Summary
Laminopathies are genetic disorders associated with alterations in nuclear envelope proteins, known as lamins. The LMNA gene encodes lamins A and C, and LMNA mutations have been linked to diseases involving fat (type 2 familial partial lipodystrophy [FPLD2]), muscle (type 2 Emery–Dreifuss muscular dystrophy [EDMD2], type 1B limb-girdle muscular dystrophy [LGMD1B], and dilated cardiomyopathy), nerves (type 2B1 Charcot–Marie–Tooth disease), and premature aging syndromes. Moreover, overlapping syndromes have been reported. This study aimed to determine the genetic basis of an overlapping syndrome in a patient with heart disease, myopathy, and features of lipodystrophy, combined with severe metabolic syndrome. We evaluated a 54-year-old woman with rheumatoid arthritis, chronic hypercortisolism (endogenous and exogenous), and a history of cured adrenal Cushing syndrome. The patient presented with a complex disorder, including metabolic syndrome associated with mild partial lipodystrophy (Köbberling-like); mild hypertrophic cardiomyopathy, with Wolff–Parkinson– White syndrome and atrial fibrillation; and limb-girdle inflammatory myopathy. Mutational analysis of the LMNA gene showed a heterozygous c.1634G>A (p.R545H) variant in exon 10 of LMNA. This variant has previously been independently associated with FPLD2, EDMD2, LGMD1B, and heart disease. We describe a new, LMNA-associated, complex overlapping syndrome in which fat, muscle, and cardiac disturbances are related to a p.R545H variant.
INTRODUCTION
Mutations in the LMNA gene (NM_170707.2) have been associated with a broad spectrum of diseases (11. Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86(3):967-1008.), including type 2 familial partial lipodystrophy (FPLD2), LMNA-related metabolic syndrome, type 2 Emery–Dreifuss muscular dystrophy (EDMD2), type 1B limb-girdle muscular dystrophy (LGMD1B), conduction-system diseases and dilated cardiomyopathy (DCM1A), and progeroid syndromes. FPLD2 begins in women during puberty, with a phenotype of fat loss in the limbs and buttocks, fat accumulation in the face and neck, well-defined musculature, phlebomegaly, insulin resistance, atherogenic dyslipidemia, and high cardiovascular risk (22. Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab. 2016;101(12):4500-11.).
The differential diagnosis includes Cushing's syndrome and truncal obesity. Previous studies have described a LMNA-associated metabolic syndrome with a Köbberling-like fat distribution (33. Guillin-Amarelle C, Sanchez-Iglesias S, Castro-Pais A, Rodriguez-Canete L, Ordonez-Mayan L, Pazos M, et al. Type 1 familial partial lipodystrophy: understanding the Kobberling syndrome. Endocrine. 2016;54(2):411-21.,44. Decaudain A, Vantyghem MC, Guerci B, Hecart AC, Auclair M, Reznik Y, et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J Clin Endocrinol Metab. 2007;92(12):4835-44.). EDMD2 is characterized as a progressive skeletal muscle weakness associated with early joint contractures. LGMD1B causes muscular weakness in the spine and pelvic girdle (11. Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86(3):967-1008.).
Cardiac muscle laminopathies can present as nonspecific alterations in electrical conduction, or they can cause malignant arrhythmias and sudden death. Two of the most intriguing features of laminopathies are their clinical heterogeneity and the prevalence of overlapping syndromes. We investigated a female patient with a double Cushing syndrome (endogenous and exogenous), metabolic syndrome, cardiomyopathy, and limb-girdle muscular dystrophy. We aimed to determine whether this syndrome was related to a LMNA mutation.
CASE AND METHODS
This study was approved by the Ethics Review Panel of the Xunta de Galicia. The patient and her relatives provided informed consent for participation in the study and for publication of their clinical, biochemical, and genetic information.
