 1991, 588, 187.
16. Norlander, B.; Gotthard, R.; Strom, M.; Aliment. Pharmacol. Ther. 1990, 4, 497.
17. Shaw, P. N.; Sivner, A. L.; Aarons, L.; Houston, J. B.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 1983, 274, 393.
18. De Vos, M.; Verdievel, H.; Schoonjans, S.; Praet, M.; Bogaert, M.; Barbier, F.; Gut 1992, 33, 1338.
19. Chungi, V. S.; Rekhi, G. S.; Shargel, L.; J. Pharm. Sci. 1989, 78, 235.
20. Palumbo, G.; Carlucci, G.; Mazzeo, P.; Frieri, G.; Pimpo, M. T.; Fanini, D.; J. Pharm. Biomed. Anal. 1995, 14, 175.
21. Palumbo, G.; Bacchi, S.; Primavera, L.; Palumbo, P.; Carlucci, G.; Biomed. Chromatogr. 2005, 19, 350.
22. Hussain, F. N.; Ajjan, R. A.; Moustafa, M.; Anderson, J. C.; Riley, S. A.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 1998, 716, 257.
23. Naganuma, M.; Iwao, Y.; Ogata, H.; Inoue, N.; Funakoshi, S.; Yamamoto, S.; Nakamura, Y.; Ishii, H.; Hibi, T.; Inflamm. Bowel. Dis. 2001, 7, 221.
24. Bystrowska, B.; Nowak, J.; Brandys, J.; J. Pharm. Biomed. Anal. 2000, 22, 341.
25. Nobilis, M.; Vybíralová, Z.; Sládková, K.; Lísa, M.; Holcapek, M.; Kvetina, J.; J. Chromatogr., A 2006, 1119, 299.
26. Pastorini, E.; Locatelli, M.; Simoni, P.; Roda, G.; Roda, E.; Roda, A.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2008, 872, 99.
27. Patel, K. M.; Patel, C. N.; Panigrahi, B.; Parikh, A. S.; Patel, H. N.; J. Young Pharmacists 2010, 2, 284.
28. Navya Sloka, S.; Gurupadayya, B. M.; Aswani Kumar, C. H.; Der Pharma Chemica 2010, 2, 389.
29. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guidelines, Validation of Analytical Procedures: Text and Methodology Q2 (R1), Current Step 4 version, Nov. 1996, Geneva, Nov. 2005.
30. Peterson, G. L.; Anal. Biochem. 1979, 100, 201.
31. Connors, K. A.; Reaction Mechanisms in Organic Analytical Chemistry, John Wiley & Sons: New York, 1973.
32. Cordes, E. H.; Jencks, W. P.; J. Am. Chem. Soc. 1962, 84, 832.
33. Siddappa, K.; Mallikarjun, M.; Reddy, P. T.; Tambe, M.; Eclet. Quím. 2008, 33, 41.
34. Srikar, A.; Channabasavaraj, K. P.; Dharmamoorty, G.; Nataraj, V.; Chinnappa, C.; Venu Babu, T.; J. Pharm. Sci. Res. 2009, 1, 13.
35. Rind, F. M. A.; Laghari, M. G. H.; Memon, A. H.; Mughal, U. R.; Almani, F.; Memon, N.; Khuhawar, M. Y.; Maheshwari, M. L.; Pak. J. Anal. Environ. Chem. 2008, 9, 43.
36. Sarika, N.; Snehal, P.; Sharda, S.; Dhanashri, S.; Yogesh, P.; J. Pharm. Sci. Res. 2010, 2, 137.
37. Gupta, K. R.; Tajne, M. R.; Wadodkar, S. G.; Indian J. Pharm. Sci. 2008, 70, 511.
Recebido em 26/10/10; aceito em 30/1/11; publicado na web em 1/4/11
- 1. Hanauer, S.; Schwartz, J.; Robinson, M.; Roufail, W.; Arora, S.; Cello, J.; Safdi, M.; Am. J. Gastroenterol. 1993, 88, 1188.
- 2. Sninsky, C. A.; Cort, D. H.; Shanahan, F.; Powers, B. J.; Sessions, J. T.; Pruitt, R. E.; Jacobs, W. H.; Lo, S. K.; Targan, S. R.; Cerda, J. J.; Ann. Int. Med. 1991, 115, 350.
- 3. Bondesen, S.; Hegnhoj, J.; Larsen, F.; Hansen, S. H.; Hansen, C. P.; Rasmussen, S. N.; Dig. Dis. Sci. 1991, 36, 1735.
- 4. Thomson, A. B. R.; Aliment. Pharmacol. Ther. 1991, 5, 449.
- 5. Scott, H.; Noah Wilson, R.; Athos, B.; Anna, Z.; Rie, M.; Peter, B.; Frederick, M.; Sarathchandra, R.; Robert, B.; Sonia, F.; Inflamm. Bowel. Dis. 2006, 12, 251.
- 6. Small, R. E.; Schraa, C. C.; Pharmacotherapy1994, 14, 385.
- 7. Sutherland, L. R.; May, G. R.; Shaffer, E. A.; Ann. Int. Med. 1993, 118, 540.
