ABSTRACT
The disease and the case reported here are relevant especially because of their varied clinical presentation, possibility of being associated with other disorders affecting several organs and possible differential diagnoses. Congenital Hepatic Fibrosis is an autosomal recessive disease due to mutation in the PKHD1 gene, which encodes the fibrocystin/polyductine protein. It is a cholangiopathy, characterized by varying degrees of periportal fibrosis and irregular proliferation of bile ducts. Affected patients are typically diagnosed in childhood, but in some cases the disease may remain asymptomatic for many years. The exact prevalence and incidence of the disease are not known, but it is consider a rare disease, with a few hundred cases described worldwide. It can affect all ethnic groups and occur associated with various hereditary and non-hereditary disorders. The clinical presentation is quite variable, with melena and hematemesis being initial symptoms in 30%-70% of the cases. More rarely, they may present episodes of cholangitis. The disease has been classified into four types: portal hypertension, cholestasis / cholangitis, mixed and latent. Diagnosis begins with imaging tests, but the definition is made by the histopathological sample. So far, there is no specific therapy that can stop or reverse the pathological process. Currently, the therapeutic strategy is to treat the complications of the disease.
HEADINGS:
Liver cirrhosis; Polycystic kidney diseases; Portal hypertension
RESUMO
A patologia e o caso aqui reportados são relevantes especialmente devido sua variada apresentação clínica, possibilidade de estar associada com outras desordens acometendo diversos órgãos e pelos possíveis diagnósticos diferenciais. A fibrose hepática congênita é uma doença autossômica recessiva, devido mutação no gene PKHD1, que codifica a proteína fibrocistina/poliductina. É uma colangiopatia, caracterizada por variados graus de fibrose periportal e proliferação irregular de ductos biliares. Os pacientes acometidos são tipicamente diagnosticados na infância, mas em alguns casos a doença pode permanecer assintomática por muitos anos. Exatas prevalência e incidência da doença não são conhecidas, mas sabe-se que é uma doença bastante rara, com algumas centenas de casos descritos no mundo. Pode afetar todos grupos étnicos e ocorrer associada com diversas desordens hereditárias e não-hereditárias. A apresentação clínica é bastante variável, com melena e hematêmese sendo sintomas iniciais em 30%-70% dos casos. Mais raramente, podem apresentar episódios de colangite. A doença tem sido classificada em quatro tipos: hipertensão portal, colestática/colangite, mista e latente. O diagnóstico inicia com exames de imagem, mas a definição é feita pela amostra histopatológica. Até o momento, não há terapia específica que possa parar ou reverter o processo patológico e a estratégia terapêutica atual é tratar as complicações da doença.