Patient clinical history
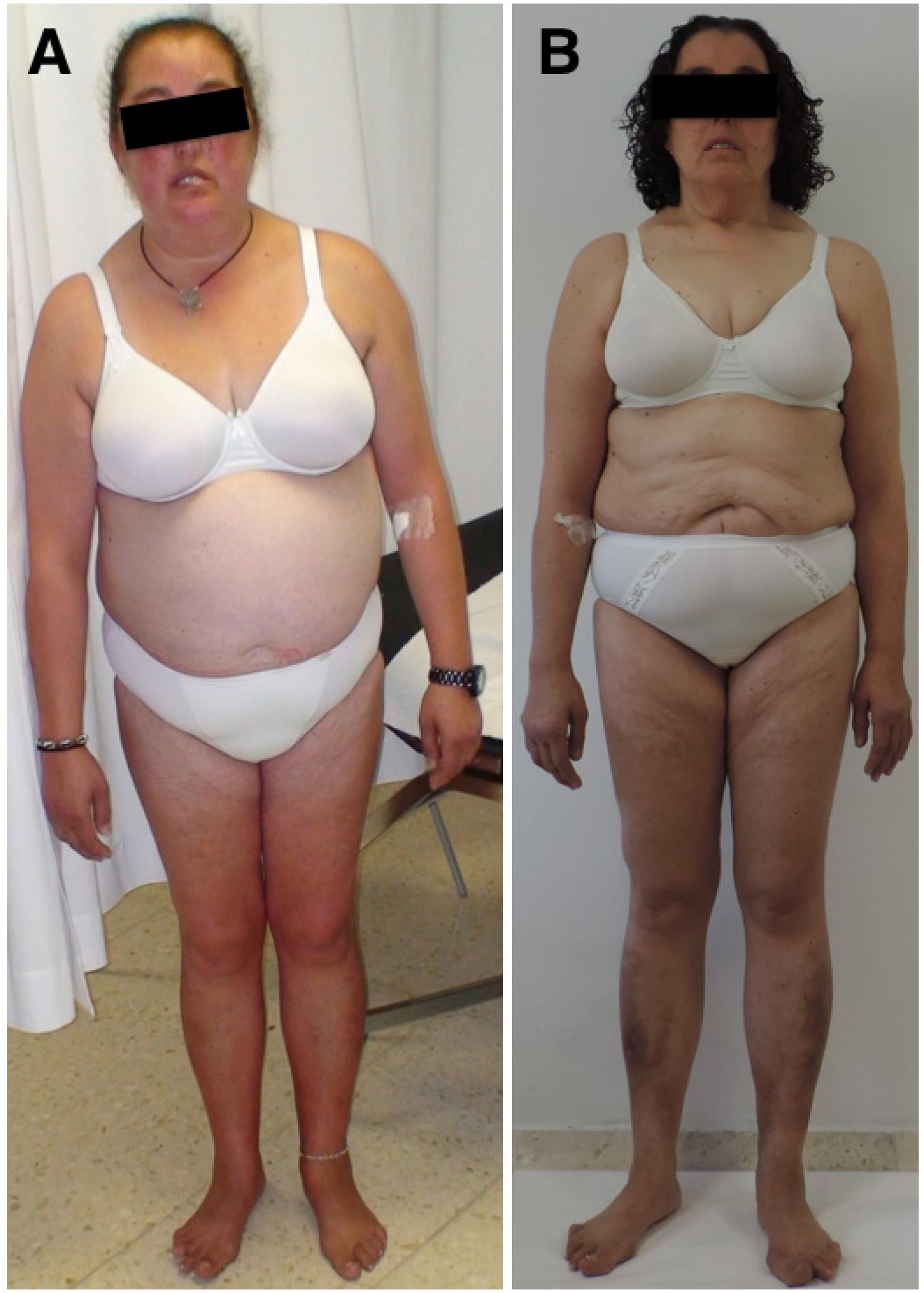
The patient (Table 1) was a 45-year-old female diagnosed with rheumatoid arthritis at age 40 y and treated with corticosteroids from that point. She was referred to the Endocrinology Division due to high blood pressure, mixed dyslipidemia, and newly diagnosed diabetes (glycated hemoglobin: 9.6%; systolic blood pressure: 150 mmHg; diastolic blood pressure: 90 mmHg; low-density lipoprotein cholesterol: 5.2 mmol/L; high-density lipoprotein cholesterol: 0.65 mmol/L; triglycerides: 3.42 mmol/L). The patient had had an android appearance since puberty, with mild lipoatrophy in the limbs and buttocks and fat accumulation in the abdomen and face (Figure 1). The patient reported that her deceased father had striking hypermuscular limbs, prominent abdominal fat, and diabetes. He had undergone a pacemaker implantation because of a third-degree atrioventricular block.
