Abstracts
The electrochemical behavior of the anti-thalassemia and anti-HIV replication drug, deferiprone, was investigated by cyclic voltammetry (CV) at a platinum electrode. In an acetate buffer solution, pH = 4.0, two irreversible anodic peaks for deferiprone, with E(0)1 = 875 mV and E(0)2 = 1235 mV (vs. Ag/AgCl) appeared at a potential sweep rate of 50 mV s-1. Cyclic voltammetric study indicated that the oxidation process is irreversible and diffusion-controlled. The diffusion and the electron transfer coefficients of deferiprone were found to be 3.50 × 10-6 cm² s-1 and 0.57, respectively. A sensitive, simple and time-saving differential pulse voltammetric procedure was developed. Using our proposed method, deferiprone can be determined with a detection limit of 1.43 × 10-5 mol L-1. The applicability of the method to direct assays of spiked human serum and urine fluids, and to commercial tablets, is described.
deferiprone; anti-thalassemia drug; platinum electrode; iron chelator; HIV-1 replication inhibitor
O comportamento eletroquímico da deferiprona, droga anti-talassêmica e anti-replicação do HIV foi investigado, por voltametria cíclica (VC), utilizando um eletrodo de platina. Observaram-se dois picos anódicos irreversíveis para a deferiprona, em tampão acetato (pH 4,0) com E(0)1 = 875 mV e E(0)2 = 1235 mV (vs. Ag/AgCl) em velocidade de varredura de 50 mV s-1. Os estudos de voltametria cíclica indicaram que o processo de oxidação é irreversível e controlado por difusão. Foram encontrados, para a deferiprona, coeficientes de difusão e de transferência de elétrons de 3,50 × 10-6 cm² s-1 e 0,57, respectivamente. Foi desenvolvido um procedimento sensível, simples e rápido, utilizando voltametria de pulso diferencial. Utilizando este método, a deferiprona pode ser determinada com um limite de detecção de 1,43 × 10-5 mol L-1. É descrita a aplicabilidade do método em ensaios diretos em soro humano enriquecido, fluidos urinários e em comprimidos comerciais.
ARTICLE
Electrochemistry of deferiprone as an orally active iron chelator and HIV-1 replication inhibitor and its determination
H. YadegariI; A. JabbariI,* * e-mail: jabbari@kntu.ac.ir ; H. HeliII; A. A. Moosavi-MovahediII; S. MajdiI
IDepartment of Chemistry, Faculty of Science, K. N. Toosi University of Technology, P.O. Box 16315-1618, Tehran, Iran
IIInstitute of Biochemistry and Biophysics, University of Tehran, Tehran, Iran
ABSTRACT
The electrochemical behavior of the anti-thalassemia and anti-HIV replication drug, deferiprone, was investigated by cyclic voltammetry (CV) at a platinum electrode. In an acetate buffer solution, pH = 4.0, two irreversible anodic peaks for deferiprone, with E01 = 875 mV and E02 = 1235 mV (vs. Ag/AgCl) appeared at a potential sweep rate of 50 mV s-1. Cyclic voltammetric study indicated that the oxidation process is irreversible and diffusion-controlled. The diffusion and the electron transfer coefficients of deferiprone were found to be 3.50 × 10-6 cm2 s-1 and 0.57, respectively. A sensitive, simple and time-saving differential pulse voltammetric procedure was developed. Using our proposed method, deferiprone can be determined with a detection limit of 1.43 × 10-5 mol L-1. The applicability of the method to direct assays of spiked human serum and urine fluids, and to commercial tablets, is described.
Keywords: deferiprone, anti-thalassemia drug, platinum electrode, iron chelator, HIV-1 replication inhibitor
RESUMO
O comportamento eletroquímico da deferiprona, droga anti-talassêmica e anti-replicação do HIV foi investigado, por voltametria cíclica (VC), utilizando um eletrodo de platina. Observaram-se dois picos anódicos irreversíveis para a deferiprona, em tampão acetato (pH 4,0) com E01 = 875 mV e E02 = 1235 mV (vs. Ag/AgCl) em velocidade de varredura de 50 mV s-1. Os estudos de voltametria cíclica indicaram que o processo de oxidação é irreversível e controlado por difusão. Foram encontrados, para a deferiprona, coeficientes de difusão e de transferência de elétrons de 3,50 × 10-6 cm2 s-1 e 0,57, respectivamente. Foi desenvolvido um procedimento sensível, simples e rápido, utilizando voltametria de pulso diferencial. Utilizando este método, a deferiprona pode ser determinada com um limite de detecção de 1,43 × 10-5 mol L-1. É descrita a aplicabilidade do método em ensaios diretos em soro humano enriquecido, fluidos urinários e em comprimidos comerciais.
