Resumos
A hiperplasia congênita das supra-renais (HCSR) representa um grupo de doenças genéticas que comprometem a síntese de cortisol devida à deficiência em uma das enzimas responsáveis pela esteroidogênese supra-renal. Na deficiência da 21-hidroxilase (D21OH), responsável por mais de 90% dos casos de HCSR, a secreção androgênica supra-renal está aumentada. A forma clássica de D21OH é tratada com glicocorticóide, repondo-se mineralocorticóide quando necessário. O uso continuado de corticosteróides pode comprometer o crescimento através de diferentes mecanismos. Por outro lado, a excessiva secreção adrenocortical dos esteróides sexuais pode levar à redução do tempo de crescimento, tanto por aceleração da idade óssea quanto pela possibilidade de indução de puberdade precoce central. A combinação dos efeitos da corticoterapia e do excesso de esteróides sexuais sobre o crescimento faz com que as crianças com HCSR estejam sob risco de baixa estatura. Daí a importância da cuidadosa avaliação e monitorização do crescimento e da evolução puberal desses pacientes. Igualmente importante é o emprego de glicocorticóides com menor capacidade em suprimir o crescimento. Desse modo, acreditamos que a melhor opção terapêutica para crianças com HCSR é representada pela hidrocortisona ou acetato de cortisona, empregadas na dose fisiológica e administrando-se a maior dose pela manhã.
Hiperplasia supra-renal congênita; Deficiência de 21-hidroxilase; Crescimento; Tratamento; Corticosteróides; Androgênios supra-renais
Congenital adrenal hyperplasia (CAH) represents a group of genetic diseases where the synthesis of cortisol is compromised by deficiency in one of the enzymes involved in adrenal steroidogenesis. In CAH owing to 21-hydroxylase deficiency (21OHD), responsible for more than 90% of all cases, the adrenal androgen secretion is increased. The classic form of 21OHD is treated with glucocorticoids, and mineralocorticoid is replaced if necessary. The chronic use of corticosteroids may compromise growth by different means. Otherwise, the excessive adrenocortical secretion of sex steroids reduces the duration of growth, by both accelerating bone age and the possible induction of central precocious puberty. The associated effects of prolonged glucocorticoid therapy and excessive amounts of sex steroids upon growth render children with CAH specially at risk for short stature. Therefore their growth and pubertal evolution must be carefully evaluated and monitored. It is also important to use glucocorticoids with less growth suppressive potential. Thus, we believe that the best therapeutic option for children with CAH is the use of hydrocortisone ou cortisone acetate in physiologic doses, the larger dose given in the morning.
Congenital adrenal hyperplasia; 21-Hydroxylase deficiency; Growth; Treatment; Corticosteroids; Adrenal androgens
revisão
Fatores Que Interferem no Crescimento e na Altura Final de Pacientes Com Hiperplasia Congênita das Supra-Renais por Deficiência da 21-Hidroxilase
Hamilton C. de Menezes Filho
Vaê Dichtchekenian
Hilton Kuperman
Thais Della Manna
Durval Damiani
Nuvarte Setian
Unidade de Endocrinologia, Instituto da Criança do Hospital das Clínicas, Faculdade de Medicina da
Universidade de São Paulo, SP.
Recebido em 03/01/00
Revisado em 12/07/01 e 16/10/01
Aceito em 19/10/01
RESUMO
A hiperplasia congênita das supra-renais (HCSR) representa um grupo de doenças genéticas que comprometem a síntese de cortisol devida à deficiência em uma das enzimas responsáveis pela esteroidogênese supra-renal. Na deficiência da 21-hidroxilase (D21OH), responsável por mais de 90% dos casos de HCSR, a secreção androgênica supra-renal está aumentada. A forma clássica de D21OH é tratada com glicocorticóide, repondo-se mineralocorticóide quando necessário. O uso continuado de corticosteróides pode comprometer o crescimento através de diferentes mecanismos. Por outro lado, a excessiva secreção adrenocortical dos esteróides sexuais pode levar à redução do tempo de crescimento, tanto por aceleração da idade óssea quanto pela possibilidade de indução de puberdade precoce central. A combinação dos efeitos da corticoterapia e do excesso de esteróides sexuais sobre o crescimento faz com que as crianças com HCSR estejam sob risco de baixa estatura. Daí a importância da cuidadosa avaliação e monitorização do crescimento e da evolução puberal desses pacientes. Igualmente importante é o emprego de glicocorticóides com menor capacidade em suprimir o crescimento. Desse modo, acreditamos que a melhor opção terapêutica para crianças com HCSR é representada pela hidrocortisona ou acetato de cortisona, empregadas na dose fisiológica e administrando-se a maior dose pela manhã.
Unitermos: Hiperplasia supra-renal congênita; Deficiência de 21-hidroxilase; Crescimento; Tratamento; Corticosteróides; Androgênios supra-renais.
ABSTRACT
Congenital adrenal hyperplasia (CAH) represents a group of genetic diseases where the synthesis of cortisol is compromised by deficiency in one of the enzymes involved in adrenal steroidogenesis. In CAH owing to 21-hydroxylase deficiency (21OHD), responsible for more than 90% of all cases, the adrenal androgen secretion is increased. The classic form of 21OHD is treated with glucocorticoids, and mineralocorticoid is replaced if necessary. The chronic use of corticosteroids may compromise growth by different means. Otherwise, the excessive adrenocortical secretion of sex steroids reduces the duration of growth, by both accelerating bone age and the possible induction of central precocious puberty. The associated effects of prolonged glucocorticoid therapy and excessive amounts of sex steroids upon growth render children with CAH specially at risk for short stature. Therefore their growth and pubertal evolution must be carefully evaluated and monitored. It is also important to use glucocorticoids with less growth suppressive potential. Thus, we believe that the best therapeutic option for children with CAH is the use of hydrocortisone ou cortisone acetate in physiologic doses, the larger dose given in the morning.
Keywords: Congenital adrenal hyperplasia; 21-Hydroxylase deficiency; Growth; Treatment; Corticosteroids; Adrenal androgens.
A HIPERPLASIA CONGÊNITA DAS SUPRA-RENAIS (HCSR) é um grupo de doenças de herança autossômica recessiva que afetam a esteroidogênese supra-renal. O termo hiperplasia refere-se ao aspecto histológico do córtex supra-renal, resultante da ação do hormônio adrenocorticotrófico (ACTH), cujos níveis na HCSR encontram-se cronicamente elevados. A elevação do ACTH é secundária à síntese deficiente de cortisol, em decorrência da diminuição da atividade em qualquer uma das enzimas envolvidas na sua produção.
Diversas enzimas participam das diferentes etapas da esteroidogênese supra-renal. O passo inicial, considerado o limitante da esteroidogênese, envolve o transporte do colesterol plasmático da porção externa da membrana mitocondrial para a sua porção interna, por meio da proteína reguladora aguda da esteroidogênese (proteína StAR)(1).
As diversas etapas da esteroidogênese são catalizadas por diferentes enzimas ou complexos enzimáticos, conforme ilustrado na figura 1. Na tabela 1 estão relacionadas as atividades dessas enzimas, bem como sua denominação antiga e atual.
A deficiência da CYP21A2 (21-hidroxilase) é responsável por mais de 90% dos casos de HCSR (2). Acredita-se que a incidência da forma clássica da HCSR por deficiência da 21-hidroxilase (D21OH) seja de um caso em 14.000 nascimentos, embora em algumas regiões a incidência seja mais elevada (3,4).
