Resumo
This review presents studies on methyl coenzyme M reductase, the biological system Factor 430 (F430) and the use of nickel(II) complexes as structural and functional models. The ability of F430 and nickel(II) macrocycle complexes to mediate the reductive dehalogenation of cyclohexyl halogens and the CH3-S bond cleavage of methyl CoM (by sodium borohydride and some intermediate species) proposed for the catalytic cycle of the biological system F430 was reviewed. The importance of the structure of the nickel complexes and the condition of the catalytic reduction reaction are also discussed.
nickel complexes; coenzyme M reductase; F430
nickel complexes; coenzyme M reductase; F430
REVISÃO
Metil coenzima M redutase (MCR) e o fator 430 (F430)
Methyl coenzyme M reductase and the factor 430
Shirley Nakagaki* * e-mail: shirley@quimica.ufpr.br ; Geraldo Roberto Friedermann; José Maurício Almeida Caiut
Departamento de Química, Universidade Federal do Paraná, CP 19081, 81531-990 Curitiba - PR, Brasil
ABSTRACT
This review presents studies on methyl coenzyme M reductase, the biological system Factor 430 (F430) and the use of nickel(II) complexes as structural and functional models. The ability of F430 and nickel(II) macrocycle complexes to mediate the reductive dehalogenation of cyclohexyl halogens and the CH3-S bond cleavage of methyl CoM (by sodium borohydride and some intermediate species) proposed for the catalytic cycle of the biological system F430 was reviewed. The importance of the structure of the nickel complexes and the condition of the catalytic reduction reaction are also discussed.
Keywords: nickel complexes; coenzyme M reductase; F430.
INTRODUÇÃO
A ocorrência de sistemas biológicos dependentes de níquel é muito inferior a outros metais, tais como ferro, cobre e zinco. No entanto, o pequeno número de biomoléculas contendo níquel encontrado nos sistemas vivos é compensado pela variedade de funções que estas moléculas desempenham, tornando o estudo bioquímico e bioinorgânico do níquel relevante e interessante1.
São conhecidos quatro tipos de enzimas dependentes de níquel (urease, monóxido de carbono desidrogenase - CODH, hidrogenase de níquel H2ase e metil coenzima M redutase MCR) e todos foram encontrados em organismos inferiores, embora uma urease tenha sido também encontrada em plantas2. O papel do níquel em organismos superiores não é ainda totalmente conhecido, embora seja um elemento traço essencial em animais3.
Enzimas de níquel do tipo MCR são conhecidas como participantes proeminentes no metabolismo de bactérias anaeróbicas, como por ex., em bactérias metanogênicas os poucos organismos conhecidos que utilizam CO2 e H2 para a produção de CH4 (metanogênese). Outros organismos podem utilizar, por ex., metanol e ácido acético em lugar de CO2.
Estima-se que juntos, estes organismos e o MCR, sejam responsáveis pela produção anual de 1 bilhão de t de metano4. Tal quantidade torna relevante o estudo destes sistemas biológicos no contexto atual de controle de emissão de carbono e sua fixação5, visto que a concentração de metano atmosférico tem aumentado durante os últimos 300 anos de 0,7 para 1,7 ppm6 (Equação 1, DGº calculado = - 130 kJ mol-1).
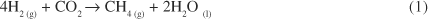
METIL COENZIMA M REDUTASE (MCR) E O FATOR 430 (F430)
A enzima MCR tem sido intensamente estudada e isolada de vários organismos. Várias revisões e trabalhos tratando do assunto foram publicadas nos últimos anos3,6,7.
O MCR é uma grande e complexa enzima de cerca de 300kDa com sua estrutura dividida em subunidades (em geral 2a, 2b e 2g)8. Tem sido observado que a enzima está associada a cofatores, sendo que apenas a localização do Cofator denominado Fator 430 (F430) é conhecida. Cada subunidade a contém uma molécula de F430, portanto, a enzima contém duas moléculas de F430 como grupo cromóforo prostético.
O fator F430 presente na metil coenzima M redutase (MCR), atuante na etapa final da metanogênese, catalisa a clivagem redutiva do metil-coenzima M.
Metil-coenzima M pode ser representado, por ex., por MeCoM ou CH3-S-CoM, tioester cofator metil coenzima-M, onde CH3-S-CoM é equivalente à forma simplificada CH3SR- (CH3-S-CH2CH2 SO3-) como mostrado na Equação (2). Este último é o produto resultante das etapas anteriores da metanogênese. Na presença de um cofator tiol, um agente redutor, produz metano9, como mostrado na Equação (2) (DGº= - 45kJ mol-1)9

cofator tiol = HS-HTP = N-7-mercapto-heptanoiltreonina fosfato, também chamado de Coenzima B e representado muitas vezes simplesmente por HS-CoB.
O fator F430, assim denominado devido à sua banda característica no espectro de absorção no UV-visível, apresenta um composto macrocíclico tetrapirrólico de Ni(II)10. A estrutura de raios-X associada a resultados obtidos com diferentes técnicas espectroscópicas, realizadas principalmente a partir da década de 90, colaboraram para o melhor entendimento da espécie F43011. Os resultados baseados na estrutura de raios-X com compostos modelos e com a enzima, somados aos estudos realizados utilizando a técnica de ressonância magnética nuclear, sugeriram que tal macrociclo assemelha-se à estrutura de uma porfirina altamente saturada12 (Figura 1) denominada níquel corfinóide, com duas moléculas de água como ligantes axiais, resultando em um sistema de configuração eletrônica S=1 (pseudooctaédrico d8)13. No entanto, muitos estudos com o cofator nativo na presença de solventes fracamente coordenantes levaram à detecção de outras espécies, indicando que o fator F430 pode acomodar geometrias de coordenação de quadrado planar a trigonal-bipiramidal, fato que sugere a facilidade da redução do Ni(II) a Ni(I) 14.
Os estudos cristalográficos de compostos-modelo, tais como níqueltetraazamacrociclos e niquelporfirinóides (Figura 2), levaram à conclusão que as distâncias maiores ou menores observadas para a ligação Ni-N podem ser diagnósticas de uma estrutura do macrociclo mais planar (quando a distância é maior) ou mais distorcida ("ruffled" quando a distância é menor)11. Estes estudos reforçaram a idéia de que tal distorção associava-se à facilidade de redução do metal de Ni(II) para Ni(I), fato relevante no ciclo catalítico a ele associado, sugerindo assim que o F430 teria uma estrutura bem flexível e concordante com o esperado para uma corfina, um macrociclo contendo menor número de duplas ligações conjugadas se comparado com uma estrutura porfirínica (Figura 2a). As corfinas apresentam quatro carbonos meso como as porfirinas, no entanto, as corfinas não apresentam conjugação entre estes quatro anéis pirrólicos do macrociclo (Figura 1). Estas mudanças de conjugação levam à ausência de bandas características no espectro de absorção UV-Vis, bem como da fluorescência encontrada na porfirina desmetalada (a camada base livre). Baseado nestes resultados concluiu-se que o macrociclo biológico poderia rapidamente acomodar mudanças estruturais e também ser modelado facilmente por sistemas macrocíclicos sintéticos de níquel altamente saturados (flexíveis) (Figura 2d).
