Resumos
Objetivo: descrever caso clínico de um lactente com insuficiência renal crônica terminal, causada por hiperoxalúria primária.Método: após revisão da literatura, verifica-se a raridade da doença; na França, a prevalência é de 1,05/milhão, com taxa de incidência de 0,12/milhão/ano. Pesquisa abordando centros especializados mundiais detectou, em 1999, 78 casos em lactentes; destes, em 14% o quadro inicial foi de uremia. A gravidade e a raridade da doença sugerem o relato deste caso.Resultados: criança de sexo feminino, com quadro de vômitos e baixo ganho de peso desde os primeiros meses de vida, desenvolveu insuficiência renal terminal aos 6 meses de idade, sendo mantida em tratamento dialítico desde então. Aos 8 meses, foi encaminhada para esclarecimento diagnóstico, apresentando déficit pôndero-estatural grave e os seguintes exames laboratoriais: uréia= 69 mg/dl, creatinina=2,2 mg/dl e clearance de creatinina= 12,5 ml/min/1.73m²SC. O exame de urina foi normal, a ultra-sonografia renal revelou tamanho normal e hiperecogenicidade de ambos os rins. A dosagem de oxalato urinário foi de 9,2mg/kg/dia ou 0,55 mmol/1.73m²SC, e a relação oxalato:creatinina, de 0,42. A biópsia renal diagnosticou presença de grande quantidade de depósitos de cristais de oxalato de cálcio no parênquima renal. A radiografia de ossos longos evidenciou sinais sugestivos de osteopatia oxalótica, e a fundoscopia indireta, sinais de retinopatia por oxalato. A criança foi mantida em diálise peritoneal ambulatorial contínua, tendo sido iniciado tratamento com piridoxina.Conclusões: a hiperoxalúria primária deve ser considerada como um dos diagnósticos diferenciais de insuficiência renal crônica em lactentes, especialmente na ausência de história sugestiva de outras patologias.
lactente; insuficiência renal crônica terminal; hiperoxalúria primária; oxalose sistêmica
Objective: to report a case of an infant with endstage renal failure caused by primary hyperoxaluria. Methods: the review of the literature showed the rarity of the disease. In France, the prevalence is about 1.05/million and the incidence rate is 0.12/million/year. A survey, performed in international specialized centers in 1999, documented 78 cases in infants; in 14% of them the initial onset symptom was uremia. The rarity and severity of the disease justify the description of this case.Results: a girl presenting vomiting and failure to thrive within the first months of life developed endstage renal failure at 6 months of age. She was being treated with dialysis. At 8 months of age, she was referred for investigation. She was undernourished and the laboratory examinations showed urea (69mg/dl), creatinine (2.2 mg/dl) and creatinine clearance (12.5 ml/min/1.73m²SA). The routine urinalysis was normal, the renal ultrasound showed increased echogenicity in both kidneys; the dosage of urinary oxalate was 9.2mg/kg/day or 0.55mmol/1.73m²SA; the urinary oxalate/creatinine ratio was 0.42. Renal biopsy showed calcium oxalate crystals throughout the renal parenchyma. The radiograph of long bones showed osteopathy and the ophthalmic examination showed flecked retinopathy. The child was treated with continuous ambulatory peritoneal dialysis and administration of pyridoxine was initiated.Conclusions: primary hyperoxaluria should be considered as a differential diagnosis for endstage renal failure in infants, especially if there are no symptoms of other diseases.