A HCSR por D21OH manifesta-se através de diferentes formas, de acordo com a intensidade de redução da atividade enzimática. Na forma clássica, a deficiência enzimática é quase completa na forma perdedora de sal (FPS), responsável por 75 a 80% dos casos, e onde as secreções deficientes de cortisol e de mineralocorticóide podem resultar em insuficiência supra-renal aguda (crise de perda de sal). O restante da forma clássica é representado pela forma virilizante simples (FVS), onde o comprometimento enzimático é menos intenso do que na FPS. Estes pacientes podem apresentar insuficiência adrenocortical diante de situações de "stress". Na forma não clássica, a deficiência enzimática é leve e os indivíduos acometidos podem ser assintomáticos.
A elevação da 17-hidroxiprogesterona (17-OHP), decorrente da D21OH, pode acentuar a perda de sal na FPS e levar à perda salina na FVS, com aumento da atividade plasmática de renina (APR). Outro efeito da elevação da 17-OHP é a maior convergência da esteroidogênese em direção à síntese dos esteróides sexuais, principalmente da androstenediona (A). O aumento da A (considerada um andrógeno fraco) resulta em elevação dos níveis plasmáticos de testosterona (T), devido à interconversão entre os esteróides na periferia, a qual é responsável pela virilização destes pacientes (5,6). Enquanto no feto feminino a virilização leva à ambiguidade genital em diversos graus de intensidade, no feto masculino não há alteração da genitália. A falta de controle adequado da HCSR após o nascimento leva à progressiva virilização, tanto nos meninos quanto nas meninas.
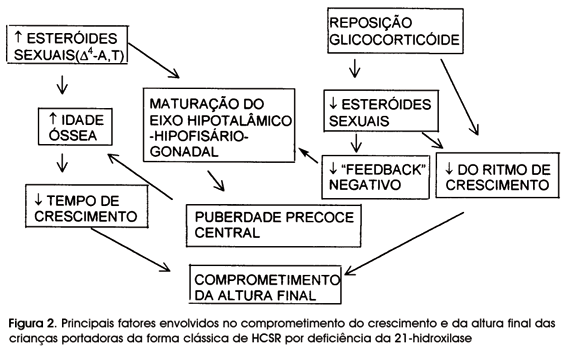
Um dos aspectos mais preocupantes em relação à virilização diz respeito à aceleração do crescimento, e principalmente da maturação epifisária, já que o fechamento precoce das cartilagens epifisárias leva à redução do tempo total de crescimento destas crianças, afetando de modo significativo e irreversível a sua altura final (AF).
O tratamento da HCSR consiste na reposição do glicocorticóide de forma a procurar imitar o padrão secretório de cortisol em indivíduos normais, com o intuito de se suprir o organismo com o esteróide, importante para o metabolismo e outras funções vitais (como a manutenção do tônus vascular), e ao mesmo tempo diminuir a hipersecreção androgênica supra-renal e suas conseqüências. A reposição de glicocorticóide para o tratamento da HCSR foi proposta inicialmente por Wilkins et al., em 1950 (7) e Bartter et al., em 1951 (8). Ambos documentaram a capacidade da cortisona em suprimir a excessiva secreção adrenocortical na HCSR.
Os diferentes corticosteróides exógenos apresentam diferentes potências anti-inflamatória e mineralocorticóide. A potência anti-inflamatória dos glicocorticóides depende da sua meia-vida biológica. Outros fatores que também determinam a sua ação anti-inflamatória são a capacidade de ligação às proteínas plasmáticas, a capacidade em atravessar a membrana plasmática, a ação intrínseca de cada molécula sobre os receptores e o"clearance" do glicocorticóide. As tabelas sobre equivalências de glicocorticóides frequentemente empregadas baseiam-se primariamente em seus efeitos anti-inflamatórios, já que estes são o principal motivo pelo qual são prescritos. É importante lembrar que o efeito do glicocorticóide varia de indivíduo para indivíduo, sendo a variabilidade maior para os compostos de maior meia-vida plasmática e biológica, como a dexametasona.
No entanto, um aspecto destes compostos permanece pouco valorizado e deveria ser considerado de maneira primordial ao se escolher a medicação a ser empregada em crianças e adolescentes e diz respeito à sua potência supressora do crescimento. Diferentes glicocorticóides suprimem o crescimento diferentemente. Os efeitos supressores do crescimento e anti-inflamatórios dos diferentes glicocorticóides não são colineares e em geral os esteróides mais potentes, com maior meia-vida biológica, têm efeito supressor do crescimento desproporcionalmente maior. A tabela 2 apresenta as diferentes potências anti-inflamatória, supressora do crescimento e retentora de sal dos principais corticóides em relação ao cortisol.
Os dados referentes às potências anti-inflamatória e retentora de sal e à meia-vida biológica foram obtidos de Haynes (9), enquanto que os referentes à potência supressora do crescimento foram obtidos de Miller (10).
Blodgett et al. (11) foram os responsáveis por uma das primeiras publicações a abordar o crescimento das crianças sob corticoterapia prolongada, verificando que a cortisona empregada por via oral reduziu os ritmos de crescimento e de maturação óssea em algumas crianças mesmo após poucas semanas de uso, e que a dose necessária para levar a estas alterações variou de acordo com a condição clínica de cada criança. Pouco tempo depois Van Metre et al. (12) descreveram as diferentes capacidades de supressão do crescimento de diferentes glicocorticóides, a partir do estudo de 19 crianças portadoras de asma perene de difícil controle, para as quais foi indicada corticoterapia. Os autores encontraram comprometimento do crescimento com o uso de prednisona ou metilprednisolona em doses superiores a 5 mg/m2/dia, ao passo que doses tão altas de cortisona quanto 55 mg/m2/dia não levaram à redução do crescimento. A conclusão foi que embora a cortisona tivesse 20% da potência anti-inflamatória da prednisona e de 17% com relação à metilprednisolona, sua potência supressora do crescimento foi de 10% em relação a estes glicocorticóides.
Os trabalhos citados anteriormente mostram evidências clínicas de muitos anos de que a utilização dos glicocorticóides em doses elevadas e por tempo prolongado leva à redução do crescimento. Atualmente sabe-se que os efeitos dos corticóides em relação à redução do crescimento são multifatoriais, e envolvem as seguintes ações:
Supressão da secreção hipofisária do hormônio de crescimento (GH)
Os glicocorticóides exercem diferentes efeitos em relação à secreção do GH, de acordo com sua administração aguda ou crônica. Quando administrados poucas horas antes de testes de estímulo de secreção do GH com o hormônio liberador do GH (GHRH) ou com a clonidina têm efeito permissivo, elevando a secreção do GH, através de aumento da sensibilidade da adenohipófise ao GHRH ou da redução da secreção hipotalâmica da somatostatina (ST). Quando administrados cronicamente, no entanto, os glicocorticóides suprimem a secreção hipofisária de GH em resposta à maior parte dos estímulos fisiológicos e farmacológicos (como exercício físico, hipoglicemia induzida pela insulina, GHRH e agonistas da dopamina), mas não a secreção do GH induzida pela arginina. Como a arginina induz a secreção do GH através da inibição da secreção de ST pelo hipotálamo, acredita-se que o uso crônico dos corticóides possa resultar em elevação do tônus somatostatinérgico e desse modo reduzir a secreção do GH (13).