Introduction
Deferiprone (1,2-dimethyl-3-hydroxypyridin-4-one, Scheme 1) is the first oral iron chelator to be used clinically, mainly in thalassemia patients.1,2 Deferiprone belongs to the family of the alpha-ketohydroxypyridines, a relatively new class of chelating agents, some of which are naturally occurring. These chelators have a high affinity for binding iron, and are able to remove it from proteins that are transporting and storing it in the body, largely sparing other biologically important metals. They are stable in conditions that exist in the human digestive system and are readily absorbed. Deferiprone can remove excess iron from various parts of the body of iron-loaded patients, including liver and, particularly, heart.1 This drug is also used worldwide to treat cancer, leukemia, in hemodialysis and other diseases. It is worthy noting that the drug deferiprone may be used in the detoxification of other metals, such as aluminum in hemodialysis patients, plutonium used in the nuclear industry and uranium used by the military.3-5
Iron is also involved in replication of the human immunodeficiency virus type 1 (HIV-1).6 Iron chelators such as deferiprone inhibit replication of HIV-1 through several routes. Deferiprone can inhibit nuclear factor-κB activation and subsequent replication of human immunodeficiency virus type 1.7 Deferiprone can also render iron-dependent ribonucleotide reductase inactive, thereby inhibiting DNA synthesis and therefore HIV replication.8
Drug analysis has an extensive impact on public health. In order to minimize the risk of side effects,9,10 to optimize further dosing intervals11 and to monitor the L1 therapy,12 methods for the accurate measurement of the drug are necessary. It was previously found that the chromatography of the 3-hydroxypyridin-4-ones is difficult on normal octadecylsilica (ODS) columns as the analytes are often characterized by broad asymmetrical and sometimes multiple peaks.13 Goddard and Kontoghiorghes14 described a HPLC based method to assay deferiprone in serum and urine. Dresow et al.15 also describe a HPLC method for the determination of deferiprone in serum and urine samples and a HPLC based assay to determine the iron excretion (due to deferiprone therapy) by measuring urinary Fe-(deferiprone)3 complex concentration. Polarography and voltammetry can be considered as convenient alternatives to routinely employed analytical methods, in that they present the great advantage of permitting a direct, simple and rapid determination requiring a minimum volume of sample. Electrochemical techniques have also been used for the determination of a wide range of drug compounds. They have the advantage of not requiring, in most instances, derivatization, and they are less sensitive to matrix effects than other analytical techniques. Additionally, electrochemical techniques allow the study of the redox mechanism of the drugs. Redox properties of drugs can provide insight into their metabolic fate, their in vivo redox processes and their pharmacological activity.16-22
Voltammetric oxidation and electrochemical determination of deferiprone on a platinum surface have not, to the best of our knowledge, been reported in the literature. In the present work, we have studied the electrochemical behavior of deferiprone at a platinum electrode, with the aim of developing an electroanalytical procedure for quantification of deferiprone in both bulk form and pharmaceutical formulation.
Experimental
All chemicals used in this work were of analytical reagent grade from Merck. Deferiprone was kindly supplied by Arasto Pharmaceutical Chemicals Inc., Tehran, Iran. The deferiprone tablets were kindly supplied by Avicenna Laboratories, Saveh, Iran.
The standard solution of deferiprone was prepared by dissolving an accurate mass of the bulk drug in an appropriate volume of 100 mmol L-1 acetate buffer solution, pH 4.0 (which was also used as supporting electrolyte), and then stored in the dark at 4 ºC. Additional diluted solutions were prepared daily by accurate dilution just before use. Deferiprone solutions were stable and their concentrations did not change with time.