infant; endstage renal failure; primary hyperoxaluria; systemic oxalosis
Hiperoxalúria primária com insuficiência renal crônica terminal em lactente
Primary hyperoxaluria with endstage renal failure in an infant
Célia S. Macedo 1 1 Célia S. Macedo Profª Adjunta Livre Docente do Dep. de Pediatria da Faculdade de Medicina de Botucatu (FMB-UNESP), disc. de Nefrologia Pediátrica. 2 Eneida M. Yoshida Residente de 3º ano em Pediatria do Dep. de Pediatria da FMB-UNESP, área de Nefrologia Pediátrica. 3 Rosa Marlene Viero Profª Drª do Dep. de Anatomia Patológica da FMB-UNESP. 4 Márcia C. Riyuzo Profª Drª do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica, Doutora em Clínica Médica: área de Nefrologia. 5 Herculano D. Bastos Prof. Adjunto Livre Docente do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica. , Eneida M. Yoshida 2 1 Célia S. Macedo Profª Adjunta Livre Docente do Dep. de Pediatria da Faculdade de Medicina de Botucatu (FMB-UNESP), disc. de Nefrologia Pediátrica. 2 Eneida M. Yoshida Residente de 3º ano em Pediatria do Dep. de Pediatria da FMB-UNESP, área de Nefrologia Pediátrica. 3 Rosa Marlene Viero Profª Drª do Dep. de Anatomia Patológica da FMB-UNESP. 4 Márcia C. Riyuzo Profª Drª do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica, Doutora em Clínica Médica: área de Nefrologia. 5 Herculano D. Bastos Prof. Adjunto Livre Docente do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica. , Rosa Marlene Viero 3 1 Célia S. Macedo Profª Adjunta Livre Docente do Dep. de Pediatria da Faculdade de Medicina de Botucatu (FMB-UNESP), disc. de Nefrologia Pediátrica. 2 Eneida M. Yoshida Residente de 3º ano em Pediatria do Dep. de Pediatria da FMB-UNESP, área de Nefrologia Pediátrica. 3 Rosa Marlene Viero Profª Drª do Dep. de Anatomia Patológica da FMB-UNESP. 4 Márcia C. Riyuzo Profª Drª do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica, Doutora em Clínica Médica: área de Nefrologia. 5 Herculano D. Bastos Prof. Adjunto Livre Docente do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica. , Márcia C. Riyuzo 4 1 Célia S. Macedo Profª Adjunta Livre Docente do Dep. de Pediatria da Faculdade de Medicina de Botucatu (FMB-UNESP), disc. de Nefrologia Pediátrica. 2 Eneida M. Yoshida Residente de 3º ano em Pediatria do Dep. de Pediatria da FMB-UNESP, área de Nefrologia Pediátrica. 3 Rosa Marlene Viero Profª Drª do Dep. de Anatomia Patológica da FMB-UNESP. 4 Márcia C. Riyuzo Profª Drª do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica, Doutora em Clínica Médica: área de Nefrologia. 5 Herculano D. Bastos Prof. Adjunto Livre Docente do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica. , Herculano D. Bastos 5 1 Célia S. Macedo Profª Adjunta Livre Docente do Dep. de Pediatria da Faculdade de Medicina de Botucatu (FMB-UNESP), disc. de Nefrologia Pediátrica. 2 Eneida M. Yoshida Residente de 3º ano em Pediatria do Dep. de Pediatria da FMB-UNESP, área de Nefrologia Pediátrica. 3 Rosa Marlene Viero Profª Drª do Dep. de Anatomia Patológica da FMB-UNESP. 4 Márcia C. Riyuzo Profª Drª do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica, Doutora em Clínica Médica: área de Nefrologia. 5 Herculano D. Bastos Prof. Adjunto Livre Docente do Dep. de Pediatria da FMB-UNESP, disciplina de Nefrologia Pediátrica.
RESUMO
ABSTRACT
Objetivo: descrever caso clínico de um lactente com insuficiência renal crônica terminal, causada por hiperoxalúria primária.
Método: após revisão da literatura, verifica-se a raridade da doença; na França, a prevalência é de 1,05/milhão, com taxa de incidência de 0,12/milhão/ano. Pesquisa abordando centros especializados mundiais detectou, em 1999, 78 casos em lactentes; destes, em 14% o quadro inicial foi de uremia. A gravidade e a raridade da doença sugerem o relato deste caso.