DESCRITORES:
Cirrose hepática; Doenças renais policísticas; Hipertensão portal
REPORT
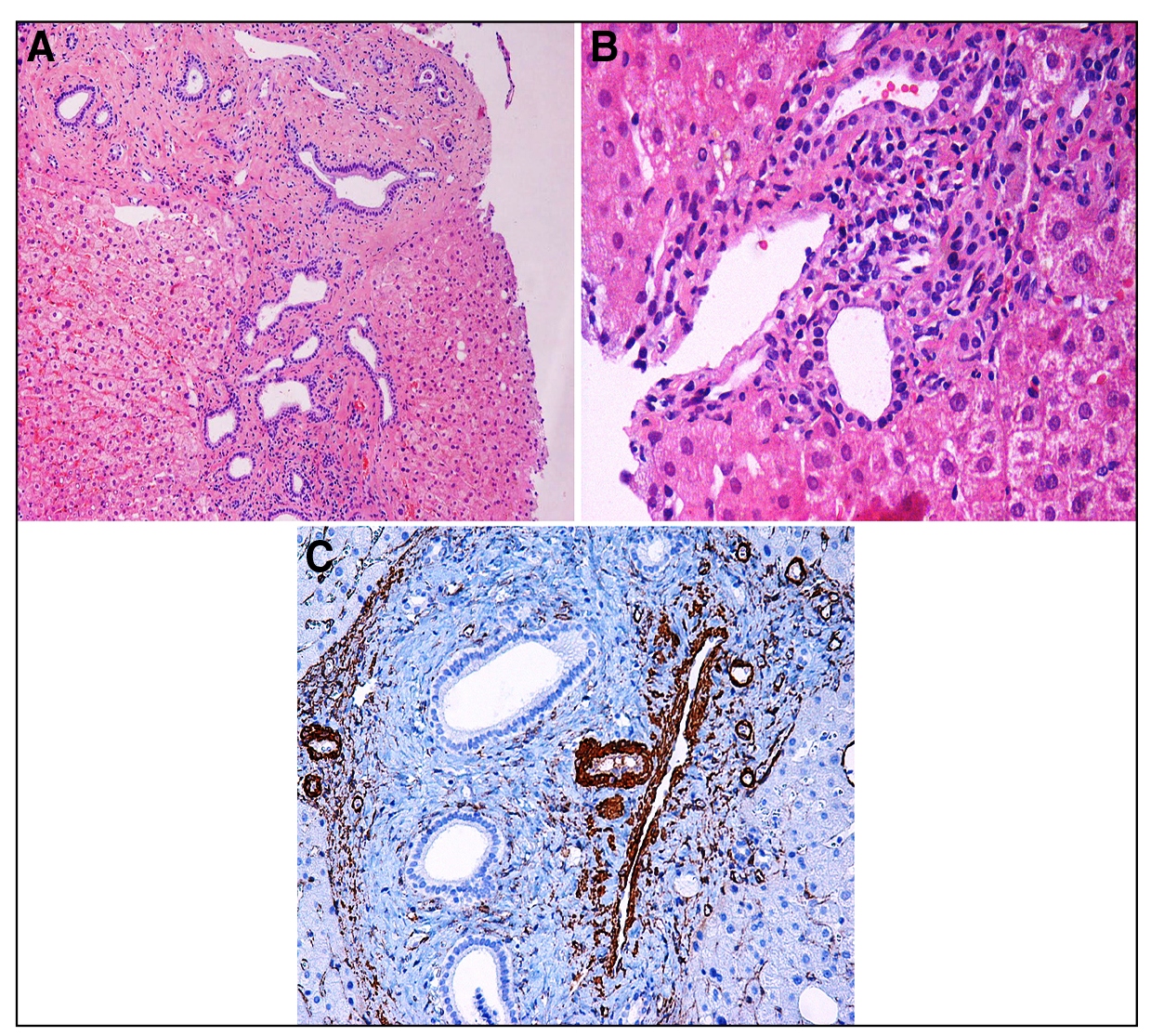
Male patient, 47 years-old, underwent an abdominal tomography after an automotive accident, when was seen a liver with irregular borders, heterogeneous density and caudate lobe hypertrophy, all findings suggestive of chronic liver disease. Thence, the patient sought specialized medical support and reported he has low platelets value. He denied previous episodes of upper gastrointestinal bleeding, ascites, encephalopathy or any other complication attributable to cirrhosis. As a previous history, he reported follow up with an urologist due to kidney stones and mild dyslipidemia. There is no history of alcohol abuse. In the family there was no reported liver disease. He was taking fibrate to treat dyslipidemia. On physical examination he had no stigmas of liver disease. The complementary tests are described in Table 1 and Figure 1. Serological tests for viral hepatitis were negative, as were autoantibodies, except for the antinuclear antibody that showed a titration of 1:320, with a speckled pattern. Serum levels of alpha-1-antitrypsin, ceruloplasmin and ferritin were within normal range. With intention to clarify diagnosis, the patient underwent liver biopsy (Figures 2 and 3). The main findings were dense portal fibrosis, sometimes leading to portal-portal septa, but without formation of portal-central septa or nodules (Figures 2A and 2B). In these portal spaces were found malformed ductal structures, reminiscent of derangement of the ductal plate (Figure 3A). Supporting the elevations of aminotransferases and especially the higher levels of gamma glutamyl transferase (GGT), several bile ducts were surrounded or even permeated by inflammatory infiltrate with lymphocytes, macrophages and some polymorphonuclear cells (Figure 3B). Attention was also drawn to the obliteration of the portal venous branch, and sometimes there were several smaller caliber veins surrounding the main obliterated vein (Figures 2B and 3C). The diagnostic conclusion was portal fibrosis with evidence of ductal plate malformation, characterizing congenital hepatic fibrosis with cholangitis component. At this moment, the patient is doing well and is has not been treated with any specific medication for congenital hepatic fibrosis.

Magnetic Resonance images of the liver - Axial T2 HASTE (A), Coronal T2 HASTE (B), Axial T1 3D GRE pre-contrast (C) and in the late hepatic arterial phase post-gadolinium (D), Coronal (E) and Axial (F) T1 3D GRE in the venous phase post-gadolinium, show liver with irregular borders, heterogeneous signal intensity and hypertrophy of the caudate lobe, findings consistent with chronic liver disease. HASTE: half-Fourier acquisition Single-Shot Turbo Spin-Echo; GRE: Gradient-Echo.