- 8. United States Pharmacopoeia, 24th ed., United States Pharmacopoeial Convention: Rockville, 2000 [CD-ROOM]
- 9. Rakesh Kumar, S.; Pankaj Singh, P.; Pragya, G.; Int. J. Pharm. Sci. Res. 2010, 1, 44.
- 10. Rafael, J. A.; Jabor, J. R.; Casagrande, R.; Georgetti, S. R.; Borin, M. F.; Fonseca, M. J. V.; Rev. Bras. Cienc. Farm. 2007, 43, 97.
- 11. Gotti, R.; Pomponio, R.; Bertucci, C.; Cavrini, V.; J. Chromatogr., A 2001, 916, 175.
- 12. Biljana, N.; Branimir, I.; J. Pharm. Biomed. Anal. 2003, 31, 169.
- 13. Haney, P. W.; Dash, A. K.; J. Chromatogr., A 1997, 765, 233.
- 14. Cendrowska, I.; Drewnowska, M.; Grzezliewicz, A.; Butkiewicz, K.; J. Chromatogr., A 1990, 509, 195.
- 15. Kersten, B. S.; Catalano, T.; Rozenman, Y.; J. Chromatogr., A 1991, 588, 187.
- 16. Norlander, B.; Gotthard, R.; Strom, M.; Aliment. Pharmacol. Ther. 1990, 4, 497.
- 17. Shaw, P. N.; Sivner, A. L.; Aarons, L.; Houston, J. B.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 1983, 274, 393.
- 18. De Vos, M.; Verdievel, H.; Schoonjans, S.; Praet, M.; Bogaert, M.; Barbier, F.; Gut 1992, 33, 1338.
- 19. Chungi, V. S.; Rekhi, G. S.; Shargel, L.; J. Pharm. Sci. 1989, 78, 235.
- 20. Palumbo, G.; Carlucci, G.; Mazzeo, P.; Frieri, G.; Pimpo, M. T.; Fanini, D.; J. Pharm. Biomed. Anal. 1995, 14, 175.
- 21. Palumbo, G.; Bacchi, S.; Primavera, L.; Palumbo, P.; Carlucci, G.; Biomed. Chromatogr. 2005, 19, 350.
- 22. Hussain, F. N.; Ajjan, R. A.; Moustafa, M.; Anderson, J. C.; Riley, S. A.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 1998, 716, 257.
- 23. Naganuma, M.; Iwao, Y.; Ogata, H.; Inoue, N.; Funakoshi, S.; Yamamoto, S.; Nakamura, Y.; Ishii, H.; Hibi, T.; Inflamm. Bowel. Dis. 2001, 7, 221.
- 24. Bystrowska, B.; Nowak, J.; Brandys, J.; J. Pharm. Biomed. Anal. 2000, 22, 341.
- 25. Nobilis, M.; Vybíralová, Z.; Sládková, K.; Lísa, M.; Holcapek, M.; Kvetina, J.; J. Chromatogr., A 2006, 1119, 299.
- 26. Pastorini, E.; Locatelli, M.; Simoni, P.; Roda, G.; Roda, E.; Roda, A.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2008, 872, 99.
- 27. Patel, K. M.; Patel, C. N.; Panigrahi, B.; Parikh, A. S.; Patel, H. N.; J. Young Pharmacists 2010, 2, 284.
- 28. Navya Sloka, S.; Gurupadayya, B. M.; Aswani Kumar, C. H.; Der Pharma Chemica 2010, 2, 389.
-
29International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guidelines, Validation of Analytical Procedures: Text and Methodology Q2 (R1), Current Step 4 version, Nov. 1996, Geneva, Nov. 2005.
- 30. Peterson, G. L.; Anal. Biochem. 1979, 100, 201.
- 31. Connors, K. A.; Reaction Mechanisms in Organic Analytical Chemistry, John Wiley & Sons: New York, 1973.
- 32. Cordes, E. H.; Jencks, W. P.; J. Am. Chem. Soc. 1962, 84, 832.
- 33. Siddappa, K.; Mallikarjun, M.; Reddy, P. T.; Tambe, M.; Eclet. Quím. 2008, 33, 41.
- 34. Srikar, A.; Channabasavaraj, K. P.; Dharmamoorty, G.; Nataraj, V.; Chinnappa, C.; Venu Babu, T.; J. Pharm. Sci. Res. 2009, 1, 13.
- 35. Rind, F. M. A.; Laghari, M. G. H.; Memon, A. H.; Mughal, U. R.; Almani, F.; Memon, N.; Khuhawar, M. Y.; Maheshwari, M. L.; Pak. J. Anal. Environ. Chem. 2008, 9, 43.
- 36. Sarika, N.; Snehal, P.; Sharda, S.; Dhanashri, S.; Yogesh, P.; J. Pharm. Sci. Res. 2010, 2, 137.
- 37. Gupta, K. R.; Tajne, M. R.; Wadodkar, S. G.; Indian J. Pharm. Sci. 2008, 70, 511.
Publication Dates
-
Publication in this collection
01 Aug 2011 -
Date of issue
2011
History
-
Accepted
30 Jan 2011 -
Received
26 Oct 2010