Resultados: criança de sexo feminino, com quadro de vômitos e baixo ganho de peso desde os primeiros meses de vida, desenvolveu insuficiência renal terminal aos 6 meses de idade, sendo mantida em tratamento dialítico desde então. Aos 8 meses, foi encaminhada para esclarecimento diagnóstico, apresentando déficit pôndero-estatural grave e os seguintes exames laboratoriais: uréia= 69 mg/dl, creatinina=2,2 mg/dl e clearance de creatinina= 12,5 ml/min/1.73m2SC. O exame de urina foi normal, a ultra-sonografia renal revelou tamanho normal e hiperecogenicidade de ambos os rins. A dosagem de oxalato urinário foi de 9,2mg/kg/dia ou 0,55 mmol/1.73m2SC, e a relação oxalato:creatinina, de 0,42. A biópsia renal diagnosticou presença de grande quantidade de depósitos de cristais de oxalato de cálcio no parênquima renal. A radiografia de ossos longos evidenciou sinais sugestivos de osteopatia oxalótica, e a fundoscopia indireta, sinais de retinopatia por oxalato. A criança foi mantida em diálise peritoneal ambulatorial contínua, tendo sido iniciado tratamento com piridoxina.
Conclusões: a hiperoxalúria primária deve ser considerada como um dos diagnósticos diferenciais de insuficiência renal crônica em lactentes, especialmente na ausência de história sugestiva de outras patologias.
lactente, insuficiência renal crônica terminal, hiperoxalúria primária, oxalose sistêmica
Objective: to report a case of an infant with endstage renal failure caused by primary hyperoxaluria.
Methods: the review of the literature showed the rarity of the disease. In France, the prevalence is about 1.05/million and the incidence rate is 0.12/million/year. A survey, performed in international specialized centers in 1999, documented 78 cases in infants; in 14% of them the initial onset symptom was uremia. The rarity and severity of the disease justify the description of this case.
Results: a girl presenting vomiting and failure to thrive within the first months of life developed endstage renal failure at 6 months of age. She was being treated with dialysis. At 8 months of age, she was referred for investigation. She was undernourished and the laboratory examinations showed urea (69mg/dl), creatinine (2.2 mg/dl) and creatinine clearance (12.5 ml/min/1.73m2SA). The routine urinalysis was normal, the renal ultrasound showed increased echogenicity in both kidneys; the dosage of urinary oxalate was 9.2mg/kg/day or 0.55mmol/1.73m2SA; the urinary oxalate/creatinine ratio was 0.42. Renal biopsy showed calcium oxalate crystals throughout the renal parenchyma. The radiograph of long bones showed osteopathy and the ophthalmic examination showed flecked retinopathy. The child was treated with continuous ambulatory peritoneal dialysis and administration of pyridoxine was initiated.
Conclusions: primary hyperoxaluria should be considered as a differential diagnosis for endstage renal failure in infants, especially if there are no symptoms of other diseases.
infant, endstage renal failure, primary hyperoxaluria, systemic oxalosis
-
Introdução
-
Relato de caso
-
Discussão
-
Referências
Introdução
Hiperoxalúrias primárias são doenças muito raras, caracterizadas pela superprodução e acúmulo de oxalato no organismo. Lepoutre, em 1925, fez a primeira descrição em caso post mortem de um lactente com múltiplos cálculos renais(1); a maior parte dos casos descritos posteriormente apresentam elevada taxa de consangüinidade(2-4).
