Resumos
A resistência ao hormônio tireoidiano (RHT) é uma síndrome que se caracteriza pela presença de níveis séricos elevados de hormônios tireoidianos (HT) e níveis séricos elevados, ou inapropriadamente normais, de hormônio estimulante da tireóide. Em geral, os pacientes apresentam resistência ao HT tanto em nível hipofisário como em tecidos periféricos. Os indivíduos afetados apresentam fenótipo variável, dependendo da severidade da mutação, da diversidade da resposta tecido-específica e outros fatores não relacionados à mutação. Na maioria dos casos, a RHT é secundária a mutações no domínio carboxiterminal do receptor ß do hormônio tireoidiano. A RHT é uma doença autossômica dominante, exceto em uma família descrita, na qual a maioria dos indivíduos é heterozigota para o alelo mutado. Novas técnicas e estudos em modelos animais têm possibilitado uma maior compreensão sobre a ação do receptor de HT; em particular, como os receptores de HT mutantes de pacientes com RHT podem bloquear a função de receptores normais (atividade dominante negativa) e como produzem efeitos diversos nos vários tecidos e entre indivíduos.
Resistência ao hormônio tireoidiano; Receptor de hormônio tireoidiano; Hormônio tireoidiano; Hormônio estimulante da tireóide; Atividade dominante negativa
Resistance to thyroid hormone (RTH) is a syndrome characterized by elevated serum thyroid hormone (TH) levels and elevated or inappropriately normal thyrotropin levels. In general, patients exhibit TH resistance in the pituitary and peripheral tissues. The phenotype of RTH is variable; the affected individuals are clinically euthyroid or even hypothyroid depending on the severity of the mutation, the variable hyposensitivity to TH among individuals as well as in different tissues. In almost all cases the genetic basis of RTH lies in mutation of the carboxyl-terminus of the ß-thyroid hormone receptor. RTH is a dominant disorder, except in one family; most individuals are heterozygous for the mutant allele. New standard techniques and genetically engineered mouse model systems have increased our understanding on TH receptor action, in particular, how mutant thyroid receptors from RTH patients can block wild-type thyroid receptor function (dominant negative activity), and how the mutant receptors can differently affect various tissues and individuals.
Resistance to thyroid hormone; Thyroid hormone receptor; Thyroid hormone; Thyrotropin; Dominant negative effect
REVISÃO
Síndrome de resistência ao hormônio tireoidiano
Thyroid hormone resistance syndrome
Gisah A. de Carvalho; Helton E. Ramos
Serviço de Endocrinologia e Metabologia da Universidade Federal do Paraná - SEMPR (GAC, HER); e Department of Medicine, Thyroid Study Unit, University of Chicago, Chicago, Illinois HER)
Endereço para correspondência Endereço para correspondência Gisah Amaral de Carvalho Serviço de Endocrinologia e Metabologia SEMPR Universidade Federal do Paraná Rua Padre Camargo 262 80060-240 Curitiba, PR Fax: (41) 264-8721 e.mail: carvalho@mais.sul.com.br
RESUMO
A resistência ao hormônio tireoidiano (RHT) é uma síndrome que se caracteriza pela presença de níveis séricos elevados de hormônios tireoidianos (HT) e níveis séricos elevados, ou inapropriadamente normais, de hormônio estimulante da tireóide. Em geral, os pacientes apresentam resistência ao HT tanto em nível hipofisário como em tecidos periféricos. Os indivíduos afetados apresentam fenótipo variável, dependendo da severidade da mutação, da diversidade da resposta tecido-específica e outros fatores não relacionados à mutação. Na maioria dos casos, a RHT é secundária a mutações no domínio carboxiterminal do receptor ß do hormônio tireoidiano. A RHT é uma doença autossômica dominante, exceto em uma família descrita, na qual a maioria dos indivíduos é heterozigota para o alelo mutado. Novas técnicas e estudos em modelos animais têm possibilitado uma maior compreensão sobre a ação do receptor de HT; em particular, como os receptores de HT mutantes de pacientes com RHT podem bloquear a função de receptores normais (atividade dominante negativa) e como produzem efeitos diversos nos vários tecidos e entre indivíduos.
Descritores: Resistência ao hormônio tireoidiano; Receptor de hormônio tireoidiano; Hormônio tireoidiano; Hormônio estimulante da tireóide; Atividade dominante negativa
ABSTRACT
Resistance to thyroid hormone (RTH) is a syndrome characterized by elevated serum thyroid hormone (TH) levels and elevated or inappropriately normal thyrotropin levels. In general, patients exhibit TH resistance in the pituitary and peripheral tissues. The phenotype of RTH is variable; the affected individuals are clinically euthyroid or even hypothyroid depending on the severity of the mutation, the variable hyposensitivity to TH among individuals as well as in different tissues. In almost all cases the genetic basis of RTH lies in mutation of the carboxyl-terminus of the ß-thyroid hormone receptor. RTH is a dominant disorder, except in one family; most individuals are heterozygous for the mutant allele. New standard techniques and genetically engineered mouse model systems have increased our understanding on TH receptor action, in particular, how mutant thyroid receptors from RTH patients can block wild-type thyroid receptor function (dominant negative activity), and how the mutant receptors can differently affect various tissues and individuals.