Photographs of the patient show body morphology due to a LMNA variant. (A) Before receiving a cure for Cushing's syndrome (46 years old). (B) Six years after receiving a cure for Cushing's syndrome (54 years old).
At age 46 y, the patient displayed poor glycemic control, and glucocorticoids were discontinued for several months, with no improvement. She was then diagnosed with adrenal Cushing's syndrome, based on a 34-mm right adrenal adenoma (urinary free cortisol: 1100 µg/ 24 h), which was histologically confirmed after a laparoscopic adrenalectomy. Additional hydrocortisone replacement was necessary, and the patient currently continues this treatment. After the adrenal tumor was resected, glycemic control, blood pressure, and lipid levels improved. She continued pharmacological treatment with antihypertensive drugs and metformin, but she was able to discontinue insulin.
Some months after the adrenalectomy, the patient was diagnosed with Wolff–Parkinson–White syndrome associated with atrial fibrillation, due to preexcitation, which was successfully ablated with radiofrequency. Transthoracic ultrasonography revealed a slightly hypertrophic non-dilated left ventricle, with preserved systolic function, type 1 diastolic dysfunction, and mild mitral regurgitation. No coronary lesions were detected with cardiac catheterization.
At age 50 y, the patient complained of muscular weakness without pain or contractures.
Creatine kinase levels ranged from 321 to 2525 IU/L, and high levels persisted after discontinuation of statins. On physical examination, muscle weakness was evident, predominantly in the pelvic girdle. She was unable to get up from the ground and had marked difficulty in rising from a chair without hand support. Electromyoneurography revealed myopathy changes and spontaneous activity (fibrillation and positive waves) in proximal muscles, without polyneuropathy.
Body composition
Skinfolds (Table 2) were measured in triplicate on the dominant extremity with a Lange skinfold caliper (Cambridge Scientific Industries, MD, USA). Segmental body fat distribution was assessed with whole-body dual-energy X-ray absorptiometry, performed with a Lunar model DPX apparatus (GE Medical Systems, Milwaukee, WI, USA).
Changes in body composition before and after Cushing cure evaluated by anthropometry and DXA
Molecular analyses
DNA was prepared from peripheral white blood cells following standard procedures (55. Sambrook J, Russell DW. The Condensed Protocols from Molecular Cloning: A Laboratory Manual: Cold Spring Harbor Laboratory Press; 2006.). LMNA exons 1–12 and the surrounding intronic sequences were amplified by PCR. Primers and conditions have been previously described (66. Araujo-Vilar D, Loidi L, Dominguez F, Cabezas-Cerrato J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Horm Metab Res. 2003;35(1):29-35.).
Histological muscle studies
Deltoid muscle biopsy samples were snap frozen in liquid nitrogen, and cryostat sections were processed and stained to exclude dystrophies and other pathological conditions (e.g., mitochondrial myopathies, metabolic diseases) as follows: PAS, modified Gomori trichrome, Oil Red O, enzyme histochemistry (muscle phosphorylase, muscle phosphofructokinase, lactate dehydrogenase, AMP deaminase, cytochrome-C oxidase, SDH, and NADH), and immunohistochemistry (beta-spectrin to check preservation of the plasma membrane; slow, fast, and fetal myosin; utrophin; alpha, beta, gamma, and delta sarcoglycan; dystrophin fractions: N- terminal, C-terminal, and rod domain; NOS; alpha and beta dystroglycan; dysferlin, caveolin-3, collagen type VI, laminin alpha2 chain of merosin (laminin-2), telethonin, myotilin, desmin, calpain-3, MHC class I antigen, and inflammatory markers (CD68/KP1, CD3 and CD20 for histiocytes and lymphocytes). The only commercial antibodies available for Lamin A/C are not helpful for diagnostic purposes (here it was used as a positive control for emerin antibody).
Immunostains were performed after antigen retrieval using a standard avidin–biotin immunoperoxidase detection technique (EnVision Systems, Dako, Glostrup, Denmark).