Os glicocorticóides também alteram a secreção basal de GH, com efeitos semelhantes aos descritos anteriormente. Assim, sua administração aguda está associada ao aumento da secreção basal do GH, decorrente de aumento da frequência dos pulsos secretórios do GH e aumento da quantidade de GH secretado em cada pulso. Por outro lado, o uso crônico dos glicocorticóides resulta em supressão da secreção espontânea do GH por diminuição da quantidade de hormônio secretado em cada pulso secretório, sem que haja interferência na frequência de pulsos de secreção do GH (13).
Apesar dos efeitos crônicos dos glicocorticóides sobre a secreção do GH, Sperling et al. (14), Vazquez et al. (15), Rappaport et al. (16) e Silva et al. (17) encontraram secreção normal do GH em crianças portadoras de HCSR tratadas com acetato de cortisona ou hidrocortisona. Com base nos resultados obtidos, Sperling et al. e Rappaport et al. propõem que na HCSR, a redução do crescimento e da maturação óssea relacionadas ao uso crônico de glicocorticóide ocorram por inibição pelo corticóide dos efeitos do GH na periferia e não por diminuição da sua secreção pela adeno-hipófise.
Redução da sensibilidade tecidual ao GH
A exposição prolongada de diversos tecidos (com destaque para o fígado e o tecido ósseo) ao glicocorticóide leva à redução da sensibilidade tecidual ao GH, decorrente de um processo de "down-regulation" do receptor tecidual de GH. Os níveis plasmáticos da proteína ligante do GH (GHBP), que em humanos é proveniente da quebra proteolítica do domínio extracelular do receptor de GH, encontram-se diminuídos e não se elevam com a administração de GH recombinante (13).
Inibição da bioatividade do fator de crescimento semelhante à insulina tipo 1 (IGF-1)
As somatomedinas, ou fatores de crescimento semelhantes à insulina (IGFs), são representadas pelo IGF-1 e IGF-2, e são responsáveis pela intermediação das ações do GH em relação ao crescimento. Os níveis sanguíneos do IGF-2 são superiores aos do IGF-1, o mesmo ocorrendo em extratos de tecido esquelético. No plasma, a maior parte dos IGFs está ligada à IGFBP-3 (proteína ligante dos IGFs, tipo 3). Ambos IGFs são importantes para o crescimento fetal, enquanto que o IGF-1 é também de fundamental importância para o crescimento pós-natal. A maior parte do IGF-1 na circulação é produzida no fígado, sob o controle do GH. Os IGFs são também produzidos nas células ósseas e em outros tecidos. Apesar do reconhecimento atual de que os IGFs são essenciais para o crescimento esquelético normal, não se determinou ainda se o IGF-1 que atua sobre o crescimento esquelético é proveniente da circulação periférica ou é sintetizado na própria placa de crescimento em resposta ao GH e a outros estímulos hormonais. Na realidade, sabe-se que o IGF-1 tem ações tanto endócrinas quanto parácrinas. Na placa de crescimento o IGF-1 estimula a proliferação dos condrócitos na zona proliferativa e aumenta a diferenciação, além de inibir a apoptose, dos condrócitos na zona hipertrófica. Não se sabe ainda se o IGF-1 é capaz por si só de estimular a proliferação dos condrócitos ou se depende de uma ação prévia do GH no sentido de transformar os pré-condrócitos em células cartilaginosas mais maduras (18).
Nos tecidos ósseos a formação dos IGFs, de seus receptores e das IGFBPs é regulada por diversos hormônios, incluindo o GH, estradiol, testosterona, proteínas ósseas morfogênicas, hormônio da paratireóide (PTH), peptídeo relacionado ao PTH (PTH-rP), calcitriol, além de citocinas e fatores de crescimento. Ao menos algumas das ações dos esteróides sexuais no osso dependem da estimulação da produção de IGF. O estímulo da proliferação dos condrócitos pelo PTH/ PTHrP é feito, ao menos em parte, às custas da indução do aumento da produção do IGF-1. Por outro lado, enquanto o IGF-1 estimula a diferenciação dos condrócitos, o PTHrP inibe este processo (18).
O IGF-1 também promove a formação do colágeno, a síntese de ácido ribonucléico e desoxiribonucléico e de proteína e é responsável pela sulfatação das proteoglicanas nas células da cartilagem.
Em 1985, Unterman e Phillips (19) verificaram que o uso da prednisona em dose farmacológica reduz a atividade do IGF-1 sem alterar seus níveis circulantes, e sugerem que os glicocorticóides não diminuem a produção do IGF-1, mas antagonizam a sua ação através do aumento da liberação hepática de inibidores do IGF-1. Atualmente sabe-se que em pacientes submetidos cronicamente a níveis elevados de glicocorticóides os valores de IGF-1 imunoreativo na circulação tendem a ser normais ou mesmo pouco elevados, e que os glicocorticóides inibem a bioatividade do IGF-1 no soro através da antagonização da ação do IGF-1, por meio de mecanismos diretos e/ou indiretos, e possivelmente por intermédio da indução de inibidores do IGF-1 (estes inibidores, ainda não caracterizados, têm peso molecular de 12-20 kDa). Os glicocorticóides podem alterar os níveis plasmáticos de algumas IGFBPs, principalmente da IGFBP-2 e da IGFBP-3, o que pode contribuir para a redução da bioatividade do IGF-1 (13).
Outros efeitos dos glicocorticóides sobre o crescimento
Durante o estirão pubertário os níveis plasmáticos de GH e IGF-1 aumentam significativamente. Em ambos sexos o aumento da produção de estradiol durante a puberdade é responsável pelo aumento da secreção do GH e IGF-1. No sexo masculino o estradiol é originado a partir da aromatização da testosterona. A dihidrotestosterona e o estradiol têm ações diretas sobre os condrócitos. O estrógeno em dose fisiológica ou em baixa dosagem aumenta o crescimento, enquanto que em doses elevadas suprime a secreção do IGF-1 e reduz o ritmo de crescimento. Também em ambos sexos o estrógeno é responsável pelas fases terminais da maturação epifisária, que leva à fusão da cartilagem de crescimento. É interessante a observação de que o IGF-1, em ausência do estrógeno, é incapaz de promover maturação da cartilagem de crescimento (ou seja, o IGF-1 isoladamente leva ao crescimento sem que ocorra aceleração indevida da maturação epifisária) (18). Os glicocorticóides levam à redução da secreção dos esteróides sexuais pelas supra-renais e pelas gônadas, sendo este outro mecanismo capaz de reduzir o ritmo de crescimento, principalmente durante a puberdade.
Os corticóides alteram a síntese do colágeno através da redução da atividade do IGF-1 e por efeito direto no metabolismo intracelular do seu precursor (pró-colágeno), inibindo a atividade das enzimas responsáveis pela hidroxilação e glicosilação do pró-colágeno. Os corticosteróides também podem aumentar o catabolismo e a excreção deste própeptídeo.
A prostaglandina E2 em baixas concentrações estimula a síntese de colágeno e de outras proteínas e promove incorporação de timidina no DNA, e em concentrações maiores a sua ação é inversa. Os lactentes tratados com prostaglandina E2 para manutenção da permeabilidade do ducto arterioso apresentam grande aumento da neoformação óssea a partir do periósteo. As prostaglandinas são produzidas pelas células ósseas, preenchendo os critérios de reguladores locais. Acredita-se que parte dos efeitos inibitórios dos glicocorticóides sobre o crescimento seja devida à inibição da síntese de prostaglandinas, através da redução da liberação do ácido araquidônico a partir dos fosfolípides (20).