Keywords: Resistance to thyroid hormone; Thyroid hormone receptor; Thyroid hormone; Thyrotropin; Dominant negative effect
A SÍNDROME DE RESISTÊNCIA AO HORMÔNIO tireoidiano (RHT) é caracterizada pela reduzida resposta dos tecidos-alvo ao hormônio tireoidiano (HT), apesar das elevadas concentrações séricas de T3 e T4 livres, associada a um TSH elevado ou inapropriadamente normal característico da sensibilidade reduzida do tireotrofo. Refetoff e cols., em 1967, foram os primeiros a descrever a síndrome clínica de RHT em dois indivíduos que apresentavam bócio, surdo-mudez e atraso na idade óssea. Os níveis séricos de HT eram elevados, contrastando com os sintomas de hipotireoidismo. A administração de HT exógeno não produziu o efeito metabólico esperado e também não suprimiu o TSH estimulado pelo TRH (1,2). Após 20 anos, a base molecular da RHT foi elucidada e se deve a anormalidades no receptor ß do hormônio tireoidiano (TRß). Desde o primeiro relato, foram identificados mais de 700 indivíduos com RHT provenientes de cerca de 250 famílias. A prevalência da RHT é de aproximadamente 1 em 50.000 recém-nascidos (3).
O padrão de herança da RHT é autossômico dominante, sendo que a transmissão recessiva foi identificada em apenas uma família (4).
O fenótipo da RHT é bastante variável, tanto entre famílias como entre membros da mesma família com mutações idênticas do TRß. A maioria dos pacientes apresenta sintomas leves a moderados. Esta variabilidade na manifestação clínica se deve, em parte, à severidade da resistência hormonal, da efetividade dos mecanismos compensatórios, da presença de fatores genéticos moduladores e dos efeitos de tratamentos anteriores (5,6). Embora a reduzida sensibilidade ao HT possa variar em severidade, existem características clínicas comumente encontradas na síndrome, que são bócio, retardo do crescimento, níveis séricos elevados de T3 e T4 livres, níveis normais ou discretamente elevados de TSH, o qual responde ao TRH e ausência dos sintomas comuns e das alterações metabólicas provocadas pelo excesso de HT (7).
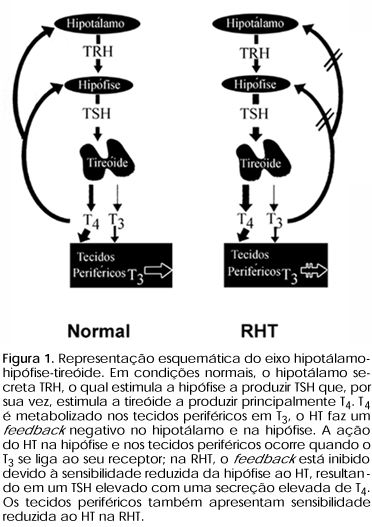
Os sinais, sintomas e anormalidades laboratoriais observadas na RHT só ocorrem devido à resposta reduzida ao HT pelos tecidos periféricos e hipófise, resultando em um feedback negativo anormal do T3 no TSH/TRH e ação intracelular do T3 diminuída (figura 1).
Mecanismo de Ação do Hormônio Tireoidiano
O hormônio tireoidiano entra na célula por um processo de difusão passiva concentração-dependente após se dissociar das proteínas transportadoras de HT. Várias moléculas têm sido apontadas como transportadoras de HT; recentemente, foi demonstrado defeito em uma proteína transportadora, causando uma apresentação clínica semelhante à da RHT. A ação do HT, em geral, é mediada por receptores nucleares específicos do HT (TRs), tendo sido descritos mecanismos de ação não genômico do HT (8).
TRs são membros da superfamília de receptores nucleares, juntamente com os receptores do ácido retinóico (RXRs), receptores do 9-cis ácido retinóico, receptores da vitamina D3 e receptores ativados pelo proliferador-peroxisoma (PPAR). Os TRs agem como fatores de transcrição dependentes de ligação que aumentam ou diminuem a expressão dos genes-alvo, dependendo se os promotores dos genes contêm elementos responsivos ao HT regulados de forma positiva ou negativa, respectivamente (9-11).
Existem dois genes que codificam os TRs, TRa e TRß, localizados nos cromossomos 17 e 3, respectivamente. Os TRs são homólogos celulares do oncogene viral erb-A. Cada gene produz, pelo menos, duas proteínas através de splicing alternativo: TRa1, TRa2, TRß1 e TRß2 (9-11). A expressão relativa dos dois TR genes e a distribuição dos seus produtos variam de tecido para tecido e nos diferentes estágios do desenvolvimento. O HT também regula, de forma diferenciada, a expressão dos genes dos TRs e de suas isoformas (9,10).
As isoformas TRa e TRß contêm um domínio de ligação ao DNA (DBD), um domínio de ligação ao ligante T3 (LBD) e um terceiro domínio de dobra que une os dois primeiros. A isoforma TRa2, devido à sua seqüência carboxiterminal alterada, não se liga ao HT e, portanto, não funciona como um receptor de HT (3,11).
Os TRs formam homodímeros (TR/TR) ou heterodímeros com RXRs (TR/RXR) e se ligam a seqüências específicas de DNA denominadas elementos responsivos ao HT (TREs), que estão localizadas próximo do ponto onde se inicia a transcrição dos genes regulados pelo HT (12).