Estudos de dicroísmo circular magnético15 e XANES ("X-ray absorption near-edge")16 do F430 no complexo enzimático sugeriram que o Ni(II) se encontrava em um ambiente hexacoordenado octaédrico, com dois oxigênios como átomos doadores de ligantes axiais.
Estudos de EXAFS ("extended X-ray fine structure absorption") sugeriram também que as formas quadrado planar de spin baixo do F430 e seu diepímero (posições 12 e 13 do anel, Figura 1) são caracterizadas por uma curta ligação N-Ni (1,9 Å). No entanto, quando ocorre uma coordenação axial e conversão para o estado de spin alto esta distância aumenta para 2,1 Å 17-19. Deformações similares do macrociclo também foram observadas durante a redução do níquel em isobacterioclorinas19 e complexos tetraazaanulenos18, enquanto que a formação de uma espécie Ni(II) p-ânion radical em clorinas e porficenos de níquel(II) não levaram a alterações da distância Ni(II)-N19.
No entanto, a informação mais relevante que a técnica de EXAFS trouxe para a elucidação do sistema F430 foi fornecer sólida evidência de que a forma ativa do MCR contém um composto Ni(I), visto que a redução do F430 levou a mudanças drásticas nas distâncias Ni-N (o valor médio da distância Ni(II)-N observada na estrutura cristalina foi de 1,86 Å, enquanto que para Ni(I)-N foram observadas dois grupos de distâncias 1,88 e 2,03 Å 17). Estes resultados sugeriram que o esqueleto do ligante no F430 deveria ser altamente flexível e poderia acomodar facilmente diferentes distâncias causadas tanto pela presença do metal nativo Ni(II) (menor distância), quanto pelo metal reduzido Ni(I) (maior distância). A redução centrada no metal foi observada também em estudos de EXAFS de sistemas-modelo saturados. No entanto, reduções centradas no ligante com a formação de espécies radicalares p aniônicas foram observadas em sistemas-modelo mais insaturados, como as porfirinas20, e tal redução não causou mudanças de geometria na vizinhança do níquel. Estes resultados reforçaram a idéia de que a habilidade do macrociclo para mudar a conformação ajuda a controlar o sítio de redução (metal X macrociclo) e pode determinar a química dos produtos de redução17,18,21. Estes efeitos estruturais podem ser parte de uma influência mais geral da conformação nas propriedades físicas e químicas dos cromóforos porfirínicos e grupos prostéticos22.
Estudos de RPE da MCR obtida da bactéria metanogênica M. marburgensis em atmosfera de H2/CO2 levaram a um sistema RPE silencioso. Este estado inativo da enzima foi denominado de MCRsilencioso23. Em atmosfera de H2 puro foi detectado a presença de dois sinais de RPE atribuídos às espécies de Ni(I)24, os quais foram designados por MCR-red1 (axial) e MCR-red2 (rômbico). A enzima no estado MCR-red1 ou red2 mostrou alta atividade. Em meio de N2/CO2 a enzima apresentou baixa atividade mas apresentou, também, um terceiro sinal de RPE denominado MCR-oxi1. Outros sinais de RPE distintos também foram observados na presença de inibidores e substratos halogenados, sugerindo que o substrato parece se ligar diretamente a uma espécie Ni(I)-F430 ou perto o suficiente para mudar o ambiente da espécie Ni(I)24. Estudos de espectroeletroquímica do Fator F430Ni(II)/Ni(I) da bactéria metanogênica em solução aquosa também sugeriram que a espécie ativa do cofator pode conter Ni(I)25. Este e outros trabalhos têm levado a muitos estudos da química catalítica redutiva do F430 e de derivados de níquel porfirina19,26.
O envolvimento de Ni(I) no MCR ocasionou grande interesse na química das formas Ni(I) de F430 e isobacterioclorinas (OeiBC), o único composto tetrapirrólico conhecido que favoreceu o isolamento de complexos de Ni(I) reduzidos (Figura 2c)26,27.
SISTEMAS-MODELO FUNCIONAIS DE F430
Na literatura existem estratégias diferentes de investigação muito utilizadas quando se buscam compostos-modelo funcionais de F430: abordagens mecanísticas, visando entender as espécies envolvidas no processo de conversão de substrato a metano e, utilização de diferentes substratos buscando catalisadores de redução.
Quanto ao substrato, tem sido observada, principalmente, a investigação da redução de haletos de alquila e arila catalisada por F430 e seus possíveis modelos e estudo da clivagem da ligação C-S de compostos semelhantes ao substrato biológico, metil-CoM (ou CH3-S-CoM). Neste caso, o substrato mais freqüentemente investigado é o sal sintético metilcoenzimaM (simplificadamente MeCoM) de amônio, obtido pela reação de iodeto de metila e o sal 2-mercaptoetanosulfônico de sódio, em solução de amônia concentrada28.
Observam-se poucos relatos de tentativas de clivagem da ligação S-CH3 da MeCoM com compostos-modelo de Ni(II) ou Ni(II)-F430, levando à produção de metano. Alguns trabalhos pioneiros foram relatados no final da década de 80 e na década de 909. Experimentos utilizando o cofator F430 previamente reduzido, contendo uma espécie Ni(I), na presença de iodeto de metila e DMF, levaram à produção de metano. No entanto, o mesmo resultado não foi observado com a simples adição da MeCoM nas mesmas condições29. O metano foi formado quando o cloreto de metila foi cataliticamente reduzido por citrato de titânio(III) na presença da coenzima F430 livre e em solução aquosa30. Drain e colaboradores31, no começo da década de 90, relataram a produção de metano a partir da utilização de MeCoM e compostos de metila correlacionados utilizando sistemas-modelo macrocíclicos de Ni(II) em solução aquosa (Figura 3a). No entanto, durante a reação de obtenção de metano, oxigênio molecular também foi formado. As evidências experimentais levaram a crer que este complexo-modelo envolvia um sistema Ni(III)/Ni(II), em vez do sistema Ni(II)/Ni(I) admitido no sistema biológico.