Drug-free serum samples were obtained from healthy male volunteers and stored frozen until the assay. The serum samples were diluted (1:7) with the supporting electrolyte and filtrated through a 30 kDalton filter to produce protein-free human serum. Various portions of stock deferiprone solution were transferred into 10 mL volumetric flasks containing 3.3 mL of the serum sample. These solutions were then diluted to the mark with the supporting electrolyte for preparation of spiked samples (final dilution of 1:20 with the supporting electrolyte). The protein-free spiked serum solutions were directly analyzed by the calibration method, according to our proposed procedure.
Urine samples taken from a healthy person were diluted (1:10) with buffer solution after adding an appropriate amount of deferiprone standard solution. The resulting solution was, then, directly analyzed, according to our proposed procedure, without any pretreatment or extraction steps.
For deferiprone determination in pharmaceutical preparations, an average mass of five tablets from the same batch was determined, then finely ground and homogenized in a mortar. The weighted amount of this powdered sample was used to prepare the sample solution, which was filtered and transferred to a volumetric flask. The volume was completed with the buffered electrolyte solution.
Electrochemical measurements were carried out in a conventional three-electrode cell (from Metrohm) powered by an electrochemical system comprising the AUTOLAB system with PGSTAT30 (Eco Chemie, Utrecht, The Netherlands). The system was run on a PC using the GPES 4.9 software. Acetate buffer solution, pH 4.0, was used as a supporting electrolyte. It should be mentioned that pH 4.0 was selected as a mean acidic medium due to the high solubility of deferiprone, in neutral pH the deferiprone solubility is very low. An Ag/AgCl with KCl 3.0 mol L-1 (from Metrohm) and a platinum disk (3.0 cm2 in surface area, from Azar Electrode Co., Iran) were used as reference and counter electrodes, respectively. The working electrode was a platinum disk (from Metrohm), exposing a surface area of 0.0314 cm2. For coulometric experiments, a low volume cell (5 mL, Goldis Co., Iran); equipped with a platinum electrode (3.0 cm2 in surface area, from Azar Electrode Co., Iran) and a glassy carbon electrode (from Azar Electrode Co., Iran) as the working and counter electrodes, respectively, were employed. The typical concentration of deferiprone in the solution was 2.1 mmol L-1. The working potentials of 851 and 1268 mV were used for the electrolysis processes. For differential pulse voltammetry (DPV) measurements, a pulse width of 25 mV, a pulse time of 50 ms, and a scan rate of 10 mV s-1 were employed. Before measurement of each voltammogram, it is necessary to perform a polishing in the working electrode with 0.05 μm suspension on alumina, on a polishing micro-cloth, followed by sonication for 3 min in an ultrasonic bath. The electrode was then introduced into the cell. Potential in the range of -300 to 1400 mV in a regime of cyclic voltammetry was applied until a stable voltammogram was achieved. The solution pH was adjusted using a Metrohm 744 pH meter. All studies were carried out at room temperature.
Results and Discussion
Figure 1 shows a typical cyclic voltammogram of 4.5 mmol L-1 deferiprone in the acetate buffer solution, pH 4.0. For comparison, the cyclic voltammogram of the blank electrolyte is also shown. Deferiprone has presented two anodic peaks (a1 and a2) in the voltammogram, with potentials of 768 mV and 1184 mV, respectively, at the potential sweep rate of 10 mV s-1. In the reverse sweep, however, no peaks appeared, indicating an irreversible heterogeneous electron transfer process for the oxidation of deferiprone at the platinum surface.
Controlled-potential coulometries were performed in the 5 mL buffer solution containing 2.1 mmol L-1 deferiprone, at 851 and 1268 mV. The consumed coulombic charge in each stage of electrolysis, calculated from coulogram (Q vs T curve) and the mole number of deferiprone electrolyzed in each stage was determined by cyclic voltammetry. Then the consumed coulombic curve versus mol number of deferiprone electrolysis was calculated and plotted for more than 15 stages of electrolysis (with an intercept of 2.097 C, R = 0.935, n = 18 for peak a1 and an intercept of 4.063 C, R = 0.971, n = 16 for peak a2). The charge consumptions for total electrolysis of the solution were derived from intercepts of these plots and the numbers of exchanged electrons were found to be 2.1 and 2.0, respectively. Therefore, deferiprone is oxidized via two two-electron steps.