Nos genes que são positivamente regulados pelo HT, os TRs não ligados suprimem a transcrição basal destes genes pela interação com uma classe de proteínas nucleares co-repressoras como o co-repressor do receptor nuclear (NcoR) e o mediador do silenciador do receptor de HT e retinóides (SMRT). Recentemente, foi demonstrado que os co-repressores podem formar complexos com outros repressores como o SIN 3 e as deacetilases de histona. Portanto, a desacetilação da histona provavelmente tem um papel crucial na repressão basal pelo complexo co-repressor/TR não ligado; isto ocorre porque a estrutura da cromatina local se mantém de uma forma que "desliga" a transcrição basal (11). A ligação do T3 no TR provoca uma alteração na conformação do TR, aumenta a ocupação do TRE pelos heterodímeros TR/RXR, libera o complexo co-repressor e recruta co-ativadores como os co-ativadores do receptor de esteróide (SRC-1, SRC-2 e SRC-3) e o complexo de proteína que interage com o receptor de vitamina D/proteína associada ao TR (complexo DRIP-TRAP), todos com atividade histona acetiltransferase (13). A estrutura da cromatina vai se modificar, vai ocorrer perda de um nucleossomo na região promotora, com isto a transcrição do gene será ativada (figura 2). Os mecanismos que envolvem os genes que são negativamente regulados pelo HT, como o TSH, são menos compreendidos, provavelmente envolvem a mesma classe de moléculas em um TRE diferente onde os co-repressores se tornam ativadores e vice-versa (11-14).
Mecanismo Molecular da RHT: Mutações no Gene do TR ß
A maioria dos indivíduos com RHT, cerca de 80-85%, possui mutações no gene do TRß; não foram descritas mutações no gene do TRa. Até o presente momento, foram identificadas cerca de 100 mutações diferentes no gene do TRß1 em mais de 250 famílias portadoras de RHT (6). A maioria das mutações ocorreu em três regiões hot spot localizadas no domínio de ligação ao ligante (LBD), entre os exons 8 e 10 do TRß (figura 3). A maioria das mutações se deve a substituições de um único nucleotídeo, mas também foram identificadas deleções ou inserções de nucleotídeos causando deleção de aminoácidos ou a formação precoce de codons de parada (stop codons) (14). A mutação no aminoácido 338 (R338W, troca da arginina por triptofano) foi encontrada em 23 famílias não relacionadas. As mutações ocorreram em 73% dos casos, quando uma citosina é seguida por uma guanina, sugerindo que estas regiões "CG" são hotspots no gene do TRß (3,14,15).
A primeira família descrita com RHT é a única com padrão de herança recessivo; em todos os outros casos descritos, o padrão de herança é autossômico dominante (1). A RHT ocorre de forma esporádica em 15% dos casos, quando a mutação aparece "de novo" ou seja, os pais apresentam um fenótipo normal.
Em geral, o receptor mutante não consegue se ligar ao HT ou possui afinidade reduzida pelo mesmo. Como conseqüência, o co-repressor não é liberado e o gene não é ativado. Alguns receptores mutantes se ligam ao T3, mas liberam o co-repressor de forma mais lenta ou têm uma interação mais fraca com o co-ativador (3,16,17).
Os TRßs mutantes interferem com a função dos TRs normais, um fenômeno denominado efeito negativo dominante (DNE). Os receptores TRß mutantes continuam capazes de se ligar ao TRE e dimerizar com TRs normais ou RXRs, desta forma competindo e bloqueando a ligação dos TRs normais ao TER. Isto explica porque indivíduos heterozigotos com deleção do TRß não manifestam o fenótipo de RHT, ou seja, o alelo mutante não esta interferindo com o alelo normal. As características de dimerização dos TRß mutantes têm um papel importante na mediação da atividade dominante negativa (18,19).
Aproximadamente 15% das famílias com RHT não possui mutações no TRß. Estes indivíduos sem mutação no TR têm um fenótipo semelhante àqueles com mutação e também apresentam um padrão de herança autossômico dominante. Provavelmente outros co-fatores de transcrição envolvidos na ação do HT são responsáveis pela resistência hormonal nestes casos (20). De fato, estudos do eixo hipotálamo/hipófise/ tireóide de camundongos knockout para os co-ativadores SRC-1 ou RXR demonstraram que a ausência de um co-fator pode causar RHT (21).
Uma observação intrigante nos pacientes com RHT é a acentuada variabilidade na resistência ao HT nos diferentes tecidos de um mesmo indivíduo ou entre indivíduos com a mesma mutação no gene do TRß. Ainda não está claro como a ação do TRß mutante é modulada, o que provavelmente explicaria estas variações. Estudos recentes demonstraram que co-fatores, como o SRC-1, podem não só modular a ação dos TRß normais, mas também a dos mutantes e de forma tecido-dependente. Portanto, a variação tecidual e individual nos pacientes com RHT ocorre devido à combinação de múltiplos fatores, como a distribuição tecido-dependente das isoformas de TR, a ação dos co-ativadores e o ambiente da região promotora dos genes regulados pelo T3 (22-25).
Foram descritas mutações no TRa e TRß em hepatomas, em câncer renal e de tireóide em humanos. Embora estas mutações possam causar RHT nestes tumores, provavelmente não são oncogênicas, considerando que pacientes com RHT e mutações germinativas no TRß não apresentam um risco aumentado de câncer (26).