Outros complexos de Ni(II) foram utilizados por Zilbermann e colaboradores14 para obtenção de Ni(I) através de radiólise de pulso. Estes macrocíclicos (Figuras 3b e c) foram escolhidos por apresentarem potenciais redox baixos para o par Ni(II)/Ni(I) em solução aquosa (-0,98 e -1,25 V, respectivamente, versus ESC) e muito similares ao potencial encontrado para o cofator nativo F430 (-0,98 V)25. Na presença de Ni(I), este sistema converteu MeCoM a metano com 10% de rendimento.
Estudos mecanísticos envolvendo o composto Ni(I) isobacterioclorina e MeCoM na presença de sais de amônio (que resultou na formação de pequena quantidade de metano) e tetrafenilfosfônio (ausência de formação de metano) sugeriram que uma possível espécie intermediária Ni-H também poderia estar envolvida na clivagem da ligação S-CH3 da MeCoM para produção de metano32, implicando em que uma espécie Ni-H semelhante também poderia estar envolvida na química do F43033. Tais estudos contribuíram para a discussão a respeito das espécies participantes no ciclo catalítico do F430 e o mecanismo envolvido quando compostos-modelo são utilizados.
Além disso, experimentos com solventes deuterados realizados por Stiles34, na desalogenação redutiva de haletos de arila catalisada por um macrocíclico de níquel (II) (Figura 3d), também sugeriram o envolvimento de uma espécie Ni-H como intermediário de reação.
Recentemente, Stolzenberg e colaboradores9 investigaram a possibilidade do envolvimento de espécies Ni-H ou Ni(I) como intermediário na clivagem da ligação S-CH3 de MeCoM e desalogenação de haletos de alquila na presença de boroidreto de sódio, utilizando uma série de oito complexos macrocíclicos de Ni(II), dentre eles o NiTMTAA, (Figura 4) e outros tetraazas (Figura 3d). Este e outros trabalhos indicam que complexos macrocíclicos de níquel são catalisadores homogêneos eficientes para redução de haletos de alquila por NaBH4 e, no entanto, mostram-se ineficientes para catálise da clivagem de ligação S-C da metil coenzima M (MeCoM). A redução de haleto de cicloexila (no caso, bromocicloexano) foi substancialmente afetada pelo solvente e pela estrutura dos catalisadores de níquel.
COMPLEXOS DE NÍQUEL(II) DIBENZOTETRAAZAANULENOS (NiTMTAA)
Complexos sintéticos do tipo dibenzotetraaza têm sido investigados como modelo do F430. Apesar da semelhança estrutural com as porfirinas, o menor número de possibilidades de ligações conjugadas encontradas nos ligantes desta família faz com que apresentem uma estrutura mais flexível, se comparada ao rígido anel relativamente planar das porfirinas. A flexibilidade do ligante é suficiente para permitir a ligação de ligantes exógenos nos sítios de coordenação axial35, além de favorecer processos redutivos centrados no metal, o que os diferencia de muitas porfirinas de níquel onde predominantemente se observam processos redutivos centrados no anel porfirínico, gerando espécies p-aniônicas26.
Os metalocomplexos denominados M(II)TMTAA (onde TMTAA é igual ao ligante di-aniônico 6,8,15,17-tetrametildibenzo[b,i][1,4,8,11]tetraaza[14]anuleno, Figura 4), ou simplesmente chamados de dibenzotetraazaanulenos, são estudados desde há muito tempo, devido à sua semelhança estrutural com metaloporfirinas. Ambos, tetraazas e porfirinas, são ligantes dianiônicos conjugados com 4 nitrogênios como pontos coordenantes ao metal36.
Devido a estas propriedades estruturais e semelhanças com o grupo prostético do sistema biológico, compostos Ni(II)TMTAA têm sido utilizados na catálise redutiva de desalogenação de haletos de alquila, visto que a enzima parece estar também envolvida na desalogenação redutiva de hidrocarbonetos clorados, por bactérias metanogênicas35.
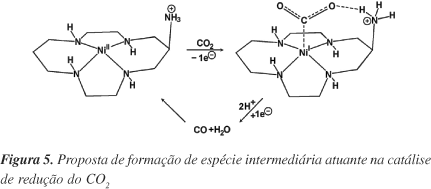
Embora compostos dibenzotetraazaanulenos de Ni(II) tenham estabilidade menor que as porfirinas (sob condições ordinárias sofrem decomposição oxidativa parcial levando à desmetalação do anel37,38), a facilidade de obtenção do composto usando o próprio íon Ni(II) como "template", a partir da reação dos precursores amina e dicetona39, pode levar a uma maior utilização como catalisador-modelo de sistema F430. Além disso, podem atuar em outras reações de redução que apresentam potencial uso industrial ou ambiental. Por ex., foi observado que modificações nas posições meso de macrocíclicos com estruturas mais simples que os da família dos NiTMTAA favoreceram a produção eletroquímica de CO. A Figura 5 representa uma reação catalisada por um composto tetraaza, que possui um grupo NH3 substituído na posição meso. Esta presença possibilitou o aumento da produção de CO, em relação ao tetraaza sem o substituinte, sugerindo uma interação efetiva do grupo NH3 com o substrato CO2, entre o oxigênio do CO2 e o hidrogênio do grupo NH340.
Na área da química bioinorgânica, complexos dibenzotetraazaanulenos de metais de transição são alvos de estudos como modelos para outros sítios ativos de certas enzimas de ocorrência natural. O complexo [CoTMTAA] (Figura 6) tem sido estudado por Moore e colaboradores como modelo da vitamina B12, devido à sua semelhança estrutural com o anel corrina desta vitamina. Além disso, os complexos tetraazas de Mn(III) têm sido investigados como compostos-modelo e relacionados ao estudo da função catalítica do citocromo P-450 na oxidação de substratos orgânicos41,42.
Verifica-se na literatura21 que a atividade catalítica de complexos macrocíclicos, bem como seu comportamento de coordenação, podem ser controlados pela estrutura do ligante. Portanto, é de grande interesse nesta área de trabalho alterar não só o sítio metálico do complexo, mas também a forma estrutural do ligante macrocíclico. A adição de substituintes no anel ou a construção do anel no interior de suportes sólidos, como por ex., zeólita, ou ainda a imobilização de metalocomplexos em suportes flexíveis (como polímeros orgânicos) ou rígidos, (como sílica gel funcionalizada ou vidros porosos) pode muitas vezes produzir distorções interessantes na molécula, causando mudanças estruturais de grande interesse nos estudos catalíticos, facilitando a obtenção de estados de oxidação do metal de relevância catalítica43.