Os diversos e significativos efeitos dos glicocorticóides em relação ao crescimento, até aqui apresentados, explicam porque crianças e adolescentes expostos por tempo prolongado a elevadas concentrações de glicocorticóides podem sofrer comprometimento do crescimento, muitas vezes de grave magnitude. Alguns autores acreditam que, como a inibição do crescimento pelos corticóides ocorre principalmente a nível tecidual, através dos seus efeitos sobre o metabolismo do colágeno e a bioatividade do IGF-1, é pouco provável que o tratamento concomitante com GH ou IGF-1 possa ser eficaz na atenuação do retardo de crescimento associado ao emprego destes esteróides (21,22). No entanto, Allen et al. (23) observaram que nos pacientes submetidos à corticoterapia em doses farmacológicas a administração concomitante de GH pode permitir o restabelecimento do ritmo normal de crescimento.
O prejuízo do crescimento produzido pelos glicocorticóides pode decorrer também de seus efeitos sobre o metabolismo mineral ósseo e a massa óssea. A perda da massa óssea induzida pelos corticóides pode vir acompanhada ou não de outros efeitos colaterais da corticoterapia e é mais grave nas regiões do esqueleto que têm elevado conteúdo de osso trabecular, como as vértebras e as costelas, sendo menos importante nos ossos longos. As crianças tratadas com corticóides estão predispostas à perda de massa óssea e ao retardo do crescimento esquelético devido à rápida renovação e remodelação ósseas que apresentam (24). Os glicocorticóides induzem perda óssea através da redução dos níveis circulantes dos esteróides gonadais e dos esteróides sexuais das supra-renais e da inibição da secreção hipofisária de gonadotrofinas em resposta ao estímulo pelo hormônio liberador do hormônio luteinizante (LHRH). Também diminuem a absorção intestinal de cálcio e aumentam sua excreção renal, induzindo quadro de hiperparatireoidismo secundário. Parece que a redução da absorção intestinal de cálcio se deve à interferência sobre os processos de transporte no intestino, através de inibição da síntese da proteína transportadora. Outros efeitos dos glicocorticóides, também implicados na perda óssea, são a inibição direta do número, função e tempo de vida dos osteoblastos e aumento do recrutamento e da atividade dos osteoclastos, além de diminuírem a atividade da calcitonina (peptídeo que inibe a atividade dos osteoclastos). Os glicocorticóides reduzem o número de precursores dos osteoblastos e a síntese de colágeno pelos mesmos. Os corticóides também inibem a síntese de outras proteínas ósseas (incluindo a osteocalcina) e de componentes da matriz óssea, como os mucopolissacárides e glicosaminoglicanas. O hiperparatireoidismo secundário é o mecanismo mais importante da osteopenia associada ao uso dos corticosteróides e a suplementação da vitamina D3 e do cálcio auxiliam na sua prevenção (25).
Nos pacientes portadores de HCSR os esteróides sexuais das supra-renais frequentemente se mantém em níveis superiores ao normal e podem compensar os efeitos dos glicocorticóides sobre o metabolismo ósteo-mineral, justificando o encontro, em alguns estudos, de densidade mineral óssea normal em crianças, adolescentes e adultos jovens portadores de HCSR (26-28).
Os diversos efeitos dos glicocorticóides sobre o crescimento tornam clara a necessidade de avaliação contínua do ritmo de crescimento e de maturação óssea das crianças sob corticoterapia. Nestas crianças, a maneira mais sensível de se detectar clinicamente a redução do crescimento consiste na avaliação do "score de desvio-padrão" (SDP) da velocidade de crescimento, que pode ser obtido a partir dos valores referenciais do estudo populacional realizado por Tanner et al. (29,30). A avaliação do SDP da velocidade de crescimento torna-se ainda mais informativa quando dispomos dos valores prévios à introdução da corticoterapia, os quais serão comparados aos valores obtidos durante o tratamento com o esteróide. Nas crianças portadoras de HCSR, onde o tratamento com glicocorticóides visa a reposição do cortisol endógeno, e não o uso de doses anti-inflamatórias (empregadas geralmente no tratamento de doenças não endócrinas, como asma, neoplasias, síndrome nefrótica, doenças reumatológicas e hematológicas), deve haver preocupação em se oferecer dose que se aproxime, dentro do possível, do ritmo de secreção fisiológica de cortisol em indivíduos normais. Em 1966, Kenny et al. (31) encontraram ritmo de produção de cortisol de 17,2±5,8 e 18,7±3,7mg/m2/dia nos cinco primeiros dias de vida em recém-nascidos de parto cesárea e normal, respectivamente, e valor de 11,8±2,5mg/m2/dia nos recém-nascidos com mais de cinco dias de vida e em todas as demais faixas etárias, inclusive a idade adulta. Anos mais tarde, Linder et al. (32) encontraram em 33 crianças e adolescentes normais com idade entre 8 e 17 anos valor de 9,5±2,5mg/dia (6,8±1,9mg/m2/dia), independentemente da idade, estádio puberal ou sexo. Portanto, o trabalho mais recente sugere que doses menores de glicocorticóide devem ser empregadas no tratamento da HCSR.
A reposição glicocorticóide na HCSR também tem por objetivo reduzir a hipersecreção do ACTH e dos andrógenos das supra-renais. O glicocorticóide administrado atua por mecanismo de "feedback" negativo sobre as células do núcleo paraventricular do hipotálamo e sobre a adeno-hipófise, reduzindo a secreção do hormônio liberador de corticotrofina (CRH) e do ACTH, respectivamente. Desse modo haverá menor estímulo do córtex supra-renal pelo ACTH, permitindo que a secreção androgênica adrenocortical seja substancialmente reduzida, ao mesmo tempo que as manifestações clínicas decorrentes do excesso de andrógenos das supra-renais, principalmente a virilização e a aceleração do crescimento e da maturação epifisária, vão aos poucos sendo controladas. A redução dos níveis plasmáticos dos esteróides sexuais das supra-renais é condição necessária para que as crianças com HCSR possam crescer normalmente e atingir altura final (AF) adequada. Os esteróides sexuais em excesso comprometem o crescimento e a AF destas crianças por: 1) promoverem avanço da idade óssea (IO), através da ação do estradiol sobre a placa de crescimento, o que implica em redução do tempo total de crescimento; 2) levarem à maturação precoce do eixo hipotalâmico-hipofisário-gonadal e portanto à puberdade precoce central, que também acelera a maturação óssea e reduz o tempo de crescimento. Por outro lado, a rápida diminuição dos níveis séricos destes esteróides após o início do tratamento leva à redução brusca do "feedback" negativo que os mesmos vinham exercendo sobre o hipotálamo e a hipófise e pode contribuir para a maturação precoce do eixo hipotálamo-hipófise-gônadas. A puberdade precoce associada somente à secreção adrenocortical excessiva deve ser abordada através da adequada reposição de glicocorticóide, que irá diminuir a secreção do ACTH e dos esteróides das supra-renais. No entanto, se houve evolução para puberdade precoce central, o prognóstico de crescimento pode ser melhorado através do seu reconhecimento a tempo e do uso de análogos do LHRH, eficazes no controle da puberdade precoce central associada à HCSR (33).