RESULTS
We identified a heterozygous c.1634G>A (p.R545H) variant of LMNA, located in exon 10. This variant had not been probed for pathogenicity with in silico approaches (PolyPhen and SIFT).
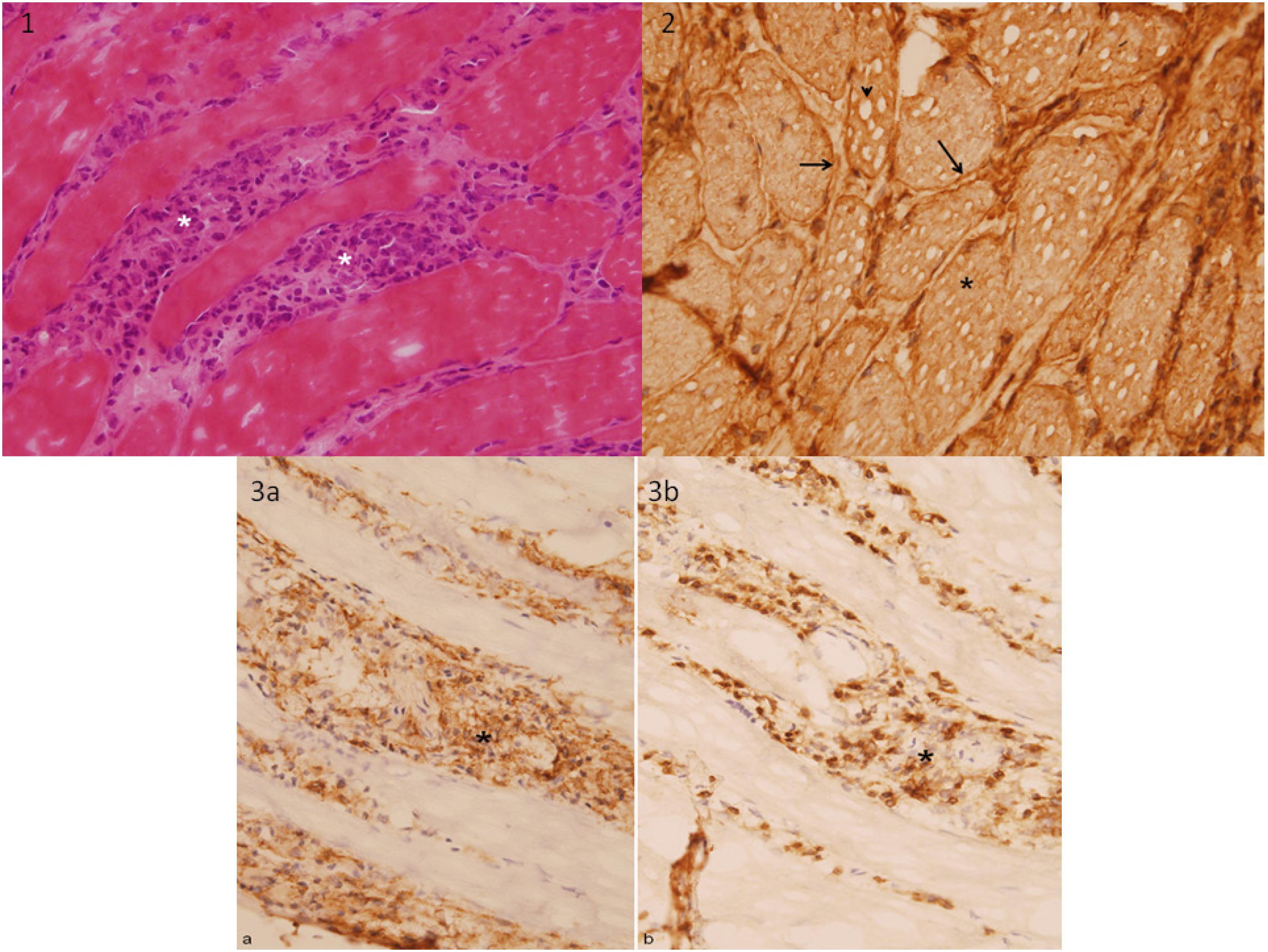
Its allelic frequency was 0.00019 (1000 Genomes Project). Of the patient's relatives whom we studied (mother, sisters, offspring), none carried this variant. Histological findings of muscle samples were consistent with acute and chronic, nonspecific, inflammatory myopathy (Figure 2).