Modelos Animais para Avaliação Específica do Receptor do HT
A importância dos TRs no mecanismo de ação do HT tem sido demonstrada através de modelos animais com ausência dos TRs e, conseqüentemente, resistência ao HT. Atualmente, existem dois diferentes modelos com similares deleções do gene TRß, um modelo com deleção especifica do TRß2 (27) e 3 linhagens de camundongos com diferentes deleções no gene do TRa. O camundongo TRß knockout (ausência de expressão das isoformas ß1 e ß2) possui TSH elevado, níveis elevados de hormônios tireoidianos, bócio e surdez neurossensorial, portanto, com fenótipo idêntico ao observado em humanos com deleção do gene TRß (28,29). Análise deste modelo permite concluir que o TRß não é necessário para a up-regulation do TSH existente na deprivação do HT (TRß não ligado), porém este receptor exerce papel fundamental na down-regulation e supressão completa do TSH (30). O camundongo TRa knockout inicialmente gerado não dispunha das isoformas TRa1 e TRa2, porém, como a região promotora interna presente no intron 7 permaneceu intacta, este modelo ainda possuía os fragmentos Da1 e Da2. Este animal possui fenótipo caracterizado por hipotireoidismo progressivo, crescimento retardado e atraso no desenvolvimento intestinal e ósseo, com morte invariavelmente por volta da quinta semana de vida (31). Este modelo ressalta a importância funcional do TRa , porém, com a expressão intacta das isoformas Da 1 e Da2, é difícil afirmar se o quadro de hipotireoidismo acentuado se deve à falta do TRa ou à ação antagonista das isoformas truncadas. Posteriormente, a avaliação do camundongo Tra0/0 (que não possui nenhum alelo TRa), que apresenta ausência simultânea da região promotora alternativa do intron 7, revelou fenótipo de hipersensibilidade ao HT (32). Um outro modelo de TRa knockout (RTa1-/-), que exibe, portanto, expressão mantida das variantes RTa2 eDa2, possui níveis mais baixos de T4 livre, crescimento, desenvolvimento e fertilidade normais, mas apresentam temperatura corporal e freqüência cardíaca reduzidas (33). Estes resultados apontam para a importância do TRa1 na ação do HT em nível de freqüência cardíaca, controle da temperatura e gasto energético. Camundongos duplo (TRa e TRß) knockout possuem níveis de hormônio tireoidiano e TSH dramaticamente elevados, bócio e crescimento retardado. Definitivamente a ausência do TRa acentua o fenótipo de resistência existente quando somente o TRß esta ausente. Porém, embora os animais sejam menores, eles são capazes de sobreviver na ausência total de TRs (com exceção dos fragmentos DTRs) (34). Outro duplo knockout (TRa1 e TRß), que ainda possuem o TRa2 intacto, tem leve atraso de crescimento, TSH marcadamente elevado e infertilidade nas fêmeas (35). Existe um terceiro tipo de duplo knockout (RTa0/0, RTß-/-) que, por sua vez, não possui nenhuma isoforma TRa ou TRDa, se apresenta com fenótipo de completa esterilidade nas fêmeas e atrofia testicular resultando em infertilidade nos machos, porém com uma sobrevida de pelo menos 6 meses (34). Estudos em modelos animais possibilitam a elucidação da ação específica de cada isoforma nos diferentes tecidos, assim como as conseqüências das diferentes mutações no fenótipo.
Manifestações Clínicas da RHT
A ausência de feedback negativo do T3 no TSH resulta em uma secreção persistente de TSH, estimulação da glândula tireoidiana e conseqüente aumento da síntese e secreção de HTs. O TSH normal ou discretamente aumentado responde ao estímulo com TRH, esta resposta distingue indivíduos com RHT daqueles com adenoma hipofisário secretor de TSH (36). O TSH de pacientes com RHT tem as mesmas propriedades imunológicas do TSH de controles normais, não contém excesso da sub-unidade a (TSHa), tipicamente encontrado no soro de pacientes com tumores hipofisários produtores de TSH (36,37). Entretanto, o TSH tem uma potência biológica aumentada in vitro, talvez isto explique como níveis séricos normais de TSH causam bócio e hipersecreção de HT pela tireóide. Uma outra explicação para este fato poderia ser o aumento do número de receptores de TSH nos tireócitos (38).
A apresentação clínica da RHT é bastante variável entre famílias e entre indivíduos da mesma família com mutações idênticas do TRß. A maioria dos indivíduos encontra-se assintomática devido ao mecanismo de compensação, ou seja, aos níveis séricos elevados de HTs. O grau de compensação à reduzida sensibilidade ao HT é variável entre indivíduos e entre diferentes tecidos. Portanto, alguns indivíduos manifestam sintomas sugestivos de privação do HT como retardo do crescimento, habilidades cognitivas reduzidas e hipercolesterolemia, enquanto que outros apresentam sintomas de excesso de HT como taquicardia, avanço da idade óssea ou hiperatividade. O mesmo indivíduo pode apresentar sintomas de ambos, excesso e deficiência de HT (7-16).
Muitas das características clínicas encontradas em alguns pacientes com RHT podem ser a manifestação da privação tecido-específica do HT durante estágios precoces do desenvolvimento. A severidade dos sintomas esta relacionada à expressão relativa do alelo mutante e dos co-fatores de forma tecido-específica. Muitos indivíduos com RHT apresentam taquicardia, pois o coração tem predominantemente TRa, que responde normalmente ao HT, o qual encontra-se elevado, resultando em taquicardia (22-24).
Indivíduos com RHT que estão aparentemente eutireóideos e com concentrações de TSH normal ou próximo do normal são classificados como tendo resistência generalizada ao HT (RHTG). Nestes indivíduos, o defeito é compensado pelos elevados níveis de HT. Em contraste, pacientes com os mesmos níveis elevados de HT encontram-se hipertireóideos e são classificados com tendo resistência hipofisária seletiva ao HT (RHTP) (16). Em alguns casos, indivíduos com a mesma mutação e, às vezes, pertencendo à mesma família foram classificados como tendo RHTG e RHTP. Posteriormente, estudos clínicos demonstraram que a resposta dos marcadores periféricos à ação do HT estava igualmente atenuada nos dois grupos. Estas observações sugerem que RHTG e RHTP são variações fenotípicas da mesma alteração genética, onde a expressão clínica é provavelmente modulada por outros fatores não relacionados à mutação (39,40).