A intensidade da supressão da secreção do ACTH depende do tipo de glicocorticóide empregado, da sua dose e do esquema de uso. Em indivíduos normais a dexametasona promove maior supressão do ACTH quando administrada às 24 horas (34). A dexametasona utilizada em dose única de 0,25 a 0,75mg às 24 horas é capaz de suprimir adequadamente a secreção adrenocortical em pacientes portadores de HCSR (35). Hansen e Loriaux, ao estudarem o uso de diferentes glicocorticóides (cortisona, hidrocortisona, prednisona e dexametasona) em doses anti-inflamatórias equivalentes, no tratamento da HCSR, observaram que a dexametasona foi a mais eficaz e a cortisona a menos eficaz em suprimir o córtex supra-renal (36). Embora a dexametasona suprima eficientemente a hipersecreção androgênica na HCSR, devemos lembrar que é também o glicocorticóide mais potente em reduzir o crescimento (vide tabela 2), o que restringe sobremaneira a sua utilização no tratamento das crianças e adolescentes portadores de HCSR.
Em relação à prednisona, Linder et al. descreveram o caso de uma menina de 11 anos portadora de HCSR tratada com prednisona em dias alternados (14mg/m2 a cada dois dias), com o intuito de se obter melhor controle de um quadro de asma, na qual a secreção androgênica supra-renal foi adequadamente controlada, o que possibilitou crescimento e maturação óssea normais durante o estudo (37). Huseman et al. mostraram que a prednisona administrada em duas ou três vezes ao dia, na dose de 4,2 a 8,8mg/m2/dia, permitiu controle adequado da secreção supra-renal em todos os pacientes avaliados, o mesmo não ocorrendo quando administrada em dose única à noite (38).
Quanto ao acetato de hidrocortisona, Winterer et al. acreditam que o seu uso em dose única pode ser tão eficaz quanto o esquema tradicional com três doses diárias no tratamento da HCSR (39). Por outro lado, Keenan et al. mostraram que a hidrocortisona em dose única noturna não foi eficaz no controle da secreção androgênica supra-renal na HCSR (40).
Além da secreção deficiente do cortisol, outros mecanismos são também capazes de manter a hipersecreção do ACTH na HCSR. Estes mecanismos têm em comum a sua origem, já que são desencadeados a partir da perda de sal. Em 1977 Rösler et al. (41) demonstraram a associação entre o grau de controle da perda de sal na HCSR, representado pela APR, e a hipersecreção de ACTH e dos andrógenos supra-renais, sugerindo inter-relação entre o sistema renina-angiotensina-aldosterona e a secreção de ACTH. Os autores acreditam que a angiotensina II pode estimular a secreção do ACTH agindo a nível hipotalâmico, provavelmente por aumentar a secreção do CRH (41). Em 1979 Horner et al. (42) observaram que a reinstituição da terapêutica mineralocorticóide em pacientes que ficaram de 3,5 a 15,5 anos sem receber a medicação levou à redução nos níveis da APR e dos esteróides supra-renais e permitiu em alguns pacientes que a dose de hidrocortisona fosse reduzida. Segundo os autores, os resultados foram devidos ao efeito direto da angiotensina II sobre a esteroidogênese supra-renal (42). Além destes mecanismos, a angiotensina II tem efeito também sobre a neuro-hipófise, onde estimula a síntese da vasopressina, a qual é capaz de aumentar a secreção do ACTH independentemente do CRH. Estes mecanismos, que mantêm elevadas as secreções do ACTH e dos andrógenos supra-renais, apesar da reposição glicocorticóide, demonstram a importância do adequado controle da perda salina na HCSR, com a finalidade não apenas de se evitar as crises agudas de perda de sal, mas também de se conseguir melhor controle da secreção adrenocortical. Justifica-se, dessa forma, a necessidade de reposição mineralocorticóide no tratamento da HCSR sempre que se detectar APR aumentada, mesmo sem haver evidência clínica de perda de sal, com o intuito de se favorecer o crescimento através tanto da utilização de doses menores de glicocorticóide quanto da redução dos efeitos dos andrógenos supra-renais sobre o crescimento e a maturação epifisária.
Entre os corticóides disponíveis, a cortisona e a hidrocortisona são consideradas as drogas de escolha para o tratamento da HCSR nas crianças porque: 1) o cortisol é o principal glicocorticóide secretado fisiologicamente pelo córtex supra-renal; 2) o cortisol contribui para a retenção de sal, enquanto que os glicocorticóides mais potentes têm pouco ou nenhum efeito mineralocorticóide (tabela 2); 3) os glicocorticóides mais potentes suprimem o crescimento mais intensamente (tabela 2).
Diante das características do tratamento da HCSR expostas anteriormente, e considerando-se que a meia-vida biológica da hidrocortisona (ou do acetato de cortisona) é de 8 a 12 horas, acreditamos que a melhor opção terapêutica para a HCSR consiste no uso da hidrocortisona (ou do acetato de cortisona), administrados três vezes ao dia, com a maior dose (50% da dose diária) pela manhã e as demais doses igualmente divididas à tarde e à noite. Em geral, doses de hidrocortisona entre 8 e 12 mg/m2/dia são suficientes para o adequado controle da doença na maior parte das crianças. O acetato de cortisona deve ser empregado em dose maior (10 a 15 mg/m2/dia), uma vez que é 20% menos potente do que a hidrocortisona. No entanto, é importante salientar que a dose do glicocorticóide deve ser sempre individualizada, evitando-se assim as conseqüências tanto do sub quanto do supertratamento. A dose diária deve ser dobrada diante de situações de "stress", como febre, extração dentária única e trauma leve, retornando-se à dose de rotina assim que o organismo voltar às condições normais. Indicamos o uso da hidrocortisona por via intramuscular (50mg) duas horas antes de cirurgias sob anestesia local ou de extrações dentárias múltiplas, ou em casos de vômitos frequentes. Nas condições clínicas associadas à elevada carga de "stress", como infecções sistêmicas e traumas graves, a hidrocortisona deve ser utilizada por via intramuscular ou endovenosa, na dose de 50-100 mg a cada seis horas. Nos casos de cirurgia sob anestesia geral a hidrocortisona é administrada por via intramuscular (50mg) duas horas antes da cirurgia, sendo infundida continuamente durante o intra-operatório. Nas primeiras 48 horas de pós-operatório usamos o triplo da dose de rotina por via intramuscular ou endovenosa, dividida em três a quatro vezes ao dia. A partir do terceiro dia a dose é reduzida para o dobro da dose habitual, e então para a dose habitual, desde que o organismo não venha a sofrer novas situações de "stress" (43).
Em alguns pacientes, com a finalidade de se facilitar a aderência ao tratamento, o acetato de cortisona (ou de hidrocortisona) pode ser administrado duas vezes ao dia, oferecendo-se a maior dose pela manhã. Apesar do maior potencial da prednisona em suprimir o crescimento, discutido anteriormente, acreditamos que possa ser empregada alternativamente quando o tratamento com acetato de cortisona (ou de hidrocortisona) não for capaz de reduzir adequadamente a secreção adrenocortical, seja por falta de aderência ao tratamento, seja pelas características intrínsecas do paciente e da doença. A meia-vida biológica da prednisona varia entre 12 e 36 horas, o que permite que seja utilizada em dose única pela manhã. A prednisona é quatro vezes mais potente do que a hidrocortisona, devendo ser empregada na dose de 2,0 a 3,0mg/m2/dia. As desvantagens do uso da prednisona são a sua maior capacidade em suprimir o crescimento (cinco vezes maior do que a hidrocortisona) e sua menor potência retentora de sal (20% menor, em relação à hidrocortisona). Por outro lado, especificamente na HCSR, o maior potencial da prednisona em suprimir o crescimento pode ser suplantado, tanto nas crianças pré-púberes quanto nas púberes, pela sua maior capacidade em reduzir a hipersecreção androgênica pelo córtex supra-renal, prevenindo assim a aceleração da maturação epifisária e melhorando o potencial de crescimento (44).