(1) Snap frozen cryostat sections of deltoid muscle showing endomisial inflammatory infiltrates composed mainly of hystiocytes (asterisks) (Hematoxylin and eosin, 400x). (2) MHC class I antigen is upregulated in all fibers, with immunolabeling at plasma membrane (arrow) and sarcoplasm (asterisk). See also “pseudovacuoles” secondary to “ice crystals” snap frozen artifact (arrow head) (Immunoperoxidase reaction, diaminobenzidine brown staining chromogen, 400x). (3a) Immunostaining of CD68 (KP1) antibody remark histiocityc inflammatory component between myofibers, with (3b) a minor population of CD3 antibody positive T lymphocytes (asterisks) (400x).
DISCUSSION
We investigated an unusual case of laminopathy and found that it was due to a p.R545H LMNA variant that overlapped with chronic hypercortisolism. Thus, after chronic hypercortisolism had been diagnosed and cured, the patient exhibited atypical partial lipodystrophy, idiopathic inflammatory myopathy, and cardiomyopathy with conduction disturbances. Although this variant has not been shown to be pathogenic with in silico approaches, it has been associated with FPLD2 (77. Chan D, McIntyre AD, Hegele RA, Don-Wauchope AC. Familial partial lipodystrophy presenting as metabolic syndrome. J Clin Lipidol. 2016;10(6):1488-91.), EDMD2 (88. Huong S. Molecular genetic studies in hereditary laminopathies of man.: Ernst- Moritz-Arndt-Universität, Greifswald; 2010.), LGMD1B (99. Maggi L, Carboni N, Bernasconi P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells. 2016;5(3):pii: E33.), and heart disease (1010. van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013;15(4):376-84.).
In FPLD2, more than 80% of cases are due to missense mutations in exon 8; however, atypical phenotypes have been related to mutations in other exons (1111. Garg A, Vinaitheerthan M, Weatherall PT, Bowcock AM. Phenotypic heterogeneity in patients with familial partial lipodystrophy (dunnigan variety) related to the site of missense mutations in lamin a/c gene. J Clin Endocrinol Metab. 2001;86(1): 59-65.). Moreover, several patients with mutations outside the immunoglobulin-like fold of lamin A have lacked the typical FPLD2 phenotype but experienced insulin resistance (44. Decaudain A, Vantyghem MC, Guerci B, Hecart AC, Auclair M, Reznik Y, et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J Clin Endocrinol Metab. 2007;92(12):4835-44.). In addition, some cases of FPLD2 have been associated with heart conduction disorders, valvulopathies, and cardiomyopathy (1212. Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med. 2002;112(7):549-55.
13. Araujo-Vilar D, Lado-Abeal J, Palos-Paz F, Lattanzi G, Bandin MA, Bellido D, et al. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin Endocrinol (Oxf). 2008;69(1):61-8.-1414. Panikkath R, Panikkath D, Sanchez-Iglesias S, Araujo-Vilar D, Lado-Abeal J. An Uncommon Association of Familial Partial Lipodystrophy, Dilated Cardiomyopathy, and Conduction System Disease. J Investig Med High Impact Case Rep. 2016;4(3):2324709616658495.). Other authors have reported LMNA-associated complex phenotypes, including heart failure and limb-girdle muscular dystrophy, due to a Ser334del variant (1515. Madej-Pilarczyk A, Niezgoda A, Janus M, Wojnicz R, Marchel M, Fidzianska A, et al. Limb-girdle muscular dystrophy with severe heart failure overlapping with lipodystrophy in a patient with LMNA mutation p.Ser334del. J Appl Genet 2017;58(1):87-91.,1616. Madej-Pilarczyk A, Niezgoda A, Janus M, Wojnicz R, Marchel M, Fidziańska A, et al. Limb-girdle muscular dystrophy with severe heart failure overlapping with lipodystrophy in a patient with LMNA mutation p.Ser334del. J Appl Genet. 2017;58(1):87-91.); muscular dystrophy, lipodystrophy, and cardiac rhythm disturbances related to a R527P variant (1717. van der Kooi AJ, Bonne G, Eymard B, Duboc D, Talim B, Van der Valk M, et al. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology. 2002;59(4):620-3.); or FPLD, early heart failure, first-degree atrioventricular block, and late proximal muscle weakness due to a R28W variant (1212. Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med. 2002;112(7):549-55.).