Anatomopathological examination. Histopathological aspects, evidenced by the red picrosirius histochemical reaction, highlighting portal fibrosis. A) Portal space expanded by dense fibrosis, highlighting the presence of small ductal structures beyond the original bile duct, located in the center of the figure. The portal venous branch is not evident (original x100). B) Portal space greatly enlarged by dense fibrosis, highlighting the presence of numerous bile ducts. The portal venous branch is elongated, with reduced lumen. In the lower face of the portal space, a branch of hepatic vein (centrolobular) is observed (original x100).

Anatomopathological examination. A) the hematoxylin-eosin staining shows the irregular contours of the various bile ducts, indicating ductal plaque remodeling disorders (he, original x100). B) mixed inflammatory infiltrate, variable in the portal spaces, is moderate in this figure, surrounding or even permeating the ductal epithelium (he, original x200). C) the immunohistochemical reactions for the detection of actin in the vessel wall demonstrate the obliteration of the portal vein branch, which is surrounded by several small venous branches (ihc, ac 1 to 4, original x100).
DISCUSSION
Congenital hepatic fibrosis (CHF) is an autosomal recessive disease11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.,22. Locatelli L, Cadamuro M, Spirli C, Fiorotto R, Lecchi S, Morell CM, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in Congenital Hepatic Fibrosis. Hepatology. 2016;63(3)., due to a mutation on PKHD1, a gene encoding fibrocystin/polyductine11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.,22. Locatelli L, Cadamuro M, Spirli C, Fiorotto R, Lecchi S, Morell CM, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in Congenital Hepatic Fibrosis. Hepatology. 2016;63(3).,33. Gunay-Aygun M, Font-Montegomery E, Lukose L, Gerstein MT, Piwnica-Worms K, Choyke P, et al. Characteristics of congenital Hepatic Fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. 2013;144:112-21., a ciliary protein expressed in cholangiocytes. CHF is defined by varying degrees of periportal fibrosis and irregular proliferation of bile ducts11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.,44. Geramizadeh B, Bahador PK, Salahi H, Nikeghbalaian S, Dehghani SM, Malek-Hosseini SA. Congenital Hepatic Fibrosis and need for liver transplantation. Int J Organ Transplant Med. 2010;1:98-100.,55. Rajekar H, Vasishta R, Chawla YK, Dhiman RK. Noncirrhotic Portal Hypertension. J Clin Exp Hepatol. 2011;1:94-108., being part of the so-called Fibropolycystic Diseases, which also include Caroli’s Disease66. Wen J. Congenital Hepatic fibrosis in autosomal recessive polycystic kidney diesease. Clin Transl Sci. 2011;4:460-5., Autosomal Dominant Renal Disease, Autosomal Recessive Polycystic Kidney Disease, Von Meyenburg Complex (bile duct hamartoma), and choledochal cyst55. Rajekar H, Vasishta R, Chawla YK, Dhiman RK. Noncirrhotic Portal Hypertension. J Clin Exp Hepatol. 2011;1:94-108.,77. Kwon JH, Kim MJ, Kim YH, Kang KJ, Kang YN, Kwon SY. Monosegmental Hepatobiliary Fibropolycystic Disease Mimicking a Mass: Report of Three Cases. Korean J Radiol 2014;15:54-60.,88. Pawar S, Zanwar V, Mohite A, Surude R, Rathi P, Balasubramani M. A family of congenital hepatic fibrosis and atypical retinitis pigmentosa. Clinics and Pratice. 2015;5:792.. Typically, CHF patients have been diagnosed in infancy or early childhood, but some recent data demonstrate that some patients remain asymptomatic for long periods44. Geramizadeh B, Bahador PK, Salahi H, Nikeghbalaian S, Dehghani SM, Malek-Hosseini SA. Congenital Hepatic Fibrosis and need for liver transplantation. Int J Organ Transplant Med. 2010;1:98-100., as the presented case here. The incidence and prevalence of CHF are not exactly known, but it is known to be a rare disease, with only a few hundred cases described in the literature99. Poala SB, Bisogon G, Colombatti R. Thrombocytopenia and splenomegaly: an unusual presentation of Congenital Hepatic Fibrosis. Orphanet J Rare Dis. 2010;5:4.. The prevalence of congenital hepatic fibrosis in portal hypertensive children in a study from north India was 3%77. Kwon JH, Kim MJ, Kim YH, Kang KJ, Kang YN, Kwon SY. Monosegmental Hepatobiliary Fibropolycystic Disease Mimicking a Mass: Report of Three Cases. Korean J Radiol 2014;15:54-60.. It is estimated that, when CHF occurs with Autosomal Recessive Polycystic Kidney Disease, the frequency is 1 in 20,000 live births, affecting all ethnic groups88. Pawar S, Zanwar V, Mohite A, Surude R, Rathi P, Balasubramani M. A family of congenital hepatic fibrosis and atypical retinitis pigmentosa. Clinics and Pratice. 2015;5:792.. The disease may occur associated with other hereditary and non-hereditary diseases, with the most frequent associations being: Joubert Syndrome and Bardet-Biedl Syndrome1010. Yönem O, Ozkayar N, Balkanci F, Harmanci O, Sökmensüer C, Ersoy O, Bayraktar Y. Is congenital hepatic fibrosis a pure liver disease? Am J Gastroenterol. 2006;101:1253-9.. The potential for involving several organs, requiring several medical specialties in differential diagnosis and treatment, reinforces the importance of this review article.