Complexos macrocíclicos tetraazaanuleno de níquel(II) foram utilizados como catalisadores homogêneos modelos do F430 em reações de redução de haletos de alquila por boroidreto de sódio, em misturas de solvente álcool/éter e acetonitrila/éter. Em ambos os casos, complexos de hidreto de níquel (Ni-H) foram sugeridos como espécies intermediárias. O NiTMTAA exibiu, no espectro de RPE, um sinal rômbico típico de Ni(I) na presença de grande excesso de NaBH4 (excesso molar de 100 vezes)35,44. Observou-se também que o sinal de RPE diminuiu rapidamente quando um substrato halogenado (bromocicloexano) foi adicionado, indicando que a espécie Ni(I) estava envolvida na catálise de desalogenação. A grande anisotropia do sinal de RPE da espécie Ni(I) comparado com amostras de [Ni(I)(TMTAA)Na(THF)3], produzidas através da redução de Ni(II) a Ni(I) por amálgama de sódio45, foi atribuída à coordenação de H- ou BH4- ao Ni(I).
Tem-se observado também que na presença de excesso do agente redutor a reação pode ocorrer de forma radicalar, sem auxílio do catalisador, e de forma catalítica com o auxílio do complexo de níquel. A via radicalar leva a uma eficiência menor na produção de cicloexano. Tal fato foi observado por Arai quando estudou os mecanismos prováveis desta reação (3) na presença de NaBH4 e na sua forma deuterada (NaBD4)35. A Equação 3 representa a adição de deutério ao substrato observado na via radicalar proposta para explicar o processo de redução do substrato.

O mecanismo radicalar utiliza o hidrogênio liberado pelo NaBH4. Para reduzir as chances de ocorrência deste processo foi adicionado, ao meio reacional, etanol anidro para que este solvente fosse usado como doador de hidrogênio.
Nesta condição considerada ótima, foram testadas as atividades catalíticas de dois novos complexos de níquel que diferem do NiTMTAA pela inserção de 2 ou 4 átomos de cloro nos anéis benzênicos (Figura 7). Tais alterações levaram a um aumento da distorção da molécula, favorecendo o processo de oxirredução do Ni(II), fato este muito desejado nas reações catalíticas modelo do F43038.
A presença de átomos de cloro na estrutura do ligante influenciou positivamente o rendimento da reação, sugerindo que a estrutura "ruffled", imposta pela interação dos grupos metila com os átomos de cloro21, pode facilitar a formação de espécies intermediárias envolvidas na reação de catálise43.
CICLO CATALÍTICO PROPOSTO PARA O MCR-F430
Encontram-se na literatura uma série de estudos anteriores a 1997, utilizando diferentes compostos como modelos estruturais de F430 para entender o sistema biológico enquanto estrutura e funcionamento do grupo prostético níquel(II) corfinóide. Muitos destes trabalhos foram citados nesta revisão. Estes estudos, em geral, usaram duas classes de macrociclos de níquel: compostos porfirinóides mais insaturados que o macrociclo do F430, (tais como clorinas e isobacterioclorinas) e ligantes macrocíclicos mais reduzidos, tais como os tetraazaciclotetradecanos (Figura 2).
Após 1997, Thauer publicou seus resultados da resolução da estrutura cristalina de MCR em diferentes condições23,46,47. Baseando-se em resultados espectroscópicos obtidos na última década, bem como em trabalhos recentes de Thauer6, o ciclo catalítico de atuação do F430 foi proposto (Figura 8).
Nesta proposta, o ciclo inicia-se quando o substrato Metilcoenzima M, MeCoM (no ciclo representado por CH3-S-CoM) se difunde até o sítio de reação, através de canais. Devem ocorrer mudanças conformacionais da proteína para permitir também a aproximação de outro substrato, a coenzima B (no ciclo representado por HS-CoB) (A). Como observado em diversos estudos espectroscópicos, a enzima ativa contém um sítio de Ni(I). A ligação carbono-enxofre da MeCoM é atacada pela espécie nucleófila Ni(I)-F430, levando a uma espécie metil-Ni(III)-F430 e um ânion coenzima M-tiolato (B). O Ni(III), por ser um oxidante forte, deve agir retirando um elétron do ânion coenzima M, ocasionando a formação de uma espécie radicalar com concomitante redução do níquel (C) e formação de metano. Para isso, é necessária a intervenção de uma espécie próton doadora. No caso desta proposta de ciclo, o resíduo de aminoácido tirosina (Tyrb367) faria este papel (D). Pela abstração de próton do substrato coenzima B, resultando no ânion tiolatocoenzima B (E), o ânion resíduo de aminoácido voltaria à sua forma neutra. A proposta desta espécie metil-Ni(III) de vida curta é compatível com os resultados recentemente apresentados por Thauer23,46, entretanto, em propostas anteriores de ciclo catalítico tal espécie não foi considerada.
Nesta etapa do ciclo, o ânion radical tiolato (na coenzima M) e o ânion tiolato (na coenzima B) levariam à formação da ligação S-S da espécie heterodissulfeto radical (F) e, finalmente, o elétron desta espécie radicalar seria responsável pela redução da espécie Ni(II)-F430 para Ni(I)-F430 (G), encerrando-se o ciclo com a produção da espécie CoB-S-S-CoM.
Além desta proposta de ciclo, um mecanismo alternativo foi proposto por Pelmenschikov48, derivado de cálculos teóricos baseados nas estruturas cristalinas conhecidas do MCR. Diferentemente da proposta de Thauer, neste mecanismo alternativo a ligação CH3-S presente na metil coenzima M seria clivada homoliticamente e um radical metila, assim como um complexo coenzima M-Ni(II), seria formado. A abstração de hidrogênio da coenzima B pelo radical metila levaria à formação do metano e as espécies restantes de coenzima M e B dariam origem ao produto dissulfeto e Ni(I), reiniciando o ciclo. A apresentação de tal ciclo alternativo, no mesmo ano do ciclo proposto por Thauer, indica o interesse intenso dos pesquisadores na área e aponta para a necessidade de novos experimentos com a enzima nativa, bem como com compostos-modelo que levem a um melhor entendimento do ciclo catalítico.
CONCLUSÃO
A elucidação completa da metanogênese nos últimos 30 anos representou um marco na bioquímica3. Este processo foi conduzido em boa parte pelos grupos do Prof. Wolfe, em Urbana Illinois (USA)7,49 e Prof. Thauer, em Marbung (Alemanha)4.