As hiperoxalúrias são um grupo raro de desordens autossômicas recessivas, tendo sido bem estudados 2 tipos: o tipo I, que se caracteriza por elevada taxa de excreção urinária de glicolato, e o tipo II, que é menos grave, e que se caracteriza pela elevada excreção urinária de L-glicerato(2,3)
A hiperoxalúria primária tipo I apresenta, na França, prevalência de 1,05/milhão e uma taxa de incidência de 0,12/milhão/ano(5). Decorre da diminuição da enzima hepática peroxissomal alanina-glioxilato aminotransferase (AGT), cujo déficit ocasiona excesso de glioxilato, que é transformado em oxalato e glicolato. A hiperoxalúria primária tipo II é causada pela deficiência da enzima citosólica glioxilato redutase D-glicerato desidrogenase, que resulta na síntese aumentada de oxalato e L-glicerato. O excesso de oxalato em ambas as entidades pode se precipitar na urina, provocando o aparecimento de litíase e nefrocalcinose(2,3,6).
A expectativa com relação à função renal na hiperoxalúria tipo I é desfavorável em crianças; há relato de desenvolvimento de insuficiência renal crônica em 50% de 16 crianças antes dos 10 anos de idade(7). Quanto ao envolvimento renal na hiperoxalúria tipo II, geralmente é menos grave, embora também ocorram nefrocalcinose e insuficiência renal terminal(3).
As manifestações clínicas são heterogêneas e variam desde sintomas relacionados a urolitíase, em qualquer idade, até a instalação precoce de insuficiência renal terminal em crianças(2,3). Estima-se que a hiperoxalúria primária tipo I represente de 1 a 2,7% das causas de insuficiência renal terminal em crianças8. Latta e Brodehl estimaram que 1 em 5 a 15.000.000, na faixa de 0 a 15 anos, apresentarão insuficiência renal devido à hiperoxalúria primária(8).
Alguns autores consideram a existência de um raro subgrupo de pacientes com hiperoxalúria primária do tipo I em lactentes, denominada oxalose infantil, que se apresenta com insuficiência renal e oxalose sistêmica avançada, geralmente sem calculose, não estando ainda estabelecido se esse grupo deva ser considerado uma entidade à parte da hiperoxalúria primária tipo I(2,3). É provável que este quadro em lactente jovem esteja mais relacionado à hiperoxalemia e suas conseqüências do que se tratar realmente de uma entidade à parte, uma vez que no recém-nascido dois mecanismos causam acúmulo de oxalato: alta taxa de produção por uma via preferencial de glioxilato a oxalato ao invés de glicolato, e baixa taxa de filtração glomerular.
O objetivo deste trabalho é relatar o caso de um lactente que apresentou insuficiência renal crônica terminal, devido à hiperoxalúria primária tipo I.
Relato de caso
C.C.S., sexo feminino, branca, primogênita de casal não consangüíneo, apresentava antecedentes de vômitos freqüentes e baixo ganho de peso desde os primeiros meses de vida. Foi internada aos 6 meses em outro hospital apresentando febre, acidose metabólica, crises convulsivas, oligúria, edema generalizado e insuficiência respiratória aguda, necessitando de ventilação mecânica por 7 dias. Os resultados iniciais dos exames bioquímicos dessa internação foram: uréia= 237 mg/dl, creatinina= 2,8 mg/dl, Na= 133mEq/L, K= 4,6 mEq/L, Ca= 9,3 mg/dl, pH=6,89, bicarbonato= 5,7 mmol/L, hemoglobina = 9,3g% e hematócrito= 23,2%. Foi instalada diálise peritoneal naquele hospital, e a paciente evoluiu com recuperação do débito urinário, sem haver recuperação dos níveis séricos de uréia e creatinina. Após 10 dias, foi transferida para outro hospital, no qual permaneceu por 1 mês, sendo então transferida para este serviço, aos 8 meses de idade, para investigação da etiologia da insuficiência renal. Ao exame físico, apresentava-se pálida, com desidratação leve, eupnéica, afebril, com freqüência respiratória de 30 ipm, freqüência cardíaca de 140 bpm e pressão arterial de 90/60 mmHg. O peso era de 4.715g (<P 2,5) e a estatura, de 63,5 cm (P2,5-10).