A pseudo-steady-state current-potential curve recorded for the first step of oxidation of deferiprone (peak a1 in Figure 1) and a typical S-shape plot was obtained. The slope of the Evs. log I plot was found to be 103.7 mV dec-1 for deferiprone (with a regression equation of y = 103.67x + 621.88, R = 0.995). Also, the electron transfer coefficient multiply by the number of exchanged electrones (αn) was determined to be 0.57.
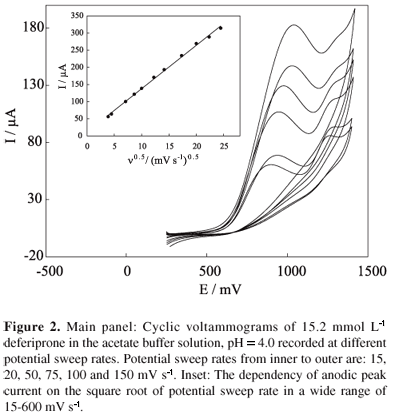
The effect of potential sweep rate was studied in the range of 15 to 600 mV s-1 (Figure 2). As the potential sweep rate increased, both peak currents increased. Also, the peak potentials shifted to more positive values and no peak appeared in the reverse sweep, indicating the irreversible nature of the reaction processes. In addition, the peak currents depend linearly on the corresponding square root of potential sweep rate (with a regression equation of y = 12.62x + 11.75, R = 0.999, n = 11, Figure 2, inset), indicating a diffusion-controlled process. From the slope of the linear dependency of current peak a1 on the square root of potential sweep rate, and using the Randles-Sevcik equation for totally irreversible electron transfer processes,23 we can calculate the diffusion coefficient of deferiprone as follows:

where a is the electron transfer coefficient, n is the number of exchanged electrons, A is the surface area of the working electrode, C* and D are the bulk concentration and diffusion coefficients of the electroreactant species, respectively, and n is the potential sweep rate. The diffusion coefficient of deferiprone was found to be 3.41 × 10-6 cm2 s-1. It should be added that the peak a2 current becomes ill-defined at high potential sweep rates due to some accumulation of reaction products generated in the course of peak a1 at the electrode surface.
The effect of pH on the electrochemical behavior of deferiprone was investigated by cyclic voltammetry using 100 mmol L-1 buffer at various pH values ranging from 2.3 to 13.1. Figure 3 shows the changes of potential peak a1 with respect to the solution pH. The plot shows that at pH < 3.2 and pH > 10.8 values, the peak a1 potential remains constant. However, in the range of 3.5 < pH < 10.2, the peak potential shifted to less positive values with pH values increases. Therefore, it is possible to estimate two pKa values for deferiprone, as pKa,1 = 3.5 and pKa,2 = 10.2, from the intersection points in the diagram according to the following acid/base reaction equilibria:

The obtained pKa values are in close agreement with those reported in the literature.24,25 Moreover, a linear displacement of the peak potential with the pH of the solution was obtained with a slope of 76 mV/pH unit in the pH range of 4.0-10.0 (with a regression equation of y = -76.35x + 1266, R = 0.993, n = 9). This result indicates the involvement of protons in the electrode reaction, and that the proton-transfer reaction precedes the electrode process.