Observa-se que, principalmente na ultima década, vários trabalhos foram apresentados na literatura com sistemas-modelo ou não, na tentativa de entender como a enzima Metilcoenzima M redutase funciona. Nestes trabalhos destacam-se algumas questões que ainda não foram definitivamente respondidas, apesar da quantidade considerável de publicações na área e das propostas recentes de ciclo catalítico9: qual é o papel da espécie Ni(I) na Ni-F430? A espécie Ni(I)-F430 pode reagir diretamente com a MeCoM ou é necessária uma ativação prévia do substrato ou do F430? Como a enzima direciona o substrato especificamente para a clivagem da ligação C-S da parte CH3-S na metil coenzima M? Qual é o papel do cofator tiol HS-HTP? Como e quando a ligação S-S do produto heterodissulfeto (CoM-S-S-HTP) se forma? Todas as espécies intermediárias presentes no ciclo catalítico foram definitivamente determinadas e caracterizadas?
Duas conclusões são evidentes quando se comparam os trabalhos na literatura, utilizando tetraazas de níquel (I), Ni(I)(OeiBC) e Ni(I)-F430 em estudos investigativos de propostas funcionais: nenhum trabalho foi conclusivo para demonstrar que complexos mononucleares de níquel podem clivar a ligação CH3-S da MeCoM para produzir metano com alto rendimento e, a facilidade de um complexo em reduzir halocarbonos ou outros eletrófilos não implica necessariamente em facilidade de clivagem da ligação C-S de MeCoM.
Embora se observe um interesse crescente por esta área, refletido pelo aumento de publicações, devido às incertezas ainda existentes a respeito dos mecanismos operantes na redução de halocarbonos e na clivagem da ligação C-S, bem como na identificação de espécies intermediárias e intermediárias ativas (como por ex., complexos de Ni(I) ou Ni-H ou Ni(III) recentemente colocada por Thauer), esta área de trabalho constitui ainda um campo de pesquisa grande e desafiante.
REFERÊNCIAS
1. Kaim, W.; Schwederski, B.; Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life. An introduction and guide, John Wiley & Sons Ltd.: England, 1994.
2. Hausinger R. P.; Biochemistry of Nickel, Plenum Press: New York, 1993.
3. Telser, J.; Struct. Bonding 1998, 31; Ghosh, A.; Wondimagegn, T.; Ryeng, H.; Curr. Opin. Chem. Biol. 2001, 5, 744; Piskorski, R.; Jaun, B.; J. Am. Chem. Soc. 2003, 125, 13120; Goenrich, M.; Mahlert, F.; Duin E. C.; Bauer, C.; Jaun, B.; Thauer, R. K.; J. Biol. Inorg. Chem. 2004, 9, 691.
4. Thauer R. K.; Biol. Hoppe-Seyler 1985, 366, 103; Ankel-Fuchs, D.; Huster, R.; Morschel, E.; Albracht S. P. J.; Thauer, R. K.; Syst. Appl. Microbiol. 1986, 7, 383.
5. Sanquetta, C. R.; Balbinot, R.; Ziliotto M. A. B., eds.; Fixação do Carbono: atualidades, projetos de pesquisa, 2 Simpósio Latino Americano sobre fixação de carbono, Ed. A M Impressos: Curitiba, 2004.
6. Shima, S.; Warkentin, E.; Thauer, R. K.; Ermler, U.; J. Biosci. Bioeng. 2002, 93, 519; Craft, J. L.; Horng, Y.-C.; Ragsdale, S. W.; Brunold, T. C.; J. Biol. Inorg. Chem. 2004, 9, 77; Duin, E. C;. Signor, L; Piskorski , R.; Mahlert, F.; Clay, M. D.; Goenrich, M.; Thauer, R. K.; Jaun, B.; Johnson, M. K.; J. Biol. Inorg. Chem. 2004, 9, 563.
7. DiMarco A. A.; Bobic T. A.; Wolfe R. S.; Annu. Rev. Biochem. 1990, 59, 355.
8. Jablonski, P. E.; Ferry J. G.; J. Bacteriol. 1991, 173, 2481.
9. Stolzenberg, A. M.; Zhang, Z.; Inorg. Chem. 1997, 36, 593; Grabarse , W.; Mahlert, F.; Shima, S.; Thauer, R.K.; J. Mol. Biol. 2000, 303, 329.
10. Gunsalus R. P.; Wolfe R. S.; FEMS Microbiol. Lett. 1978, 3, 191.
11. Farber, G.; Keller, W.; Kratky, C.; Jaun, B.; Pfalz, A.; Spinner, C.; Kobelt. A.; Eschenmoser, A.; Helv. Chim. Acta 1991, 74, 697; Won, H.; Olson, K. D.; Hare, D. R.; Wolfe, R. S.; Kratky, C.; Summers, M. F.; J. Am. Chem. Soc. 1992, 114, 6880.
12. Fassler, A.; Kobelt, A. Pfaltz, A.; Eschenmoser, A.; Bladon, C.; Battersby, A. R.; Thauer, R. K.; Helv. Chim. Acta 1985, 68, 2287; AnkelFuchs, D.; Thauer R. K.; The Bioinorganic Chemistry of Nickel, J. R. Lancaster, ed., VCH Publishers: New York, 1988, p. 93.
13. Pfaltz, A.; Livinston, D.A.; Jaun, B.; Diekert, G.; Thauer, R. K.; Eschenmose, A.; Helv. Chim. Acta 1985, 68, 1338.
14. Zilbermann, I.; Golub, G.; Cohen, H.; Meyerstein, D.; Inorg. Chim. Acta 1994, 227,1.
15. Cheesman, M. R.; Ankel-Fuchs, D.; Thauer, R. K.; Thompson, A.; J. Biochem. 1989, 260, 613; Hamilton, C. L.; Scott, R. A.; Johnson, M. K.; J. Biol. Chem. 1989, 264, 11605.
16. Shiemke, A. K.; Kaplan, W. A.; Hamilton, C. L.; Shelnutt, J. A.; Scott, R. A.; J. Biol. Chem. 1989, 264, 7276.
17. Furenlid, L. R.; Renner, M. W.; Fajer, J.; J. Am. Chem. Soc.1990, 112, 8987.
18. Furenlid, L. R.; Renner, M. W.; Szalda, D. J.; Fujita, E.; J. Am. Chem. Soc. 1991, 113, 883.
19. Furenlid, L. R.; Renner, M. W.; Smith, K. M.; Fajer, J.; J. Am. Chem. Soc. 1990, 112, 1634.
20. Kadish, K. M.; Franzen, M. M.; Han, B. C.; Araullo-McAdams, C.; Sazou, D.; J. Am. Chem. Soc. 1991, 113, 512.
21. Renner, M. W.; Furenlid L. R.; Barkigia, K. M.; Forman A.; Shim, H. K.; Simpson, D. J.; Smith, K. M.; Fajer, J.; J. Am. Chem. Soc. 1991, 113, 6891.