A paciente foi mantida em diálise peritoneal ambulatorial contínua, com os seguintes resultados de exames laboratoriais séricos: uréia = 69 mg/dl; creatinina =2,20 mg/dl; fósforo de 3,3 mg/dl, cálcio = 8,8 mg/dl, sódio= 136 mEq/L, potássio= 4,5 mEq/L e clearance de creatinina de 12,5 ml/min/1.73m2SC.
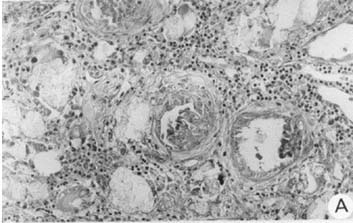
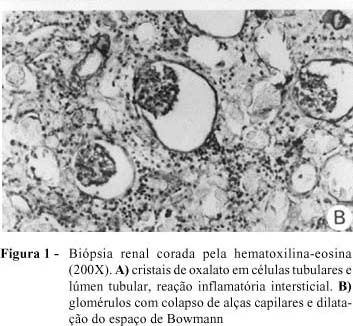
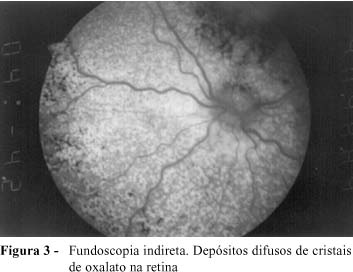
Na investigação, foram obtidos os seguintes resultados: exame de urina normal; ultra-sonografia renal apresentando hiperecogenicidade de ambos os rins, com tamanho compatível para a idade; radiografia de abdômen sem alterações. As dosagens de oxalato urinário de 24 horas apresentaram os seguintes resultados: 9,2 mg/kg/dia ou 0,55 mmol/1.73m2SC, e relação oxalato:creatinina de 0,42. Foi realizada biópsia renal no 3º dia de internação. A análise do material obtido revelou presença de grande quantidade de cristais de oxalato em células tubulares, interstício renal e luz tubular com reação granulomatosa do tipo corpo estranho ao redor dos cristais (Figura 1-A). Os glomérulos apresentavam-se afetados secundariamente pelo processo obstrutivo, com colapso glomerular e dilatação do espaço de Bowmann (Figura 1-B). A radiografia de ossos longos mostrou osteopenia e linha transversa de densidade radiológica aumentada na região da placa de crescimento (Figura 2). A fundoscopia indireta evidenciou presença de lesões hiperpigmentadas puntiformes adjacentes a áreas hipopigmentadas na retina, compatíveis com o diagnóstico de retinopatia por oxalato (Figura 3).
Com esses resultados, foi iniciado tratamento com vitamina B6 na dose de 10mg/dia, e a criança continuou em diálise peritoneal ambulatorial contínua, além de receber suplementações de ferro, calcitriol e eritropoetina, bicarbonato de sódio e dieta hipercalórica, evoluindo satisfatoriamente, recebendo alta hospitalar após 2 meses de internação e realizando consultas na unidade de diálise.
Discussão
Descrevemos caso de um lactente com hiperoxalúria primária tipo I, que apresentou insuficiência renal terminal aos 6 meses de idade, elevada excreção de oxalato na urina e biópsia renal demonstrando intensa deposição de cristais de oxalato em luz, células tubulares e interstício renal, com afecção glomerular secundária a esse processo, deposição maciça de oxalato na retina e lesão óssea sugestiva de oxalose sistêmica.
Há controvérsias com relação aos valores normais de oxalato urinário em lactentes. Em 1991, Barratt, estudando 137 crianças normais incluindo lactentes, obteve os seguintes valores da relação oxalato/creatina (mmol/mmol): média geométrica de 0,061, variando de 0,015 a 0,26(9). No presente caso, obtivemos uma relação molar de 0,42, configurando uma hiperoxalúria, comparando com esses valores. Embora não tenham sido dosados os níveis de oxalemia, estes certamente estariam elevados, pois considera-se que a concentração do oxalato se eleva à medida que a taxa de filtração glomerular diminui, sendo a hiperoxalemia o fator responsável não apenas pela deposição de oxalato renal, mas também pela oxalose sistêmica, caracterizada pela osteopatia, envolvimento ocular, neurológico, arteriopatia e cardiopatia(10). Essa maciça deposição de oxalato é um dos fatores a ser considerado na decisão de realização de transplante renal.