Exon 10 of LMNA corresponds to the C-terminal domain, common to both lamins A and C, which forms an immunoglobulin-like, three-dimensional structure (11. Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86(3):967-1008.). The conformation of this domain is well defined for residues 430–545 (1818. Krimm I, Ostlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, et al. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure. 2002;10(6):811-23.). Arginine 545 is at the external surface of the structure. Mutations in the Ig-fold can affect either head-to-tail polymerization, which destabilizes the three-dimensional structure of the C-terminal domain, or lamin A/C interactions with other proteins (11. Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86(3):967-1008.). At least 13 missense mutations at the C-terminal domain are related to EDMD2 (1818. Krimm I, Ostlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, et al. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure. 2002;10(6):811-23.), including the nearby R541H. Although the R545 residue has not been specifically studied, it does not seem to influence the stability of the 3D structure. However, the exchange of arginine (positive polar) for histidine (neutral polar) could alter interactions with other proteins at the nuclear lamina.
This study was particularly challenging because of the presence of Cushing's syndrome and the autoimmune background. Chronic hypercortisolism causes a characteristic fat distribution, in particular, excess abdominal fat (1919. Garrapa GG, Pantanetti P, Arnaldi G, Mantero F, Faloia E. Body composition and metabolic features in women with adrenal incidentaloma or Cushing's syndrome. J Clin Endocrinol Metab. 2001;86(11):5301-6.); however, no particular changes in limb fat have been reported (2020. Rockall AG, Sohaib SA, Evans D, Kaltsas G, Isidori AM, Monson JP, et al. Computed tomography assessment of fat distribution in male and female patients with Cushing's syndrome. Eur J Endocrinol. 2003;149(6):561-7.). Strikingly, in this patient, once hypercortisolism was cured, an abnormal fat distribution became more evident, although it was not as severe as observed in classical Dunnigan disease. This change in fat distribution was particularly intriguing because it highlighted important differences among LMNA mutations that cause FPLD (1313. Araujo-Vilar D, Lado-Abeal J, Palos-Paz F, Lattanzi G, Bandin MA, Bellido D, et al. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin Endocrinol (Oxf). 2008;69(1):61-8.). The R545H variant, previously associated with FPLD (77. Chan D, McIntyre AD, Hegele RA, Don-Wauchope AC. Familial partial lipodystrophy presenting as metabolic syndrome. J Clin Lipidol. 2016;10(6):1488-91.), caused severe metabolic syndrome with android fat distribution in this patient.
The patient was initially diagnosed with polymyositis, based on girdle weakness, high creatine kinase, and muscle lymphocyte infiltration. Because of her autoimmune background, this diagnosis was probably the most parsimonious. However, the R545H variant has been associated with LGMD1B (99. Maggi L, Carboni N, Bernasconi P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells. 2016;5(3):pii: E33.), which clinically overlaps with polymyositis. Other LMNA mutations that cause muscular and/or cardiac laminopathies have been related to inflammatory changes in muscle specimen biopsies. For example, in biopsies from patients with infantile- onset LMNA-associated myopathy, Komaki and cols. (2121. Komaki H, Hayashi YK, Tsuburaya R, Sugie K, Kato M, Nagai T, et al. Inflammatory changes in infantile-onset LMNA-associated myopathy. Neuromuscul Disord. 2011;21(8):563-8.) reported mononuclear cell infiltrations that were positive for lymphocyte markers CD4, CD8, or CD20, active necrosis, and regeneration. Additionally, they observed elevated sarcolemmal HLA staining in many fibers. Similarly, HLA class I antigens were upregulated in our samples and in samples described previously in a study on LGMD1B (1515. Madej-Pilarczyk A, Niezgoda A, Janus M, Wojnicz R, Marchel M, Fidzianska A, et al. Limb-girdle muscular dystrophy with severe heart failure overlapping with lipodystrophy in a patient with LMNA mutation p.Ser334del. J Appl Genet 2017;58(1):87-91.,1616. Madej-Pilarczyk A, Niezgoda A, Janus M, Wojnicz R, Marchel M, Fidziańska A, et al. Limb-girdle muscular dystrophy with severe heart failure overlapping with lipodystrophy in a patient with LMNA mutation p.Ser334del. J Appl Genet. 2017;58(1):87-91.). It cannot be ruled out that chronic hypercortisolism had influenced the myopathy. However, there are some clues to differences between the corticoid-myopathy case and the case described here.