Embora na HCSR a intenção é que sejam utilizadas doses fisiológicas do corticóide, não sabemos com certeza qual a dose fisiológica quando a medicação é empregada por via oral. Além disso, a necessidade fisiológica pode não ser a mesma para todos, e algumas crianças podem estar recebendo doses maiores do que realmente necessitam. A hipercortisolemia que se segue à administração do corticosteróide pode ser outro fator adverso ao crescimento destas crianças (45). Somam-se ainda as dificuldades próprias que uma doença crônica representa na faixa etária pediátrica, que leva muitas vezes à não aderência ao tratamento ideal, que exige que o corticóide seja administrado diáriamente em duas ou três vezes.
A necessidade de tratamento crônico com corticóides e o aumento da secreção adrenocortical dos esteróides sexuais fazem com que as crianças portadoras de HCSR estejam especialmente sob risco de crescimento desordenado, do qual resulta baixa estatura em grande porcentagem delas. A figura 2 demonstra como se relacionam os diversos fatores que afetam o crescimento e a altura final destes pacientes.
A preocupação com o crescimento das crianças portadoras de HCSR levou à realização de diversos estudos abordando o assunto. Desses, possivelmente o pioneiro foi o trabalho de Blizzard e Wilkins (46), que descreveram o crescimento de quatro crianças tratadas com cortisona. Os autores encontraram crescimento e maturação epifisária normais quando o tratamento foi apropriado e precoce, enquanto que doses excessivas de cortisona levaram à redução significativa do ritmo de crescimento e ao aparecimento de sinais clínicos da síndrome de Cushing. Por outro lado, o tratamento insuficiente resultou em aceleração da maturação esquelética (46).
Alguns autores estudaram o crescimento destas crianças durante os primeiros anos de vida. Sperling et al. e Rappaport et al. encontraram comprometimento da altura aos dois anos de idade (14,47). As crianças avaliadas por Gussinyé et al. tiveram crescimento diminuído durante o primeiro ano de vida (48). Nestes estudos o crescimento inadequado nos primeiros anos de vida foi atribuído ao emprego de doses elevadas de hidrocortisona ou de acetato de cortisona. Jääskeläinen e Voutilainen também responsabilizaram as altas doses de hidrocortisona pela redução do crescimento durante o primeiro ano de vida nos lactentes diagnosticados precocemente (49). Segundo Rappaport et al. parte das crianças tratadas com as maiores doses de hidrocortisona durante o primeiro ano de vida mantiveram crescimento deficiente durante a infância e atraso da IO, mesmo após a redução da dose do glicocorticóide (50). Em 1963 Tanner já havia descrito que o déficit de crescimento associado a níveis elevados de glicocorticóides circulantes é parcialmente corrigido com a retirada ou redução do corticosteróide, mas raramente o potencial genético de crescimento é restabelecido, sugerindo "catch-up" incompleto nas crianças submetidas ao excesso de glicocorticóides (51). Gasparini et al. (52) acreditam que as crianças que estudaram tiveram crescimento preservado até os 7 anos por terem sido tratadas com doses menores (variando de 13,5 a 17,8 mg/m2/dia) de acetato de cortisona durante o primeiro ano de vida. Atualmente, procurando-se evitar que doses elevadas de glicocorticóides sejam utilizadas, principalmente durante os primeiros anos de vida, recomenda-se que a dose diária do corticóide nas crianças portadoras de HCSR não seja aumentada durante infecções de vias aéreas superiores ou após imunizações, a menos que evoluam com febre (10).
A AF atingida pelos pacientes portadores de HCSR foi avaliada em diversos estudos. Bergstrand (53) observou que em alguns pacientes nunca tratados, ou tratados somente após os cinco anos de idade ou inadequadamente tratados, a AF situou-se dentro dos limites normais. Em alguns estudos a AF foi prejudicada somente nos pacientes tratados após o primeiro ano de vida ou naqueles não tratados até o término do crescimento (54-56). Knorr e de Lienau (57) encontraram melhor AF nos pacientes tratados até os dois anos com doses menores de hidrocortisona. No estudo de Jääskeläinen e Voutilainen (49) a AF foi prejudicada nas meninas diagnosticadas com menos de um ano e nos meninos diagnosticados após um ano. A pior AF observada nas pacientes com menores níveis plasmáticos de 17-OHP foi atribuída por Helleday et al. (58) ao super-tratamento e à redução dos andrógenos circulantes. Yu e Grant (59) atribuíram a pior AF das pacientes portadoras da FVS, em comparação às portadoras da FPS, à diferença da altura parental média entre os dois grupos. Os pacientes estudados por Clayton (60) tiveram AF maior ou igual à altura parental média. No entanto, de forma geral tem predominado na literatura a descrição de comprometimento do crescimento dos pacientes portadores de HCSR, com prejuízo da sua AF em relação à altura parental ou à média de altura da população referencial (48,61-68). Eugster et al. (69) realizaram meta-análise envolvendo 18 estudos a respeito do crescimento em pacientes com deficiência da 21-hidroxilase, encontrando média de SDP de AF de ¹1,37. Os autores acreditam que o diagnóstico precoce e a aderência ao tratamento são capazes de fazer com que a AF destes pacientes não se distancie mais do que um DP da altura-alvo (69).
Estudo realizado na Unidade de Endocrinologia do Instituto da Criança do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, a respeito do crescimento de pacientes com a forma clássica de HCSR por deficiência da 21-hidroxilase, revelou resultados coincidentes com a maior parte dos estudos publicados a esse respeito. Foram estudados retrospectivamente 20 pacientes (14 meninas e 6 meninos) que atingiram a altura final. Durante os anos pré-puberais a média de utilização do acetato de cortisona foi de 61% e da prednisona de 39%. Durante a puberdade a média de utilização da prednisona foi de 72% e do acetato de cortisona de 25% (neste período duas pacientes receberam dexametasona por 1,3 a 2 anos e uma delas recebeu metil-prednisolona de depósito por um ano). Geralmente o glicocorticóide foi administrado em duas a três vezes ao dia, utilizando-se a maior dose pela manhã ou dose igual pela manhã e à tarde e dose menor à noite. A dose média de hidrocortisona, expressa em mg/m2/dia, foi de 19,61 no período pré-puberal e de 18,73 durante a puberdade. Com a exceção de um paciente, todos requereram reposição mineralocorticóide, desde o início do tratamento ou durante o período de seguimento, tendo sido utilizada a 9a-flúorhidrocortisona na dose de 25 a 200mcg ao dia, em dose única pela manhã. Encontramos prejuízo da AF em 19 dos 20 pacientes avaliados, nos quais o SDP da AF foi inferior ao SDP da altura-alvo. A média da diferença entre o SDP da altura-alvo e o SDP da AF foi 1,50. Esta diferença significou perda média de 9,36cm na AF, em comparação à altura-alvo. Nestes pacientes o SDP da altura aos dois anos não diferiu significativamente do SDP da altura-alvo, sugerindo preservação do crescimento durante os dois primeiros anos de vida (44).