A alteração básica dessa doença é um defeito enzimático que ocorre no interior do hepatócito. A hiperoxalúria tipo I é causada por uma deficiência da enzima aminotransferase alanina:glioxilato do hepatócito, que se apresenta com grande heterogeneidade com relação ao nível enzimático. Dois terços dos pacientes têm níveis indetectáveis da atividade catalítica da enzima aminotransferase alanina, glioxilato, enquanto que um terço apresenta atividade que varia de 3 a 50% da atividade normal média. Em um terço dos pacientes com níveis indetectáveis de atividade da aminotransferase alanina:glioxilato, detecta-se a proteína dessa enzima através de métodos imunorreativos. Nestes pacientes, pode existir um defeito na distribuição celular da enzima, que se localiza no interior da mitocôndria, e não dos peroxissomos do hepatócito, com conseqüente inadequada função metabólica(2,3). O diagnóstico do defeito enzimático é feito pela obtenção de tecido hepático por biópsia e estudo da atividade catalítica da enzima aminotransferase alanina:glioxilato(2,3,11), não realizado neste caso, pelo fato de a paciente já ter desenvolvido insuficiência renal. Esse procedimento é importante em fase anterior ao desenvolvimento da oxalose sitêmica, pois encontrando-se atividade catalítica da enzima alanina:glioxilato aminotransferase (AGT), o uso da piridoxina poderá ser adequadamente indicado(12), uma vez que o fosfato de piridoxina é a coenzima das aminotransferases, incluindo a AGT. A piridoxina (vitamina B6) tem sido utilizada em casos de hiperoxalúria tipo I, com respostas variáveis em crianças a partir de 1 a 2 meses de idade. Tem sido realizado esse tratamento em irmãos de lactentes com oxalose infantil, com resposta satisfatória(3).
Cinco a 10% dos pacientes mostram uma resposta parcial ou completa com a administração de doses farmacológicas de piridoxina(13). O restante dos pacientes (90%) não se beneficiam do tratamento.
No presente caso, foi introduzida a piridoxina na dose de 10 mg/dia com o objetivo de se reduzir o acúmulo de oxalato, como teste terapêutico. Sabe-se que a taxa de remoção de oxalato por diálise peritoneal ambulatorial contínua (DPAC) é menor do que a síntese na hiperoxalúria tipo I(14,15) e, embora haja melhora do quadro de insuficiência renal, a oxalose sistêmica progride. No presente caso, deverão ser realizadas novas coletas urinárias controles e/ou dosagens séricas de oxalato para continuidade ou suspensão do tratamento com piridoxina.
Em relação ao transplante renal na hiperoxalúria tipo I, existe uma controvérsia do ponto de vista ético quando se refere a transplante realizado com doador vivo; entretanto tem-se realizado este procedimento em crianças(16,17). Em 10 crianças que receberam transplante renal com rim de doador vivo, 7 tiveram sucesso quanto à função renal, não sendo observado evidência de recorrência da hiperoxalúria em 6 crianças(13).
Em 1995, o Estudo Cooperativo de Transplante Renal Pediátrico Norte-americano (NAPRTCS) publicou relato de 22 crianças portadoras de hiperoxalúria tipo I transplantadas, sendo que 17 receberam rim de doador vivo, relacionado ou não, com 50% dos casos mantendo função renal adequada pós-transplante. Não houve sucesso nas crianças que receberam transplante com rim de cadáver(16). Em 1999, esse mesmo grupo relatou a evolução de 128 pacientes com hiperoxalúria, concluindo que o transplante de rim é um procedimento benéfico e o que ofereceu melhor sobrevida aos pacientes, quando comparado ao transplante combinado de rim e fígado ou quando não era realizado(18). Neste paciente que estamos descrevendo, o transplante renal é uma possibilidade terapêutica indicada com restrições devido ao quadro de oxalose sistêmica intensa. O momento ideal para a realização do transplante renal ou hepático é antes do desenvolvimento da oxalose sistêmica avançada(19).