Em crianças, a investigação levando ao diagnóstico de RHT ocorre, em geral, devido à presença de bócio, hiperatividade, dificuldades no aprendizado ou atraso no desenvolvimento. Nos adultos, o diagnóstico é feito pela presença de bócio e taquicardia sem as demais características clínicas de hipertireoidismo. Atualmente, o diagnóstico de RHT é feito precocemente através dos programas de detecção de hipotireoidismo congênito que usam a dosagem de TSH e de T4 total e em filhos de pais portadores de RHT que são submetidos à avaliação hormonal e genética já na primeira infância (14-16). Em adultos, o diagnóstico de RHT também ocorre por ocasião de uma avaliação de rotina, em que são encontradas anormalidades nos testes de função tireoidiana características desta síndrome.
Bócio é a anormalidade mais comumente encontrada no exame físico, ocorrendo em 85% dos casos. Taquicardia, que ocorre em 90% dos indivíduos com RHT, é causada pela ação do HT em níveis elevados no TRa (16). Metade dos pacientes com RHT tem dificuldade no aprendizado, freqüentemente associada com hiperatividade e déficit de atenção e, em geral, baixo quociente de inteligência (QI). Entretanto, retardo mental (QI < 60) ocorre em apenas 3% dos casos. Discreto ou moderado retardo no crescimento e atraso na idade óssea estão presentes em 25% dos casos. Defeitos auditivos também estão presentes em um quarto dos pacientes (tabela 1). Outras manifestações foram observadas, como infecções repetidas nas vias aéreas superiores, diminuição da massa óssea, surdez, hipotonia e convulsão (41,42).
O curso da doença, assim como a sua apresentação, pode ser variável. Alguns indivíduos têm o crescimento e desenvolvimento normal, outros apresentam graus variáveis de retardo mental e/ou de crescimento. Estudos em humanos e em camundongos TRß knockout demonstraram que a severidade da doença tende a melhorar com o envelhecimento (16-20).
Diagnóstico
Níveis elevados de T3 e T4 livres em combinação com um TSH não suprimido é indicativo de RHT. Deve ser eliminada qualquer condição que possa apresentar as mesmas características laboratoriais de RHT, como anormalidades nas proteínas transportadoras de HT, drogas e a presença de anticorpos que interagem com os ensaios que medem os hormônios em questão (16).
Os níveis de T3 e T4 séricos variam desde elevação discreta, pouco acima, até várias vezes acima do limite normal para ensaio. A conversão extratireoidiana de T4 para T3 é normal, resultando numa proporção T3/T4 normal; a concentração de T3 reverso também está elevada em pacientes com RHT. A concentração sérica de tireoglobulina reflete o grau de estimulação da glândula tireóide induzida pelo TSH (43,44). A captação de radioiodo pela tireóide está elevada (2 e 24hs), e o iodo é normalmente organificado, ou seja, o teste do perclorato é normal (1).
O sinal patognomômico da síndrome de RHT é a presença de elevados níveis séricos de TSH e da resposta ao TRH preservada a despeito dos elevados níveis de HT. A resposta do TSH ao TRH esta normal ou exagerada, isto ocorre mais comumente em pacientes que estão sendo tratados com drogas antitireoidianas ou com história prévia de cirurgia ou de terapia com radioiodo (45). A bioatividade do TSH é normal ou aumentada e a sub-unidade a não está desproporcionalmente elevada, como ocorre nos tumores produtores de TSH (38).
A resistência ao HT tem o papel principal na patogênese da síndrome, os pacientes recebem HT exógeno para estabelecer a presença e o grau de sensibilidade reduzida ao HT. Para tanto, recomenda-se um protocolo padrão que utiliza a administração de doses crescentes de L-T3. São utilizadas três doses em seqüência, uma dose de reposição de 50µg/dia e duas doses suprafisiológicas de 100 e 200µg/dia. O hormônio (L-T3) é administrado a cada 12 horas e a dose é incrementada a cada três dias. As doses são ajustadas em crianças e adultos com peso fora do padrão para manter o mesmo nível sérico de T3. O L-T3 é utilizado preferencialmente ao L-T4, devido ao seu efeito direto nos tecidos sobrepondo potenciais defeitos no transporte ou no metabolismo do T4. Em adição, o início mais rápido e a curta duração da ação do T3 encurtam o período da avaliação (9).
A administração de doses suprafisiológicas de HT causa a supressão da secreção do TSH, resultando na diminuição ou abolição da resposta do TSH ao TRH (37). Várias respostas dos tecidos periféricos ao HT foram quantificadas, como medida da taxa de metabolismo basal, pulso, tempo de relaxamento dos reflexos tendinosos, lipídeos, globulina ligadora do hormônio sexual (SHBG) e excreção urinária de hidroxiprolina, creatina e carnitina. Não foram observadas alterações significativas nestes parâmetros ou as alterações foram pequenas considerando a quantidade de HT utilizada (46).
Quando o diagnóstico de RHT é suspeitado, a realização dos testes que avaliam função tireoidiana nos parentes pode ajudar no diagnóstico. O mesmo fenótipo em vários indivíduos da mesma família torna mais provável o diagnóstico de RHT do que o de tumor secretor de TSH, visto que este não apresenta padrão de herança genética.