In the first case, serum values of muscle enzymes are typically normal or slightly elevated, electromyoneurography is usually normal, and there are no inflammatory infiltrates or necrosis on muscle biopsy samples (2222. Gupta A, Gupta Y. Glucocorticoid-induced myopathy: Pathophysiology, diagnosis, and treatment. Indian J Endocrinol Metab. 2013;17(5):913-6.). Although unusual, polymyositis has been associated with cardiac involvement, manifested as rhythm disturbances, conduction defects, and heart failure (2323. Eisen A, Arnson Y, Dovrish Z, Hadary R, Amital H. Arrhythmias and conduction defects in rheumatological diseases--a comprehensive review. Semin Arthritis Rheum. 2009;39(3):145-56.). Similarly, laminopathies, like FPLD2 or LGMD1B (99. Maggi L, Carboni N, Bernasconi P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells. 2016;5(3):pii: E33.,1414. Panikkath R, Panikkath D, Sanchez-Iglesias S, Araujo-Vilar D, Lado-Abeal J. An Uncommon Association of Familial Partial Lipodystrophy, Dilated Cardiomyopathy, and Conduction System Disease. J Investig Med High Impact Case Rep. 2016;4(3):2324709616658495.), and particularly the R545H variant, have also been related to heart disease (1010. van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013;15(4):376-84.). Patients with cardiac compromise related to LMNA typically show initial signs of nonspecific rhythm disturbances after age 30 y (2424. van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl). 2005;83(1):79-83.), and they frequently need a permanent pacemaker. Later, cardiac compromise may be further complicated with cardiomyopathy (2525. Carboni N, Mateddu A, Marrosu G, Cocco E, Marrosu MG. Genetic and clinical characteristics of skeletal and cardiac muscle in patients with lamin A/C gene mutations. Muscle Nerve. 2013;48(2):161-70.). Our patient was diagnosed with Wolff–Parkinson–White syndrome, atrial fibrillation, mitral regurgitation, and mild hypertrophic cardiomyopathy. Taken together, the diagnoses of severe metabolic syndrome, girdle weakness appearing in the fourth decade, and heart involvement in a patient that carried the R545H variant in LMNA resembled a complex laminopathic disorder. However, the presence of chronic hypercortisolism represented a confounding factor, and rheumatoid arthritis prevented us from definitely ruling out a random association of different unrelated autoimmune entities.
In summary, this case emphasizes the need for actively searching for LMNA mutations in patients with clinical features compatible with partial lipodystrophy, cardiac conduction abnormalities, cardiomyopathy, and/or certain types of myopathy. A correct diagnosis can facilitate early interventions to prevent the consequences of these pathologies, which can be lethal.
-
Ethical Publication Statement: we confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Acknowledgments:
we are indebted to the patients of this study for their collaboration. This work was funded by the Instituto de Salud Carlos III (grant number: PI081449) and the European Regional Development Fund, FEDER. In addition, SRG was awarded a Research Fellowship granted by the Asociación Española de Familiares y Afectados de Lipodistrofias (AELIP). None of the authors has any conflict of interest to disclose.
REFERENCES
-
1Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86(3):967-1008.
-
2Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab. 2016;101(12):4500-11.
-
3Guillin-Amarelle C, Sanchez-Iglesias S, Castro-Pais A, Rodriguez-Canete L, Ordonez-Mayan L, Pazos M, et al. Type 1 familial partial lipodystrophy: understanding the Kobberling syndrome. Endocrine. 2016;54(2):411-21.
-
4Decaudain A, Vantyghem MC, Guerci B, Hecart AC, Auclair M, Reznik Y, et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J Clin Endocrinol Metab. 2007;92(12):4835-44.