Como a enzima aminotransferase alanina:glioxilato é encontrada em quantidade significativa somente no fígado, e está localizada dentro dos peroxissomas hepáticos, o transplante hepático há 10 anos tem sido empregado como meio terapêutico mais eficiente nessa patologia, bem como transplantes combinados de fígado e rim, com resultados favoráveis(3,20,21). Um total de 98 transplantes combinados (rim e fígado) em 93 pacientes foram relatados na Europa, com taxa de sobrevida em 10 anos de 65% (22,23).
Os sintomas relatados na apresentação inicial de hiperoxalúria primária tipo I são: falta de desenvolvimento pôndero-estatural (22%), infecção urinária (21%) e uremia (14%)(24). O paciente em questão apresentou déficit de desenvolvimento pôndero-estatural nos primeiros meses de vida, porém, sendo essa doença infreqüente, e em nosso meio muitos outros fatores poderem contribuir para esse dado, não houve suspeição diagnóstica. A criança em questão pode ter desenvolvido desidratação com hipovolemia, e conseqüente deposição maciça de oxalato nos túbulos renais, precipitando a irreversibilidade do quadro renal aos seis meses de idade.
Concluímos que a hiperoxalúria primária tipo I deve ser considerada como um dos diagnósticos diferenciais de insuficiência renal nos primeiros meses de vida, especialmente na ausência de história sugestiva de outras patologias.
Agradecimentos
Agradecemos à Prof. Eliane Chaves Jorge, do Departamento de Oftalmologia da Faculdade de Medicina de Botucatu-UNESP, pela realização da fotografia do exame da retina.
Sobe
Endereço para correspondência:
Drª Célia S. Macedo
Dep. de Pediatria da Faculdade de Medicina de Botucatu-UNESP
Rubião Jr., s/n
CEP 18618-970 Botucatu, SP
Fone/Fax: 014 6802.6274 / 6802.6083
E-mail: pediatri@fmb.unesp.br
Sobe
Artigo submetido em 09.10.01, aceito em 09.01.02.
- 1. Lepoutre C. Calculs multiples chez un enfant: Infiltration du parenchyme rénal par des dépots cristallins. J Urol 1925;20:424.
- 2. Van Acker KJ, Wolff ED. Hyperoxaluria and oxalosis. In: Edelman Jr CM, Bernstein J, Meadow SR, Spitzer A, Travis LB, eds. Pediatric kidney disease. 2ª ed. Boston: Little, Brown and Company; 1992. p.1687-93.
- 3. Barratt TM, Danpure CJ. Hyperoxaluria. In: Barratt TM, Avner ED, Harmon WE, eds. Pediatric nephrology. 4ª ed. Baltimore: Lippincott Williams & Wilkins; 1999. p.609-19.
- 4. von Schnakenburg C, Hulton AS, Melford DV, Roper HD, Rumsby G. Variable presentation of primary hyperoxaluria type I in 2 patients homozigous for a novel combined deletion and insertion in Exon 8 of AGXT gene. Nephron 1998;78:485-8.
- 5. Milliner DS, Wilson DM, Smithson LH. Phenotipic expression of primary hyperoxaluria: Comparative features of types I and II. Kidney Int 2001;59:31-6.
- 6. Nishimura JL, Heilberg IP, Schor N. Hiperoxalúria. In: Schor N, Heilberg IP, eds. Calculose renal - fisiopatologia, diagnóstico e tratamento. São Paulo: Sarvier; 1995. p.61-9.