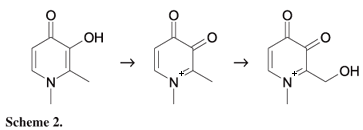
On the basis of our results (and assuming one can be proposed), we have depicted a mechanism for the oxidation of deferiprone in Scheme 2. In the first step, deferiprone is oxidized to corresponding dione. Then the dione undergoes a probable anodic hydroxylation of the methyl side chain.26
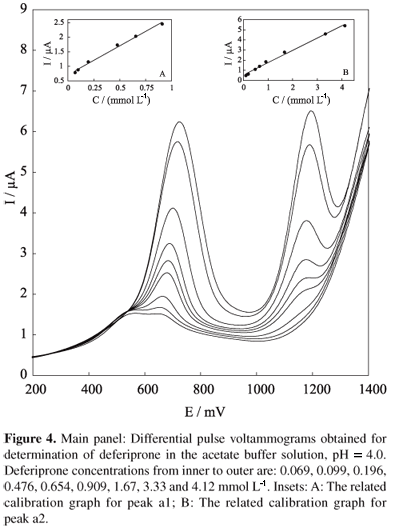
The calibration curve for deferiprone in buffer solution, pH 4.0 was obtained by differential pulse voltammetry (DPV). Figure 4 shows typical DPV curves for different concentrations of deferiprone in buffer solution. The dependency between peak current and drug concentration was rectilinear for peak a1 within the range of 0.07 to 0.91 mmol L-1 (with a regression equation of y = 1.98x + 0.71, R = 0.996, n = 6, Figure 4, inset A), and for peak a2 within the range of 0.10 to 4.12 mmol L-1 (with a regression equation of y = 1.22x + 0.54, R = 0.996, n = 8, Figure 4, inset B). The limits of detection (LOD) and quantitation (LOQ) of the procedure were calculated according to the 3 S.D./m and 10 S.D./m criteria, respectively, where S.D. is the standard deviation of the blank and m is the slope of the calibration curves.27 The limits of detection and quantitation were found to be 1.79 × 10-6 mol L-1 and 4.71 × 10-6 mol L-1 for peak a1, and 2.51 × 10-5 mol L-1 and 8.22 × 10-5 mol L-1 for peak a2, respectively. Precision and accuracy were assessed by performing replicate analyses of deferiprone samples. The precision of the method was calculated as the relative standard deviation (RSD). The procedure was repeated on the same day on the same spiked solutions at concentrations in the range of the standard series. The intra-assay RSDs of the proposed method, determined on the basis of peak current for 10 replications, were 1.6% and 1.7% for peaks a1 and a2, respectively, and showed good repeatability.
The accuracy of the proposed method was determined by spiking serum and urine samples with different concentrations of deferiprone. Good percentage recoveries were obtained from both samples. The analytical parameters obtained for the analyzed samples are summarized in Table 1.
The applicability of the proposed method for the determination of deferiprone in biological fluids was examined by measuring the peak a1 current as a function of the bulk concentration of the drug in urine and serum (Figure 5) samples. The urine and serum samples were diluted 10 and 20 times with buffer solution of pH 4.0 prior to performing the measurements, to prevent the matrix effect of real samples. The generally poor selectivity of voltammetric techniques can pose problems in the analysis of biological samples, if they contain oxidizable substances. However, no current due to oxidation of the compounds in either the serum or the urine samples appeared. The results obtained from our voltammetric technique for determining deferiprone in serum and urine samples are listed in Table 1. The percentage recovery of deferiprone was determined by comparing the peak currents of a known drug concentration in both media with their equivalents in calibration curves; these results are also summarized in Table 1. Good recoveries of deferiprone were achieved from these matrices, meaning that application of our proposed voltammetric method to the analysis of deferiprone in biological fluids could be easily assessed.
In order to evaluate our method in practical analytical applications, quantification of deferiprone in tablet form was attempted. The sample solution prepared as described in Experimental and voltammograms were recorded similar to the case of standard solution of deferiprone. The real sample showed peaks for deferiprone quite similar to the pure sample. The content of deferiprone was calculated from the regression equation, with a 0.64% difference with respect to the labeled amount.
Conclusions
The electrochemical behavior of deferiprone was studied in buffer solution on a platinum electrode, using cyclic voltammetry. The kinetic parameters such as the electron transfer coefficient for oxidation and the diffusion coefficient of deferiprone, were determined. A differential pulse voltammetry procedure was optimized and successfully applied for quantification of deferiprone in bulk form, human biological fluids and commercial tablets. The simplicity, sensitivity, selectivity and short time of analysis are the main advantages of these procedures, making them useful for routine analysis.
Acknowledgments
The financial support of Research Council of K. N. Toosi Univerity of Technology and University of Tehran is gratefully acknowledged. The authors are also grateful to Dr A. Parsaye for his fruitful comments.
Received: April 25, 2007/
Web Release Date: June 5, 2008
- 1. Kontoghiorghes, G. J.; Pattichis, K.; Neocleous, K.; Kolnagou, A.; Curr. Med. Chem. 2004, 11, 2161.