Pathophysiology
CHF is a genetic cholangiopathy caused by mutations in PKHD1, the gene located on chromosome 6p21-231111. Masyuk T, Larusso N. Policistic liver disease: New insights into disease pathogenesis. Hepatology. 2006;43(5)., which encodes fibrocystin/polyductine (FPC), a protein of yet unknown function, expressed in the cilia and centromers of the bile ducts and renal tubule cells22. Locatelli L, Cadamuro M, Spirli C, Fiorotto R, Lecchi S, Morell CM, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in Congenital Hepatic Fibrosis. Hepatology. 2016;63(3).. In others chronic hepatobiliary diseases, progression to fibrosis is due to a repair response against necroinflammatory damage in hepatocytes and/or cholangiocytes. In CHF, the essential pathological processes appear to be genetically based, leading to ductal plate malformation, possibly beginning with maturation arrest and lack of ductal plate remodeling77. Kwon JH, Kim MJ, Kim YH, Kang KJ, Kang YN, Kwon SY. Monosegmental Hepatobiliary Fibropolycystic Disease Mimicking a Mass: Report of Three Cases. Korean J Radiol 2014;15:54-60.. The ductal plate is an embryological structure formed by a cylindrical layer of epithelial cells that involve a branch of the portal vein11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90. and is the embryological precursor of intrahepatic bile ducts1212. Strazzabosco M, Fabris L. Development of the bile ducts: Essentials for the clinical hepatologist. J Hepatol. 2012;56:1159-70.. As a result of this defect in remodeling the ductal plate, a persistence of immature embryological ductile structures occurs, which lead to the formation of portal fibrosis tissues. This portal fibrosis would ultimately lead to recurrent cholangitis or portal hypertension and associated symptoms11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.. Interestingly, our reported patient had no history of symptoms and complications at the time of diagnosis.
Although it is well established that chronic portal hypertension can lead to portal vein thrombosis with eventual cavernomatous transformation (CTPV)1313. Yusuf B. Portal ductopathy: clinical importance and nomenclature. World J Gastroenterol . 2011;17:1410-5., it is believed that in the case of the CHF the lesions of the portal vein branches are a component of the disease, present from the beginning. The main support for this theory is based on the embryological relationship of the hepatic vessels and ducts1212. Strazzabosco M, Fabris L. Development of the bile ducts: Essentials for the clinical hepatologist. J Hepatol. 2012;56:1159-70.. Malformation of the ductal plate has a strong association with malformation of the portal vein, resulting in excessive small and branched branches of the portal vein11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.,1414. Yonem O, Bayraktar Y. Is portal vein cavernous transformation a component of congenital hepatic fibrosis? World J Gastroenterol . 2007;13:1928-9.. A study from Barayktar et al. showed that almost 50% of CHF patients present CTPV and that patients with CTPV presented more prominent splenomegaly and a greater chance of varicose digestive hemorrhage1515. Bayraktar Y, Balkanci F, Kayhan B, Uzunalimoglu B, Ozenc A, Ozdemir A, Dündar S, Arslan S, Sivri B, Telatar H. Congenital hepatic fibrosis associated with cavernous transformation of the portal vein. Hepatogastroenterology. 1997;44:1588-94.. Our patient also had no signs of CTPV.