- 7. Barratt TM, von Sperling V, Dillon MJ. Primary oxaluria in children. In: Rose GA, ed. Oxalate metabolism in relation to urinary stone. London: Springer- Verlag; 1988. p.83-101.
- 8. Latta K, Brodehl J. Primary hyperoxaluria type I. Eur J Pediatr 1990;149:518-22.
- 9. Barratt TM, Kasidas GP, Murdoch I, Rose GA. Urinary oxalate and glycolate excretion and plasma oxalate concentration. Arch Dis Child 1991;66:501-3.
- 10. Barratt TM, Van't Hohh WG. Are there guidelines for a strategy according to glomerular filtration rate, plasma oxalate determination and the risk of oxalate accumulation? Nephrol Dial Transplant 1995;10 Suppl 8:22-3.
- 11. Danpure CJ, Jennings PR, Watts RW. Enzymological diagnosis of primary hyperoxaluria type I by measurement of hepatic alanine: glyoxylate aminotransferase activity. Lancet 1987;1:289-91.
- 12. Broyer M, Jouvet P, Niaudet P, Dauon M, Revillon Y. Management of oxalosis. Kidney Int 1996;49 Suppl 53:S93.
- 13. Latta K, Jamieson NV, Scheinman JI, Schärer K, Bensman A, Cochat P, et al. Selection of transplantation procedures and perioperative management in primary hyperoxaluria type I. Nephrol Dial Transplant 1995;10 Suppl 8:53-7.
- 14. Watts RWE, Veall N, Purkins P. Oxalate dynamics and removal rates during haemodialysis and peritoneal dialysis in patients with primary hyperoxaluria and severe erenal failure. Clin Sci 1984;66:591-7.
- 15. Watts RWE. Treatment of renal failure in the primary hyperoxalurias. Nephron 1990;56:1-5.
- 16. Scheinmam J, Alexander M, Campbell ED, Transplantation for primary hyperoxaluria in the USA. Nephrol Dial Transplant 1995;10 Suppl 8:42-6.
- 17. Warady BA, Fivush BA, Alexander SR. Peritoneal Dialysis. In: Barrat TM, Avner ED, Harmon WE eds. Pediatric nephrology. 4ª ed. Baltimore: Lippincott Williams & Wilkins; 1999. p.1251-65.
- 18. Saborio P, Scheinman JI. Transplantation for primary hyperoxaluria in United States. Kidney Int 1999;56:1094-100.
- 19. Cochat P. Primary hyperoxaluria type I. Kidney Int 1999;55:2533-47.
- 20. Jamieson NV. The European Primary Hyperoxaluria Type I Transplant Registry report on the results of combined liver/kidney transplantation for primary hyperoxaluria 1984-1994. European PH 1 Transplantation Study Group. Nephrol Dial Transplant 1995;10 Suppl 8:33-7.
- 21. Shapiro R, Weisman I, Mandel H, Eisenstein B, Bem-Ari Z, Bar-Nathan N, et al. Primary hyperoxaluria type I: improved outcome with timely liver transplantation: a single-center report of 36 children. Transplantation 2001;72:428-32.
- 22. 5th Workshop on primary hyperoxalurias. Nephrol Dial Transplant 1999;14:2784-89.
- 23. Jamieson NV. The results of combined liver/kidney transplantation for primary hyperoxaluria (PH1) 1984-1997. The European PH1 transplant registry report. European PH1 Transplantation Study Group. J Nephrol 1998;11 Suppl 1:36-41.
- 24. Cochat P, Nogueira PCK, Mahamoud MA, Jamieson N, Scheinman JI, Rolland MD. Primary hyperoxaluria in infants: medical, ethical and economic issues. J Pediatr 1999;135:746-50.
Datas de Publicação
-
Publicação nesta coleção
26 Nov 2002 -
Data do Fascículo
Abr 2002
Histórico
-
Aceito
09 Jan 2002 -
Recebido
09 Out 2001