O teste genético pode ser realizado no caso índice após o diagnóstico clínico de RHT estar confirmado. Uma vez identificada a mutação, o rastreamento dos outros membros da família para a mutação pode ser mais sensível do que o rastreamento pelo fenótipo, devido à natureza variável do mesmo na RHT. A confirmação da mutação em recém-nascidos pode eliminar a necessidade de tratamento ou tratamentos errôneos. No feto, a confirmação da mutação pode permitir o tratamento intra-uterino se necessário (14).
Avaliação diagnóstica recomendada:
Níveis séricos elevados de T3 e T4 livres com TSH não suprimido;
Confirmar resultados e excluir defeito de transporte de HT ou interferência por anticorpos nos ensaios;
Demonstrar a não supressão do TSH e a resposta metabólica diminuída à administração de doses suprafisiológicas de HT;
Avaliar os testes de função tireoidiana em outros membros da família;
Em casos esporádicos, medir sub-unidade a do TSH para excluir adenoma de hipófise;
Análise genética se necessário.
Tratamento
Não existe um tratamento específico para corrigir completamente o defeito na RHT. Na maioria dos casos, a resistência parcial ao HT parece ser adequadamente compensada pelo aumento endógeno de HT, não é necessário nestes casos nenhum tratamento. O tratamento com HT deve ser instituído nos casos em que foi feito um diagnóstico prévio errôneo resultando em hipotireoidismo pós-cirúrgico ou pós-radioiodo. O TSH pode ser usado como diretriz para ajuste na dose de L-T4, nestes casos as doses podem chegar a 1000µg/dia (47).
Alguns pacientes com RHT apresentam uma resistência maior nos tecidos periféricos do que na hipófise. Nestes casos, a administração de doses suprafisiológicas está indicada e deve ser determinada individualmente através da resposta tecidual (1,16). Pacientes com maior resistência em nível hipofisário e sintomas de hipertireoidismo também necessitam de tratamento, geralmente sintomático com agente bloqueador ß adrenérgico (47). Tratamento com L-T3 pode melhorar os sintomas de hiperatividade e déficit de atenção em crianças com RHT (48). Doenças tireoidianas comuns, como hipotireoidismo e hipertireoidismo, podem ocorrer em pacientes com RHT. A ocorrência de doença de Graves e RHT altera de maneira significativa a abordagem diagnóstica e terapêutica; o objetivo, nestes casos, é o eutireoidismo clínico com um TSH sérico normal (14,47).
O ácido triiodotiroacético (TRIAC®) tem sido usado com sucesso na redução dos níveis séricos de TSH e de HT para reduzir o tamanho do bócio e melhorar alguns sintomas secundários ao excesso de HT nos tecidos periféricos. O ácido triiodotiroacético suprime o TSH sem aumentar o efeito tireomimético nos tecidos periféricos, isto se deve à sua maior afinidade pelo TRß e não pelo TRa quando comparado com T3, e a degradação mais rápida. A D-T4 possui efeito semelhante ao TRIAC®, porém o seu mecanismo de ação ainda não é compreendido (47,49,50). Recentemente, o ácido triiodotiroacético foi utilizado intra-útero para reduzir o tamanho do bócio de um feto com mutação no TRß. O tratamento foi bem sucedido na redução do tamanho do bócio, porém existem controvérsias pelo fato de que foi necessário realizar cordocentese repetidas vezes e de não haver conhecimento sobre o transporte placentário e o metabolismo fetal do TRIAC® (51). Drogas dopaminérgicas e análogos da somatostatina não são indicados, por apresentarem efeitos colaterais e não serem efetivas em manter o TSH supresso (47).
As diretrizes para o tratamento com HT, em geral L-T4, são níveis elevados de TSH, sinais ou sintomas de hipotireoidismo, presença de convulsão, atraso no desenvolvimento e história de retardo mental ou atraso no desenvolvimento em membros afetados da família (1,16,47).
O tratamento da RHT, no futuro, será com o uso de drogas que irão interagir especificamente com o TRß mutante, este análogo do HT deverá se ligar com uma maior afinidade ao TRß mutante e vai eliminar o seu efeito dominante negativo (14). Outras drogas, CG-1 e KB-141, que estão sendo desenvolvidas, são isoformas agonistas do TRß. Estas drogas não estimulam o TRa, protegendo o coração da ação deletéria do HT (52).
O manejo de gestantes com RHT é, ainda, questionável. Os níveis elevados de HT materno podem ser necessários para o desenvolvimento do feto se este for portador de RHT, mas podem ser deletérios para um feto normal. Por outro lado, uma mãe normal provavelmente não irá fornecer a quantidade adequada de HT para um feto afetado. Os poucos dados de literatura indicam que filhos, afetados ou não, de mães normais ou afetadas não apresentaram morbidades (51-53). Futuras investigações serão importantes para uma melhor avaliação destas questões.
Recebido em 17/10/03
Aceito em 15/11/03
- 1. Refetoff S, DeWind LT, DeGroot LJ. Familial syndrome combining deaf-mutism, stippled epiphyses, goiter and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab 1967;27:279-94.
- 2. Refetoff S, DeGroot LJ, Bernard B, DeWind LT. Studies of a sibship with apparent hereditary resistance to the intracellular action of thyroid hormone. Metabolism 1972;21:723-56.
- 3. Usala SJ. Thyroid Hormone Resistance. In: Weintraub BD, editor. Molecular endocrinology. Basic concepts and clinical correlations 1st ed. New York: Raven Press, 1995
- 4. Takeda K, Sakurai A, DeGroot LJ, Refetoff S. Recessive inheritance of thyroid hormone resistance caused by complete deletion of the protein-coding region of the thyroid hormone receptor-beta gene. J Clin Endocrinol Metab 1992;74:49-55.