22. Barkigia, K. M.; Berber, M. D.; Fajer, J.; Medforth, C. J.; Renner, M. W.; Smith, K. M.; J. Am. Chem. Soc. 1990,112, 8851; Renner, M. W.; Cheng, R.-J.; Chang, C. K.; Fajer, J.; J . Phys. Chem. 1990, 94, 8508.
23. Ermler, U.; Grabarse, W.; Shima, S.; Goubeaud, M.; Thauer, R. K.; Science 1997, 278, 1457.
24. Albracht, S. P. J.; Ankel-Fuchs, D.; Van der Zwaan, J. W.; Fontjin, R. D.; Thauer, R. K. Biochim. Biophys. Acta 1986, 870, 50; Albracht, S. P. J.; Ankel-Fuchs, D.; Bkher, R.; Ellerman, J.; Moll, J.; Van der Zwaan, J. W.; Thauer, R. K.; Biochim. Biophys. Acta 1988, 955, 86; Rospert, S.; Bocher, R.; Albracht, S. P. J.; Thauer, R. K.; FEBS Lett. 1992, 291, 371.
25. Hollinges, C.; Pierich, A. J.; Reijerse E. J.; J. Am. Chem. Soc. 1993, 115, 5651.
26. Stolzenberg, A. M.; Stershic, M. T.; Inorg. Chem. 1987, 26, 1970; Stolzenberg, A. M.; Stershic, M.; J. Am. Chem. Soc. 1988, 110, 5397; Renner, M. W.; Forman, A.; Fajer, J.; Simpson, D.; Smith, K. M.; Barkigia, K. M.; J. Biophys. 1988, 53, 277; Renner, M. W.; Forman, A.; Wu, W.; Chang, C. K.; Fajer, J.; J. Am. Chem. Soc. 1989, 111, 8618; Latos-Grazynski, L.; Olmstead, M. M.; Balch, A. L.; Inorg. Chem. 1989, 28, 4066; Lexa, D.; Momenteau, M.; Mispelter. J.; Saveant, J. M.; Inorg. Chem. 1989, 28, 30; Kadish, K. M.; Sazou, D.; Maiya, G. B.; Han, B. C.; Liu, Y. M.; Saoiabi, A.; Ferhat, M.; Guilard, R.; Inorg. Chem. 1989, 28, 2542; Nahor, G. S.; Neta, P.; Hambright, P.; Robinson, L. R.; Harriman, A.; J. Phys. Chem. 1990, 94, 6659; Kadish. K. M.; Franzen, M. M.; Han, B. C.; Araullo-McAdams, C.; Sazou, D.; J. Am. Chem. Soc. 1991, 113, 512; Craft, J. L.; Horng, Y.-C.; Ragsdale, S. W.; Brunold, T. C.; J. Am. Chem. Soc. 2004, 126, 4068.
27. Furenlid, L. R.; Renner, M. W.; Barkigia, K. M.; Forman, A., Shim, H. K.; Simpson, D. J.; Smith, K. M.; Fajer, J. J.; J. Am. Chem. Soc. 1991, 113, 6891.
28. Taylor, C. D.; Wolfe, R. S.; Biochem. 1978, 17, 2374.
29. Jaun, B.; Pfaltz, A.; J. Chem. Soc. Chem. Commun. 1988, 293.
30. Krone, U. E.; Laufer, K.; Thauer, R. K.; Hogenkam, H. P. C.; Biochem. 1989, 28, 10061.
31. Drain, C. M.; Sable, D. B.; Corden, B. B.; Inorg. Chem. 1990, 29, 1429.
32. Lahiri, G. K.; Schussel, L. J.; Stolzenberg, A. M.; Inorg. Chem. 1992, 31, 4991; Lahiri, G. K.; Stolzenberg, A M.; Inorg. Chem. 1993, 32, 4409.
33. Lin, S. K.; Jaun, B.; Helv. Chim. Acta 1992, 75, 1478.
34. Stiles, M.; J. Org. Chem. 1994, 59, 5381.
35. Arai, T.; Kashitani, K.; Kondo, H.; Sakaki, S.; Bull. Chem. Soc. Jpn. 1994, 67, 705; Todd, L. N.; Zimmer, M.; Inorg. Chem. 2002, 41, 6831.
36. Ram, M. S.; Riordan, C. G.; Ostrander, R.; Rheingold, A. L.; Inorg. Chem. 1995, 34, 5884.
37. Friedermann, G. R.; Dissertação de Mestrado, Universidade Federal do Paraná, Brasil, 2001.
38. Caiut, J. M.; Dissertação de Mestrado, Departamento de Química, Universidade Federal do Paraná, Brasil, 2002.
39. Goedken, V. L.; Weiss, M. C.; Inorg. Synt. 1976, 20, 115; Cotton, F. A.; Czuchajowska, J.; Polyhedron 1990, 9, 2553; Wang, Z.; Sakata, K.; Hashimoto, M.; Polyhedron 1989, 17, 4451; Wang, Z.; Hu, N.; Sakata, K.; Hashimoto, M.; J. Chem. Soc. Dalton Trans. 1999, 1695; Jager, E. G.; Z. Anorg. Allg. Chemic. 1969, 364, 177; Purrington, S., Knight, B., Bereman, R.; Inorg. Chim. Acta 1994, 223, 187; Hochesang, P. J.; Bereman, R. D.; Inorg. Chim. Acta 1989, 162,191.
40. Kimura, E.; Haruta, M.; Koike, T.; Shionaya, M.; Takenouchi, K.; Itaka, Y.; Inorg. Chem. 1993, 32, 2779.
41. Mountford, P.; Chem. Soc. Rev. 1998, 27, 105.
42. Eilmes, J.; Polyhedron 1992,11, 581.
43. Caiut, J. M. A.; Nakagaki, S.; Friedermann, G. R.; Drechsel, S. M.; Zarbin, A. J. G.; J. Mol. Catal. A: Chem. 2004, 222, 213.
44. Arai, T.; Kondo, H.; Sakaki, S.; J. Chem. Soc. Dalton Trans. 1992, 2753.
45. Ciurli, S.; Meter, E. M.; Floriani, C.; Chiesi-Villa, A. Guastini, C.; J. Chem. Soc. Chem. Commun. 1987, 281.
46. Grabarse, W.; Mahlert, F.; Duin, E. C.; Goubeaud, M.; Shimia, S.; Thauer, R. K.; Lamzin, V.; Ermler, U.; J. Mol. Biol. 2001, 309, 315.