-
5Sambrook J, Russell DW. The Condensed Protocols from Molecular Cloning: A Laboratory Manual: Cold Spring Harbor Laboratory Press; 2006.
-
6Araujo-Vilar D, Loidi L, Dominguez F, Cabezas-Cerrato J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Horm Metab Res. 2003;35(1):29-35.
-
7Chan D, McIntyre AD, Hegele RA, Don-Wauchope AC. Familial partial lipodystrophy presenting as metabolic syndrome. J Clin Lipidol. 2016;10(6):1488-91.
-
8Huong S. Molecular genetic studies in hereditary laminopathies of man.: Ernst- Moritz-Arndt-Universität, Greifswald; 2010.
-
9Maggi L, Carboni N, Bernasconi P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells. 2016;5(3):pii: E33.
-
10van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013;15(4):376-84.
-
11Garg A, Vinaitheerthan M, Weatherall PT, Bowcock AM. Phenotypic heterogeneity in patients with familial partial lipodystrophy (dunnigan variety) related to the site of missense mutations in lamin a/c gene. J Clin Endocrinol Metab. 2001;86(1): 59-65.
-
12Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med. 2002;112(7):549-55.
-
13Araujo-Vilar D, Lado-Abeal J, Palos-Paz F, Lattanzi G, Bandin MA, Bellido D, et al. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin Endocrinol (Oxf). 2008;69(1):61-8.
-
14Panikkath R, Panikkath D, Sanchez-Iglesias S, Araujo-Vilar D, Lado-Abeal J. An Uncommon Association of Familial Partial Lipodystrophy, Dilated Cardiomyopathy, and Conduction System Disease. J Investig Med High Impact Case Rep. 2016;4(3):2324709616658495.
-
15Madej-Pilarczyk A, Niezgoda A, Janus M, Wojnicz R, Marchel M, Fidzianska A, et al. Limb-girdle muscular dystrophy with severe heart failure overlapping with lipodystrophy in a patient with LMNA mutation p.Ser334del. J Appl Genet 2017;58(1):87-91.
-
16Madej-Pilarczyk A, Niezgoda A, Janus M, Wojnicz R, Marchel M, Fidziańska A, et al. Limb-girdle muscular dystrophy with severe heart failure overlapping with lipodystrophy in a patient with LMNA mutation p.Ser334del. J Appl Genet. 2017;58(1):87-91.
-
17van der Kooi AJ, Bonne G, Eymard B, Duboc D, Talim B, Van der Valk M, et al. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology. 2002;59(4):620-3.
-
18Krimm I, Ostlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, et al. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure. 2002;10(6):811-23.
-
19Garrapa GG, Pantanetti P, Arnaldi G, Mantero F, Faloia E. Body composition and metabolic features in women with adrenal incidentaloma or Cushing's syndrome. J Clin Endocrinol Metab. 2001;86(11):5301-6.
-
20Rockall AG, Sohaib SA, Evans D, Kaltsas G, Isidori AM, Monson JP, et al. Computed tomography assessment of fat distribution in male and female patients with Cushing's syndrome. Eur J Endocrinol. 2003;149(6):561-7.
-
21Komaki H, Hayashi YK, Tsuburaya R, Sugie K, Kato M, Nagai T, et al. Inflammatory changes in infantile-onset LMNA-associated myopathy. Neuromuscul Disord. 2011;21(8):563-8.
-
22Gupta A, Gupta Y. Glucocorticoid-induced myopathy: Pathophysiology, diagnosis, and treatment. Indian J Endocrinol Metab. 2013;17(5):913-6.
-
23Eisen A, Arnson Y, Dovrish Z, Hadary R, Amital H. Arrhythmias and conduction defects in rheumatological diseases--a comprehensive review. Semin Arthritis Rheum. 2009;39(3):145-56.
-
24van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl). 2005;83(1):79-83.
-
25Carboni N, Mateddu A, Marrosu G, Cocco E, Marrosu MG. Genetic and clinical characteristics of skeletal and cardiac muscle in patients with lamin A/C gene mutations. Muscle Nerve. 2013;48(2):161-70.
Publication Dates
-
Publication in this collection
17 May 2018 -
Date of issue
June 2018
History
-
Received
19 July 2017 -
Accepted
07 Feb 2018