- 5. Weiss RE, Tunca H, Knapple WL, Faas FH, Refetoff S. Phenotype differences of resistance to thyroid hormone in two unrelated families with identical mutation in the thyroid hormone receptor ß gene (R320). Thyroid 1997;7:35-8.
- 6. Phillips SA, Rotman-Pikielny P, Lazar J, Ando S, Hauser P, Skarulis MC, et al. Extreme thyroid hormone resistance in a patient with a novel truncated TR mutant. J Clin Endocrinol Metab 2001;86:5142-7.
- 7. Beck-Peccoz P, Chatterjee VK. The variable clinical phenotype in thyroid hormone resistance syndrome. Thyroid 1994;4:225-32.
- 8. Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev 2001;81:1097-142.
- 9. Hu I, Lazar MA. Transcriptional repression by nuclear hormone receptors. Trends Endocrinol Metab 2000;11:6-10.
- 10. Zhang J, Lazar MA. The mechanism of action of thyroid hormones. Ann Rev Physiol 2000;62:439-66.
- 11. Yen PM. Molecular basis of resistance to thyroid hormone. Trends Endocrinol Metab 2003;14:327-33.
- 12. Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimmers and heterodimers. Endocr Rev 1994;15:391-407.
- 13. McKenna NJ. Nuclear receptor co-regulators: cellular and molecular biology. Endocr Rev 1999;20:321-44.
- 14. Weiss RE, Carvalho GA. Resistance to thyroid hormone. In: Martini L, editor. Encyclopedia of endocrine diseases 1st ed. Amsterdan:Elsevier/Academic Press, 2004 (in press).
- 15. Weiss RE, Weinberg M, Refetoff S. Identical mutations in unrelated families with generalized resistance to thyroid hormone occur in CG-rich areas of the thyroid hormone receptor beta gene: Analysis of fifteen families. J Clin Invest 1993;91:2408-15.
- 16. Refetoff S, Weiss RE, Usala S. The syndromes of resistance to thyroid hormone. Endocr Rev 1993;14:348-99.
- 17. Yoh SM, Chatterjee VK, Privalsky ML. Thyroid hormone resistance syndrome manifests as an aberrant interaction between mutant T3 receptors and transcriptional co-repressors. Mol Endocrinol 1997;11:470-80.
- 18. Collingwood TN, Adams M, Tone Y, Chatterjee VK. Spectrum of transcriptional, dimerization, and dominant negative properties of twenty different mutant thyroid hormone ß-receptors in thyroid hormone resistance syndrome. Mol Endocrinol 1994;8:1261-77.
- 19. Yen PM, Sugawara A, Refetoff S, Chin WW. New insights on the mechanism(s) of the dominant negative effect of mutant thyroid hormone receptor in generalized resistance to thyroid hormone. J Clin Invest 1992;90:1825-31.
- 20. Reutrakul S, Sadow PM, Pannain S, Pohlenz J, Carvalho GA, Macchia PE, et al. Search for abnormalities of nuclear co-repressors, co-activators, and a co-regulator in families with resistance to thyroid hormone without mutations in TRß or a genes. J Clin Endocrinol Metab 2000;85:3609-17.
- 21. Xu J, Qiu Y, DeMayo FJ, Tsai SY, Tsai MJ, O'Malley BW. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science 1998;279:1922-5.
- 22. Kamiya Y, Zhang XY, Ying H, Kato Y, Willingham MC, Xu J, et al. Modulation by steroid receptor coactivator-1 of target-tissue responsiveness in resistance to thyroid hormone. Endocrinology 2003;144:4144-53.
- 23. McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and co-regulators. Cell 2002;108:465-74.
- 24. Sadow PM, Chassande O, Koo EK, Gauthier K, Samarut J, Xu J, et al. Regulation of expression of thyroid hormone receptor isoforms and co-activators in liver and heart by thyroid hormone. Mol Cell Endocrinol 2003;203:65-75.
- 25. Forrest D, Hanebuth E, Smeyne RJ, Evereds N, Stewart CL, Wehner JM, et al. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor ß: evidence for tissue-specific modulation of receptor function. EMBO 1996;15:3006-15.
- 26. Ando S, Sarlis NJ, Oldfield EH, Yen PM. Somatic mutation of TRß can cause a defect in negative regulation of TSH in a TSH-secreting pituitary tumor. J Clin Endocrinol Metab 2001;86:5572-6.
- 27. Abel Ed, Kaulbach HC, Campos-Barros A, Ahima RS, Boers ME, Hashimoto K, et al. Novel insight from transgenic mice into thyroid hormone resistance and the regulation of thyrotropin. J Clin Invest 1999;103:271-9.
- 28. Takeda K, Sakurai A, DeGroot LJ, Refetoff S. Recessive inheritance of thyroid hormone resistance caused by complete deletion of the protein-coding region of the thyroid hormone receptor-beta gene. J Clin Endocrinol Metab 1992;74:49-55.
- 29. Forrest D, Erway LC, Ng L, Altschuler R, Curran T. Thyroid hormone receptor beta is essential for development of auditory function. Nat Genet 1996;13:354-7.
- 30. Weiss RE, Forrest D, Pohlenz J, Cua K, Curan T, Refetoff S. Thyrotropin regulation by thyroid hormone receptor beta-deficient mice. Endocrinology 1997;138:3624-9.
- 31. Fraichard A, Chassandre O, Plateroti M, Roux JP, Trouillas J, Dehay C, et al. The T3R alpha gene encoding a thyroid hormone receptor is essential for post-natal development and thyroid hormone production. EMBO J 1997;16:4412-20.