47. Shima, S.; Goubeaud, M.; Vinzenz, D.; Thauer, R. K.; Ermler, U.; J. Biochem. 1997, 121, 829.
48. Pelmeschikov, V.; Blomberg, M. R. A.; Siegbahn, P. E. M.; Crabtree, R. H.; J. Am. Chem. Soc. 2002, 24, 946.
49. Hartzel P. L.; Wolfe, R. S.; Syst. Appl. Microbiol. 1986, 7, 376; Wolfe, R. S.; Ann. Rev. Microbiol. 1991, 45, 1.
Recebido em 10/2/05; aceito em 27/9/05; publicado na web em 31/3/06
- 1. Kaim, W.; Schwederski, B.; Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life An introduction and guide, John Wiley & Sons Ltd.: England, 1994.
- 2. Hausinger R. P.; Biochemistry of Nickel, Plenum Press: New York, 1993.
- 3. Telser, J.; Struct. Bonding 1998, 31;
- Ghosh, A.; Wondimagegn, T.; Ryeng, H.; Curr. Opin. Chem. Biol. 2001, 5, 744;
- Piskorski, R.; Jaun, B.; J. Am. Chem. Soc 2003, 125, 13120;
- Goenrich, M.; Mahlert, F.; Duin E. C.; Bauer, C.; Jaun, B.; Thauer, R. K.; J. Biol. Inorg. Chem 2004, 9, 691.
- 4. Thauer R. K.; Biol. Hoppe-Seyler 1985, 366, 103;
- Ankel-Fuchs, D.; Huster, R.; Morschel, E.; Albracht S. P. J.; Thauer, R. K.; Syst. Appl. Microbiol 1986, 7, 383.
- 5. Sanquetta, C. R.; Balbinot, R.; Ziliotto M. A. B., eds.; Fixação do Carbono: atualidades, projetos de pesquisa, 2 Simpósio Latino Americano sobre fixação de carbono, Ed. A M Impressos: Curitiba, 2004.
- 6. Shima, S.; Warkentin, E.; Thauer, R. K.; Ermler, U.; J Biosci. Bioeng. 2002, 93, 519;
- Craft, J. L.; Horng, Y.-C.; Ragsdale, S. W.; Brunold, T. C.; J. Biol. Inorg. Chem 2004, 9, 77;
- Duin, E. C;. Signor, L; Piskorski , R.; Mahlert, F.; Clay, M. D.; Goenrich, M.; Thauer, R. K.; Jaun, B.; Johnson, M. K.; J. Biol. Inorg. Chem 2004, 9, 563.
- 7. DiMarco A. A.; Bobic T. A.; Wolfe R. S.; Annu. Rev. Biochem 1990, 59, 355.
- 8. Jablonski, P. E.; Ferry J. G.; J. Bacteriol 1991, 173, 2481.
- 9. Stolzenberg, A. M.; Zhang, Z.; Inorg. Chem 1997, 36, 593;
- Grabarse , W.; Mahlert, F.; Shima, S.; Thauer, R.K.; J. Mol. Biol. 2000, 303, 329.
- 10. Gunsalus R. P.; Wolfe R. S.; FEMS Microbiol. Lett 1978, 3, 191.
- 11. Farber, G.; Keller, W.; Kratky, C.; Jaun, B.; Pfalz, A.; Spinner, C.; Kobelt. A.; Eschenmoser, A.; Helv. Chim. Acta 1991, 74, 697;
- Won, H.; Olson, K. D.; Hare, D. R.; Wolfe, R. S.; Kratky, C.; Summers, M. F.; J. Am. Chem. Soc 1992, 114, 6880.
- 12. Fassler, A.; Kobelt, A. Pfaltz, A.; Eschenmoser, A.; Bladon, C.; Battersby, A. R.; Thauer, R. K.; Helv. Chim. Acta 1985, 68, 2287;
- AnkelFuchs, D.; Thauer R. K.; The Bioinorganic Chemistry of Nickel, J. R. Lancaster, ed., VCH Publishers: New York, 1988, p. 93.
- 13. Pfaltz, A.; Livinston, D.A.; Jaun, B.; Diekert, G.; Thauer, R. K.; Eschenmose, A.; Helv. Chim. Acta 1985, 68, 1338.
- 14. Zilbermann, I.; Golub, G.; Cohen, H.; Meyerstein, D.; Inorg. Chim. Acta 1994, 227,1.
- 15. Cheesman, M. R.; Ankel-Fuchs, D.; Thauer, R. K.; Thompson, A.; J. Biochem 1989, 260, 613;
- Hamilton, C. L.; Scott, R. A.; Johnson, M. K.; J. Biol. Chem 1989, 264, 11605.
- 16. Shiemke, A. K.; Kaplan, W. A.; Hamilton, C. L.; Shelnutt, J. A.; Scott, R. A.; J. Biol. Chem 1989, 264, 7276.
- 17. Furenlid, L. R.; Renner, M. W.; Fajer, J.; J. Am. Chem. Soc.1990, 112, 8987.
- 18. Furenlid, L. R.; Renner, M. W.; Szalda, D. J.; Fujita, E.; J. Am. Chem. Soc 1991, 113, 883.
- 19. Furenlid, L. R.; Renner, M. W.; Smith, K. M.; Fajer, J.; J. Am. Chem. Soc 1990, 112, 1634.
- 20. Kadish, K. M.; Franzen, M. M.; Han, B. C.; Araullo-McAdams, C.; Sazou, D.; J. Am. Chem. Soc 1991, 113, 512.
- 21. Renner, M. W.; Furenlid L. R.; Barkigia, K. M.; Forman A.; Shim, H. K.; Simpson, D. J.; Smith, K. M.; Fajer, J.; J. Am. Chem. Soc. 1991, 113, 6891.
- 22. Barkigia, K. M.; Berber, M. D.; Fajer, J.; Medforth, C. J.; Renner, M. W.; Smith, K. M.; J. Am. Chem. Soc. 1990,112, 8851;
- Renner, M. W.; Cheng, R.-J.; Chang, C. K.; Fajer, J.; J . Phys. Chem. 1990, 94, 8508.
- 23. Ermler, U.; Grabarse, W.; Shima, S.; Goubeaud, M.; Thauer, R. K.; Science 1997, 278, 1457.
- 24. Albracht, S. P. J.; Ankel-Fuchs, D.; Van der Zwaan, J. W.; Fontjin, R. D.; Thauer, R. K. Biochim. Biophys. Acta 1986, 870, 50;
- Albracht, S. P. J.; Ankel-Fuchs, D.; Bkher, R.; Ellerman, J.; Moll, J.; Van der Zwaan, J. W.; Thauer, R. K.; Biochim. Biophys. Acta 1988, 955, 86;
- Rospert, S.; Bocher, R.; Albracht, S. P. J.; Thauer, R. K.; FEBS Lett 1992, 291, 371.