Em resumo, a obtenção de ritmo de crescimento normal, capaz de permitir que as crianças portadoras da forma clássica de HCSR por D21OH desenvolvam todo o seu potencial de crescimento, e dessa forma atinjam AF adequada em relação à sua população, e principalmente em relação à sua altura-alvo, continua a representar um verdadeiro desafio no campo da endocrinologia pediátrica. Acreditamos que a preocupação em se utilizar doses menores de glicocorticóide durante todo o período de crescimento, e com especial atenção para os dois primeiros anos de vida, deve conduzir a resultados melhores em relação ao crescimento destas crianças. Igualmente importante é a monitorização cuidadosa e permanente da velocidade de crescimento, do ritmo de maturação epifisária e da secreção adrenocortical, procurando-se através da reposição glicocorticóide e mineralocorticóide atingir-se para cada paciente o ponto mais adequado de equilíbrio entre todos estes fatores. O adequado reconhecimento e tratamento da puberdade precoce central também tem importante papel no desafio de se garantir AF próxima do normal a estas crianças.
Endereço para correspondência:
Hamilton Cabral de Menezes Filho
Rua Joinville, 637 - Apto 701
04008-011 São Paulo, SP
e-mail: hamiltoncm@uol.com.br
- 1. Stocco DM, Clark BJ. The role of the steroidogenic acute regulatory protein in steroidogenesis. Steroids 1997;62:29-36.
- 2. Donohoue PA, Parker K, Migeon CJ. Congenital adrenal hyperplasia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. 7th ed. McGraw-Hill, 1995:2929-66.
- 3. Pang S, Wallace MA, Hofman L, Thuline HC, Dorche C, Lyon ICT, et al. Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 1988;81:866-74.
- 4. Hirschfeld AJ, Fleshman JK. An unusually high incidence of salt-losing congenital adrenal hyperplasia in the alaskan eskimo. J Pediatr 1969;75:492-4.
- 5. Migeon CJ. Adrenal androgens in man. Am J Med 1972;53:606-26.
- 6. Rivarola MA, Saez JM, Migeon CJ. Studies of androgens in patients with congenital adrenal hyperplasia. J Clin Endocrinol 1967;27:624-30.
- 7. Wilkins L, Lewis RA, Klein R, Rosemberg E. The suppression of androgen secretion by cortisone in a case of congenital adrenal hyperplasia. Bull Johns Hopkins Hosp 1950;86:249-52.
- 8. Bartter FC, Albright F, Forbes AP, Leaf A, Dempsey E, Carroll E. The effects of adrenocorticotropic hormone and cortisone in the adrenogenital syndrome associated with congenital adrenal hyperplasia: an attempt to explain and correct its disordered hormonal pattern. J Clin Invest 1951;30:237-51.
- 9. Haynes RC Jr. Adrenocorticotropic hormone; adrenocortical steroids and their synthetic analogs; inhibitors of the synthesis and actions of adrenocortical hormones. In: Gilman AG, Rall TW, Nies AS, Taylor P, eds. Goodman and Gilmanăs The Pharmacological Basis of Therapeutics. 8th ed. New York:Pergamon Press, 1990:1431-62.
- 10. Miller WL. Clinical review 54. Genetics, diagnosis and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab 1994;78:241-6.
- 11. Blodgett FM, Burgin L, Iezzoni D, Gribetz D, Talbot NB. Effects of prolonged cortisone therapy on the statural growth, skeletal maturation and metabolic status of children. N Engl J Med 1956;254:636-41.
- 12. Van Metre TE JR, Niermann WA, Rosen LJ. A comparison of the growth supressive effect of cortisone, prednisone, and other adrenal cortical hormones. J Allergy 1960;31:531-42.
- 13. Tönshoff B, Mehls O. Interaction between glucocorticoids and the somatotropic axis. Acta Paediatr Suppl 1996;417:72-5.
- 14. Sperling MA, Kenny FM, Schutt-Aine JC, Drash AL. Linear growth and growth hormonal responsiveness in treated congenital adrenal hyperplasia. Amer J Dis Child 1971;122:408-13.
- 15. Vazquez AM, Schutt-Aine JC, Kenny FM. Effect of cortisone therapy on the diurnal pattern of growth hormone secretion in congenital adrenal hyperplasia. J Pediatr 1972;80:433-40.
- 16. Rappaport R, Bouthreuil E, Marti-Henneberg C, Basmaciogullari A. Linear growth rate, bone maturation and growth hormone secretion in prepubertal children with congenital adrenal hyperplasia. Acta Paediatr Scand 1973;62:513-9.
- 17. Silva IN, Kater CE, Cunha CF, Viana MB. Randomised controlled trial of growth effect of hydrocortisone in congenital adrenal hyperplasia. Arch Dis Child 1997;77:214-8.
- 18. Van Wyk JJ, Smith EP. Insulin-like growth factors and skeletal growth: possibilities for therapeutic interventions. J Clin Endocrinol Metab 1999;84:4349-54.
- 19. Unterman TG, Phillips LS. Glucocorticoid effects on somatomedins and somatomedin inhibitors. J Clin Endocrinol Metab 1985;61:618-26.
- 20. Raisz LG, Kream BE. Regulation of bone formation (second of two parts). N Engl J Med 1983;309:83-9.
- 21. Hyams JS, Carey DE. Corticosteroids and growth. J Pediatr 1988;113:249-54.
- 22. Hughes IA. Steroids and growth. Br Med J 1987;295:683-4.
- 23. Allen DB, Julius JR, Breen TJ, Attie KM. Treatment of glucocorticoid-induced growth suppression with growth hormone. J Clin Endocrinol Metab 1998;83:2824-9.
- 24. Peck W, Gennari C, Raisz L, Meunier P, Ritz E, Krane S, et al. Corticosteroids and bone. Calcif Tissue Int 1984;36:4-7.
- 25. Lane NE, Lukert B. The science and therapy of glucocorticoid-induced bone loss. Endocrinol Metab Clin North Am 1998;27:465-83.
- 26. Cameron FJ, Kaymakci B, Byrt EA, Ebeling PR, Warne GL, Wark JD. Bone mineral density and body composition in congenital adrenal hyperplasia. J Clin Endocrinol Metab 1995;80:2238-43.
- 27. Girgis R, Winter JSD. The effects of glucocorticoid replacement therapy on growth, bone mineral density, and bone turnover markers in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab 1997;82:3926-9.
- 28. Silva IN, Kater CE, Cunha CF, Viana MB. Análise da massa óssea em crianças com hiperplasia adrenal congęnita em uso de glicocorticóides. Arq Bras Endcrinol Metab 1997;41:24-30.
- 29. Tanner JM, Whitehouse RH, Takaishi M. Standards from birth to maturity for height, weight, height velocity, and weight velocity: british children, 1965. Part I. Arch Dis Child 1966;41:454-71.
- 30. Tanner JM, Whitehouse RH, Takaishi M. Standards from birth to maturity for height, weight, height velocity, and weight velocity: british children, 1965. Part II. Arch Dis Child 1966;41:613-35.
- 31. Kenny FM, Preeyasombat C, Migeon CJ. Cortisol production rate II. Normal infants, children, and adults. Pediatrics 1966;v.37:34-42.
- 32. Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. J Pediatr 1990;117:892-6.
- 33. Pescovitz OH, Comite F, Cassorla F, Dwyer AJ, Poth MA, Sperling MA et al. True precocius puberty complicating congenital adrenal hyperplasia: treatment with a luteinizing hormone-releasing hormone analog. J Clin Endocrinol Metab 1984;58:857-61.
- 34. Nichols T, Nugent CA, Tyler FH. Diurnal variation in suppression of adrenal function by glucocorticoids. J Clin Endocrinol 1965;25:343-9.
- 35. Hayek A, Crawford JD, Bode HH. Single dose dexamethasone in treatment of congenital adrenocortical hyperplasia. Metabolism 1971;20:897-901.