- 32. Macchia PE, Takeuchi Y, Kawai T, Cua K, Gauthier K, Chassande O, et al. Increased sensitivity to thyroid hormone in mice with complete deficiency of thyroid hormone receptor alpha. Proc Natl Acad Sci USA 2001;98:349-54.
- 33. Wikstrom L, Johansson C, Salto C, Barlow C, Campos-Barros A, Baas F, et al. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor alpha 1. EMBO J 1998;17:455-61.
- 34. Gauthier K, Chassande O, Plateroti M, Roux JP, Legrand C, Pain B, et al. Different functions for the thyroid hormone receptors TRa and TRß in the control of thyroid hormone production and post-natal development. EMBO J 1999;18:623-31.
- 35. Johansson C, Gothe S, Forrest D, Vennstrom B, Thoren P. Cardiovascular phenotype and temperature control in mice lacking thyroid hormone receptor-beta or both alpha1 and beta. Am J Physiol Regul Integr Comp Physiol 2000;278:598-603.
- 36. Bode HH, Danon M, Weintraub BD. Partial target organ resistance to thyroid hormone. J Clin Invest 1973;52:776-80.
- 37. Vandalem JL, Pirens G, Hennen G. Familial inappropriate TSH secretion: evidence suggesting a dissociated pituitary resistance to T3 and T4 J Clin Endocrinol Invest 1981;4:413-7.
- 38. Persani L, Asteria C, Tonacchera M, Vitti P, Chatterjee KV. Evidence for secretion of thyrotropin with enhanced bioactivity in syndromes of thyroid hormone resistance. J Clin Endocrinol Metab 1994;78:1034-9.
- 39. Safer JD, O'Connor MG, Colan SD, Srinivasan S, Tollin SR, Wondisford FE. The thyroid hormone receptor-ß gene mutation R383H is associated with isolated central resistance to thyroid hormone. J Clin Endocrinol Metab 1999;84:3099-109.
- 40. Adams M, Matthews C, Collingwood TN, Tone Y, Beck-Peccoz P, Chatterjee KV. Genetic analysis of 29 kindreds with generalized and pituitary resistance to thyroid hormone. J Clin Invest 1994;94:506-15.
- 41. Brucker-Davis I, Skarulis MC, Grace MB, Benichou J, Hauser P, Wiggs E, et al. Genetic and clinical features of 42 kindreds with resistance to thyroid hormone: the National Institutes of Health prospective study. Ann Intern Med 1995;123:572-83.
- 42. Weiss RE, Stein MA, Trommer B, Refetoff S. Attention-deficit hyperactivity disorder and thyroid function. J Pediatr 1993;123:539-45.
- 43. Bantle JP, Seeling S, Mariash CN, Ulstrom RA, Oppenheimer JH. Resistance to thyroid hormone a disorder frequently confused with Graves´ disease. Arch Intern Med 1982;142:1867-71.
- 44. Lamberg BA, Liewendahl K. Thyroid hormone resistance. Ann Clin Res 1980;12:243-53.
- 45. Sarne DH, Sobieszczyk S, Ain KB, Refetoff S. Serum thyrotropin and prolactin in the syndrome of generalized resistance to thyroid hormone: responses to thyrotropin-releasing hormone stimulation and triiodothyronine suppression. J Clin Endocrinol Metab 1990;70:1305-11.
- 46. Refetoff S, Salazar A, Smith TJ, Scherberg NH. The consequences of inappropriate treatment due to failure to recognize the syndrome of pituitary and peripheral tissue resistance to thyroid hormone. Metabolism 1983;32:822-34.
- 47. Weiss RE. Management of resistance to thyroid hormone. Thyroid Today 1999;22:1-11.
- 48. Weiss RE, Stein MA, Refetoff S. Behavioral effects of liothyronine (L-T3) in children with attention deficit hyperactivity disorder in the presence and absence of resistance to thyroid hormone. Thyroid 1997;7:389-93.
- 49. Radetti G, Persani L, Molinaro G, Mannavola D, Cortelazzi D, Chatterjee VK, et al. Clinical and hormonal outcome after two years of triiodothyroacetic acid treatment in a child with thyroid hormone resistance. Thyroid 1997;7:775-8.
- 50. Takeda T, Suzuki S, Liu RT, DeGroot LJ. Triiodothyroacetic acid has a unique potential for therapy of resistance to thyroid hormone. J Clin Endocrinol Metab 1995;80:2033-40.
- 51. Asteria C, Rajanayagam O, Collingwood TN, Persani L, Romoli R, Mannavola D, et al. Prenatal diagnosis of thyroid hormone resistance. J Clin Endocrinol Metab 1999,84:405-10.
- 52. Grover GJ, Mellstrom K, Ye L, Malm J, Li YL, Bladh LG, et al. Selective thyroid hormone receptor-beta activation: a strategy for reduction of weight, cholesterol, and lipoprotein(a) with reduced cardiovascular liability. Proc Natl Acad Sci USA 2003;100:10067-72.
- 53. Kane S, Fox R, Close C. Thyroid hormone resistance and pregnancy. BJOG 2003;110:633-4.
- 54. Refetoff S, Weiss RE. Molecular basis of the resistance to thyroid hormone. In: Thakker RV, editor. Molecular genetics of endocrine disorders 1st ed. London: Chapman & Hall Medical, 1997;85-122.
Endereço para correspondência
Datas de Publicação
-
Publicação nesta coleção
31 Maio 2004 -
Data do Fascículo
Fev 2004
Histórico
-
Recebido
17 Out 2003 -
Aceito
15 Nov 2003