- 25. Hollinges, C.; Pierich, A. J.; Reijerse E. J.; J. Am. Chem. Soc. 1993, 115, 5651.
- 26. Stolzenberg, A. M.; Stershic, M. T.; Inorg. Chem. 1987, 26, 1970;
- Stolzenberg, A. M.; Stershic, M.; J. Am. Chem. Soc. 1988, 110, 5397;
- Renner, M. W.; Forman, A.; Fajer, J.; Simpson, D.; Smith, K. M.; Barkigia, K. M.; J. Biophys. 1988, 53, 277;
- Renner, M. W.; Forman, A.; Wu, W.; Chang, C. K.; Fajer, J.; J. Am. Chem. Soc. 1989, 111, 8618;
- Latos-Grazynski, L.; Olmstead, M. M.; Balch, A. L.; Inorg. Chem. 1989, 28, 4066;
- Lexa, D.; Momenteau, M.; Mispelter. J.; Saveant, J. M.; Inorg. Chem. 1989, 28, 30;
- Kadish, K. M.; Sazou, D.; Maiya, G. B.; Han, B. C.; Liu, Y. M.; Saoiabi, A.; Ferhat, M.; Guilard, R.; Inorg. Chem. 1989, 28, 2542;
- Nahor, G. S.; Neta, P.; Hambright, P.; Robinson, L. R.; Harriman, A.; J. Phys. Chem. 1990, 94, 6659;
- Kadish. K. M.; Franzen, M. M.; Han, B. C.; Araullo-McAdams, C.; Sazou, D.; J. Am. Chem. Soc. 1991, 113, 512;
- Craft, J. L.; Horng, Y.-C.; Ragsdale, S. W.; Brunold, T. C.; J. Am. Chem. Soc. 2004, 126, 4068.
- 27. Furenlid, L. R.; Renner, M. W.; Barkigia, K. M.; Forman, A., Shim, H. K.; Simpson, D. J.; Smith, K. M.; Fajer, J. J.; J. Am. Chem. Soc. 1991, 113, 6891.
- 28. Taylor, C. D.; Wolfe, R. S.; Biochem. 1978, 17, 2374.
- 29. Jaun, B.; Pfaltz, A.; J. Chem. Soc. Chem. Commun. 1988, 293.
- 30. Krone, U. E.; Laufer, K.; Thauer, R. K.; Hogenkam, H. P. C.; Biochem 1989, 28, 10061.
- 31. Drain, C. M.; Sable, D. B.; Corden, B. B.; Inorg. Chem. 1990, 29, 1429.
- 32. Lahiri, G. K.; Schussel, L. J.; Stolzenberg, A. M.; Inorg. Chem. 1992, 31, 4991;
- Lahiri, G. K.; Stolzenberg, A M.; Inorg. Chem. 1993, 32, 4409.
- 33. Lin, S. K.; Jaun, B.; Helv. Chim. Acta 1992, 75, 1478.
- 34. Stiles, M.; J. Org. Chem. 1994, 59, 5381.
- 35. Arai, T.; Kashitani, K.; Kondo, H.; Sakaki, S.; Bull. Chem. Soc. Jpn. 1994, 67, 705;
- Todd, L. N.; Zimmer, M.; Inorg. Chem 2002, 41, 6831.
- 36. Ram, M. S.; Riordan, C. G.; Ostrander, R.; Rheingold, A. L.; Inorg. Chem. 1995, 34, 5884.
- 37. Friedermann, G. R.; Dissertação de Mestrado, Universidade Federal do Paraná, Brasil, 2001.
- 38. Caiut, J. M.; Dissertação de Mestrado, Departamento de Química, Universidade Federal do Paraná, Brasil, 2002.
- 39. Goedken, V. L.; Weiss, M. C.; Inorg. Synt. 1976, 20, 115;
- Cotton, F. A.; Czuchajowska, J.; Polyhedron 1990, 9, 2553;
- Wang, Z.; Sakata, K.; Hashimoto, M.; Polyhedron 1989, 17, 4451;
- Wang, Z.; Hu, N.; Sakata, K.; Hashimoto, M.; J. Chem. Soc. Dalton Trans. 1999, 1695;
- Jager, E. G.; Z. Anorg. Allg. Chemic. 1969, 364, 177;
- Purrington, S., Knight, B., Bereman, R.; Inorg. Chim. Acta 1994, 223, 187;
- Hochesang, P. J.; Bereman, R. D.; Inorg. Chim. Acta 1989, 162,191.
- 40. Kimura, E.; Haruta, M.; Koike, T.; Shionaya, M.; Takenouchi, K.; Itaka, Y.; Inorg. Chem 1993, 32, 2779.
- 41. Mountford, P.; Chem. Soc. Rev 1998, 27, 105.
- 42. Eilmes, J.; Polyhedron 1992,11, 581.
- 43. Caiut, J. M. A.; Nakagaki, S.; Friedermann, G. R.; Drechsel, S. M.; Zarbin, A. J. G.; J. Mol. Catal. A: Chem. 2004, 222, 213.
- 44. Arai, T.; Kondo, H.; Sakaki, S.; J. Chem. Soc. Dalton Trans 1992, 2753.
- 45. Ciurli, S.; Meter, E. M.; Floriani, C.; Chiesi-Villa, A. Guastini, C.; J. Chem. Soc. Chem. Commun. 1987, 281.
- 46. Grabarse, W.; Mahlert, F.; Duin, E. C.; Goubeaud, M.; Shimia, S.; Thauer, R. K.; Lamzin, V.; Ermler, U.; J. Mol. Biol 2001, 309, 315.
- 47. Shima, S.; Goubeaud, M.; Vinzenz, D.; Thauer, R. K.; Ermler, U.; J. Biochem. 1997, 121, 829.
- 48. Pelmeschikov, V.; Blomberg, M. R. A.; Siegbahn, P. E. M.; Crabtree, R. H.; J. Am. Chem. Soc. 2002, 24, 946.
- 49. Hartzel P. L.; Wolfe, R. S.; Syst. Appl. Microbiol. 1986, 7, 376;
- Wolfe, R. S.; Ann. Rev. Microbiol. 1991, 45, 1.
Datas de Publicação
-
Publicação nesta coleção
06 Set 2011 -
Data do Fascículo
Out 2006
Histórico
-
Recebido
10 Fev 2005 -
Aceito
27 Set 2005