- 2. Kontoghiorghes, G. J.; Drugs Today 2001, 37, 23.
- 3. Paschalidis, I.; Kontoghiorghes, G. J.; J. Radioanal. Nucl. Chem. 1999, 242, 181.
- 4. Pashalidis, I.; Kontoghiorghes, G. J.; Biomarkers Environ. 2001, 4, 80.
- 5. Di, J.; Zhang, F.; Zhang, M.; Bi,, S.; Electroanalysis 2004, 16, 644.
- 6. Georgiou, N. A.; van der Bruggen, T.; Oudshoorn, M.; Nottet, H. S. L. M.; Marx, J. J. M.; van Asbeck, B. S.; J. Infect. Diseases 2000, 181, 484.
- 7. Sappey, C.; Boelaert, J. R.; Legrand Poels, S.; Forceille, C.; Favier, A.; Piette, J.; AIDS Res. Hum. Retroviruses 1995, 11, 1049.
- 8. Hoffbrand, A. V.; Ganeshaguru, K.; Hooton, J. W. L.; Tattersall, M. H. N.; Br. J. Haematol. 1976, 33, 517.
- 9. Barlett, A. N.; Hoffbrand A. V.; Kontoghiorghes, G. J.; Br. J. Haematol. 1990, 76, 301.
- 10. A1-Refaie, F. N.; Wonke, B.; Hoffbrand, A. V.; Wickens, D. G.; Nortey, P.; Kontoghiorghes, G. J.; Blood 1992, 80, 593.
- 11. Fassos, F. F.; Klein, J.; Fernandes, D.; Matsui, D.; Olivieri, N. F.; Koren, G.; Clin. Pharmacol. Ther. 1994, 55, 70.
- 12. Hoffbrand, A. V.; Kontoghiorghes, G. J.; Drugs Today 1992, 28, 149.
- 13. Epemolu, R. O.; Singh, S.; Hider, R. C.; Damani, L. A.; J. Chromatogr 1990, 519, 171.
- 14. Goddard, J. G.; Kontoghiorghes, G. J.; Clin. Chem. 1990, 36, 5.
- 15. Dresow, B.; Fischer, R.; Janka, G. E.; Gabbe, E. E.; Fresenius J. Anal. Chem. 1995, 352, 562.
- 16. Wang, J., ed. In Electroanalytical Techniques in Clinical Chemistry and Laboratory Medicine; VCH Publishers: New York, 1996.
- 17. Kissenger, P. T.; Heineman, W. R., eds. In Laboratory Techniques in Electroanalytical Chemistry; Marcel Dekker: New York, 1996.
- 18. Kauffmann, J. M.; Vire, J. C.; Anal. Chim. Acta 1993, 273, 329.
- 19. Ozkan, S. A.; Uslu, B.; Sentürk, Z.; Electroanalysis 2004, 16, 231.
- 20. Ozkan, S. A.; Uslu, B.; Aboul-Enein, H. Y.; Crit. Rev. Anal. Chem 2003, 33, 155.
- 21. Smyth, M. R.; Vos, J. G., eds. In Analytical Voltammetry; Elsevier Science Ltd.: Amsterdam, 1992.
- 22. Yardimci, C.; Ozaltin, N.; Analyst 2001, 126, 361.
- 23. Bard, A. J.; Faulkner, L. R.; Electrochemical Methods, Wiley: New York, 2001, p. 236.
- 24. Motekaitis, R. J.; Martell, A. E.; Inorg. Chim. Acta 1991, 183, 71.
- 25. Kline, M. A.; Orvig, C.; Clin. Chem 1992, 38, 562.
- 26. Lund, H.; Hammerich, O. In Organic Electrochemistry; Hammerich, O.; Utley, J. H. P.; Eberson, L., eds.; Marcel Dekker: New York, 1991, ch. 24.
- 27. Miller, J. C.; Miller, J. N.; Statistics for Analytical Chemistry, 4th ed., Ellis-Howood: New York, 1994, p. 115.
Publication Dates
-
Publication in this collection
05 Aug 2008 -
Date of issue
2008
History
-
Accepted
05 June 2008 -
Received
25 Apr 2007