- 36. Hansen JW, Loriaux DL. Variable efficacy of glucocorticoids in congenital adrenal hyperplasia. Pediatrics 1976;57:942-7.
- 37. Linder BL, Feuillan P, Chrousos GP. Alternate day prednisone therapy in congenital adrenal hyperplasia: adrenal androgen suppression and normal growth. J Clin Endocrinol Metab 1989;69:191-5.
- 38. Huseman CA, Varma MM, Blizzard RM, Johanson A. Treatment of congenital virilizing adrenal hyperplasia patients with single and multiple daily doses of prednisone. J Pediatr 1977;90:538-42.
- 39. Winterer J, Chrousos GP, Loriaux DL, Cutler GB. Effect of hydrocortisone dose schedule on adrenal steroid secretion in congenital adrenal hyperplasia. J Pediatr 1985;106:137-42.
- 40. Keenan BS, Eberle AE, Tsu-Hui L, Clayton GW. Inappropriate adrenal androgen secretion with once-a-day corticosteroid therapy for congenital adrenal hyperplasia. J Pediatr 1990;116:133-6.
- 41. R¸sler A, Levine LS, Schneider B, Novogroder M, New MI. The interrelationship of sodium balance, plasma renin activity and ACTH in congenital adrenal hyperplasia. J Clin Endocrinol Metab 1977;45: 500-12.
- 42. Horner JM, Hintz RL, Luetscher JA. The role of renin and angiotensin in salt-losing, 21-hydroxylase-deficient congenital adrenal hyperplasia. J Clin Endocrinol Metab 1979;48:776-83.
- 43. Kuperman H. Corticoterapia. In: Setian N, ed. Endocrinologia Pediátrica. 1a ed. Săo Paulo:Sarvier, 1989:324-31.
- 44. Menezes Filho HC. Estudo do crescimento e da altura final dos pacientes portadores da forma clássica de hiperplasia congęnita das supra-renais por deficięncia da 21-hidroxilase. Săo paulo, 1999. 167p. Dissertaăo (Mestrado) - Faculdade de Medicina, Universidade de Săo Paulo.
- 45. Frisch H, Parth K, Schober E, Swoboda W. Circadian patterns of plasma cortisol, 17-hydroxyprogesterone, and testosterone in congenital adrenal hyperplasia. Arch Dis Child 1981;56:208-13.
- 46. Blizzard RM, Wilkins L. Present concepts of steroid therapy in virilizing adrenal hyperplasia. Arch Intern Med 1957;100:729-38.
- 47. Rappaport R, Cornu G, Royer P. Statural growth in congenital adrenal hyperplasia treated with hydrocortisone. J Pediatr 1968;73:760-6.
- 48. Gussinyé M, Potau N, Vicens-Calvet E, Albisu MA, Yeste D, Ibáńez L et al. Talla adulta, patrón de crecimiento y desarrollo puberal en pacientes con hiperplasia suprarrenal congęnita, forma perdedora de sal. Med Clin (Barc) 1997;108:87-90.
- 49. Jääskeläinen J, Voutilainen R. Growth of patients with 21-hydroxylase deficiency: an analysis of the factors influencing adult height. Pediatr Res 1997;41:30-3.
- 50. Rappaport R, Bouthreuil E, Marti-Henneberg C, Basmaciogullari A. Linear growth rate, bone maturation and growth hormone secretion in prepubertal children with congenital adrenal hyperplasia. Acta Paediatr Scand 1973;62:513-9.
- 51. Tanner JM. Regulation of growth in size in mammals. Nature 1963;199:845-50.
- 52. Gasparini N, Di Maio S, Salerno M, Argenziano A, Franzese A, Tenore A. Growth pattern during the first 36 months of life in congenital adrenal hyperplasia (21-hydroxylase deficiency). Horm Res 1997;47:17-22.
- 53. Bergstrand CG. Growth in congenital adrenal hyperplasia. Acta Paediatr Scand 1966;55:463-72.
- 54. Brook CGD, Zachmann M, Prader A, Mürset G. Experience with long-term therapy in congenital adrenal hyperplasia. J Pediatr 1974;85:12-9.
- 55. Klingensmith JG, Garcia SC, Jones Jr HW, Migeon CJ, Blizzard RM. Glucocorticoid treatment of girls with congenital adrenal hyperplasia: effects on height, sexual maturation, and fertility. J Pediatr 1977;90:996-1004.
- 56. Kirkland RT, Keenan BS, Holcombe JH, Kirkland JL, Clayton GW. The effect of therapy on mature height in congenital adrenal hyperplasia. J Clin Endocrinol Metab 1978;47:1320-4.
- 57. Knorr D, de Lienau SGCH. Persistent obesity and short final height after corticoid overtreatment for congenital adrenal hyperplasia (CAH) in infancy. Acta Paediatr Jpn 1988;30:89-92. Supplement.
- 58. Helleday J, Siwers B, Ritz┌n EM, Carlstr¸m K. Subnormal androgen and elevated progesterone levels in women treated for congenital virilizing 21-hydroxylase deficiency. J Clin Endocrinol Metab 1993;76: 933-6.
- 59. Yu ACM, Grant DB. Adult height in women with early-treated congenital adrenal hyperplasia (21-hydroxylase type): relation to body mass index in earlier childhood. Acta Paediatr 1995;84:899-903.
- 60. Clayton GW. Patterns of growth from birth to maturity in infants and children with congenital adrenal hyperplasia. Acta Endocrinol (Copenh) 1986;279(suppl):295-304.
- 61. Di Martino-Nardi J, Stoner E, OăConnel A, New MI. The effect of treatment on final height in classical congenital adrenal hyperplasia (CAH). Acta Endocrinol (Copenh) 1986;279(suppl):305-14.
- 62. Urban MD, Lee PA, Migeon CJ. Adult height and fertility in men with congenital virilizing adrenal hyperplasia. N Engl J Med 1978;299:1392-6.
- 63. New MI, Gertner JM, Chir B, Speiser PW, del Balzo P. Growth and final height in classical and nonclassical 21-hydroxylase deficiency. Acta Paediatr Jpn 1988;30:79-88. Supplement.
- 64. Tanae A, Hibi I. Unresolved problems in the treatment of congenital adrenal hyperplasia. Acta Paediatr Jpn 1988;30:93-8. Supplement.
- 65. Young MC, Ribeiro J, Hughes IA. Growth and body proportions in congenital adrenal hyperplasia. Arch Dis Child 1989;64:1554-8.
- 66. Thilén A, Larsson A. Congenital adrenal hyperplasia in Sweden 1969-1986. Prevalence, symptoms and age at diagnosis. Acta Paediatr Scand;1990;79:168-75.
- 67. Jocham A, Kuhnle U, Knorr D, Schwarz HP. Final height and long-term follow-up of 108 adult patients with congenital adrenal hyperplasia. Pediatr Res 1993;33:15. Supplement 1.
- 68. Kandemir N, Yordam N. Congenital adrenal hyperplasia in Turkey: a review of 273 patients. Acta Paediatr 1997;86:22-5.
- 69. Eugster EA, DiMeglio LA, Wright JC, Freidenberg GR, Seshadri R, Pescovitz OH. Height outcome in congenital adrenal hyperplasia caused by 21-hydroxylase deficiency: a meta-analysis. J Pediatr 2001;138:26-32.
Datas de Publicação
-
Publicação nesta coleção
14 Mar 2002 -
Data do Fascículo
Dez 2001
Histórico
-
Recebido
03 Jan 2000 -
Revisado
16 Out 2001 -
Aceito
19 Out 2001