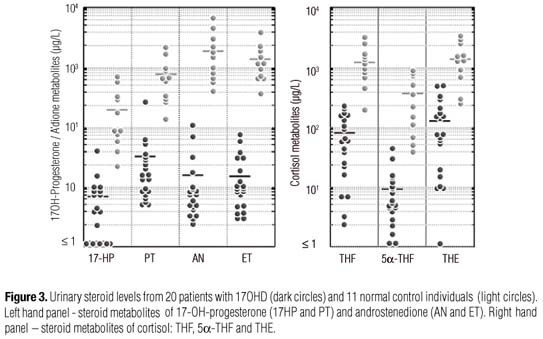
OBJECTIVES: (1) Characterize serum (S) and urinary (U) steroid metabolites in complete CYP17 deficiency (cCYP17D); (2) analyze the relative 17α-hydroxylase (17OH) and 17,20-lyase (17,20L) activities in vivo; and (3) comparedata from the two most prevalent mutations in Brazil. SUBJECTS AND METHODS: 20 genotyped cCYP17D patients from a previously reported cohort were homozygous for W406R or R362C; 11 controls were CYP17 wild types (WT). WT and cCYP17D patients had S and U samples drawn to measure: cortisol (F), corticosterone (B), deoxycorticosterone (DOC), 18OH-B, 18OH-DOC, and 17OHP; and tetrahydro (TH)-B, THA, THDOC, THF+5α-THF, TH-cortisone, androsterone, etiocholanolone, 5-pregnenediol, 17OH-pregnenolone and pregnanetriol. RESULTS: Compared to WT, cCYP17D patients had marked elevations of B, DOC, 18OH-B and 18OH-DOC, whereas 17OHP, F and adrenal androgens (AA) were reduced; U steroids parallel S findings. Metabolite ratios revealed that both 17OH and 17,20L activities were impaired in cCYP17D. There were nodifferences between W406R andR362C mutations. CONCLUSIONS: cCYP17D patients show parallel overproduction/overexcretion of 17-deoxysteroids, and marked reduction of F and AA. In addition to 17OH, 17,20-L activity was also impaired in cCYP17D. W406 and R362C mutations disclose similar Sand U patterns.
Congenital adrenal hyperplasia; CYP17; 17-hydroxylase deficiency; 17,20-lyase deficiency; urinary steroid metabolome; corticosterone