The maturation stage of the ductal plate arrest would determine the final disease: either it involves the small interlobular bile ducts leading to CHF or involves the middle intrahepatic bile ducts causing Caroli’s Disease55. Rajekar H, Vasishta R, Chawla YK, Dhiman RK. Noncirrhotic Portal Hypertension. J Clin Exp Hepatol. 2011;1:94-108.. The occurrence of simultaneous involvement of the two types of bile ducts (small and medium) results in Caroli Syndrome (FHC associated with Caroli Disease). In these cases, the clinic of Caroli’s disease (recurrent cholangitis episodes) will be predominant, with CHF often underdiagnosed. Thus, hepatic biopsy becomes essential for differential diagnosis between Caroli’s Disease or Syndrome11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90..
In a more recent study, Locatelli et al. described that FPC deficiency would lead to the formation of microhamartomas and segmental dilations of the bile ducts with portal hypertension. Other studies have also identified various defects in intracellular signaling of FPC-deficient cells, including increased adenosine cyclic monophosphate (cAMP)/protein kinase A signaling and activation of β-catenin-dependent protein kinase A1616. Spirli C, Locatelli L, Morell C, Fiorotto R, Morton SD, Cadamuro M, Fabris L, Strazzabosco M. Protein Kinase A-dependent pSer 675 -Beta-catenin, a Novel Signaling in a mouse model of Congenital Hepatic Fibrosis. Hepatology 2013;58:1713-23.. This, in turn, has appeared as a new regulator of inflammation, capable of influencing secretion of cytokines in liver cancer in experimental models. Hence, it has been proposed that persistent and unresolved cellular dysfunction may promote a chronic and low-grade inflammatory response and may lead to scarring (called “parainflammation”)22. Locatelli L, Cadamuro M, Spirli C, Fiorotto R, Lecchi S, Morell CM, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in Congenital Hepatic Fibrosis. Hepatology. 2016;63(3).. In this same study, the author showed evidence that this chronic and mild inflammatory response, generated by FPC-deficient cholangiocytes, secretes chemokines capable of recruiting macrophages derived from bone marrow, and responds to them through the activation of αvβ6 integrin, a local activator of TGFβ1. This theory proposes a new paradigm of biliary fibrosis and would be a model for understanding the relationship between cellular dysfunction, inflammation, hepatic fibrosis and macrophage polarization over time. This theory, however, contradicts the previously mentioned that the process of hepatic fibrosis in the CHF would occur without the presence of chronic inflammation.
Other authors have demonstrated and established that TGFβ is a potent growth inhibitory factor and a pro-fibrotic cytokine, with a central role in the physiological healing process as well as in the pathogenesis of organ fibrosis. The onset of activation of hepatic stellate cells (which are also at the center of the liver fibrosis process)1717. Alcolado R, Arthur MJ, Iredale JP. Pathogenesis of liver fibrosis. Clin Sci (Lond). 1997;92:103-12.,1818. Rockey DC. Current and future anti-fibrotic therapies for chronic liver disease. Clin Liver Dis. 2008;12:939-62, xi. is primarily induced by TGFβ1 derived from Kupfer cells11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.. TGFβ1 would mediate its pro-fibrotic action through stimulation of fibroblasts and related cell types, including stellate cells, to secrete various extracellular matrix proteins1919. Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med 1994;331:1286-92.. Therefore, in pathological situations would lead to the accumulation of fibrotic matrix and in physiological situations would lead to efficient wound healing11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.. There are other studies that intend to establish the pathophysiological mechanism behind this abnormal and excessive fibrotic response associated with CHF, in which the possible involved ones would be plasminogen and tissue plasminogen activator (tPA), osteopontin and microRNA11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.,2020. Chu AS, Friedman JR. A role for microRNA in cystic liver and kidney diseases. J Clin Invest. 2008;118:3585-7..
Clinical course
The onset of presentation and severity of symptoms is very variable. The majority manifests even in childhood77. Kwon JH, Kim MJ, Kim YH, Kang KJ, Kang YN, Kwon SY. Monosegmental Hepatobiliary Fibropolycystic Disease Mimicking a Mass: Report of Three Cases. Korean J Radiol 2014;15:54-60.,2121. Ghadir MR, Bagheri M, Ghanoon AH. Congenital Hepatic Fibrosis leading to cirrhosis and hepatocelular carcinoma: a case report. J Med Case Reports. 2011;5:160.. In most patients, the first manifestations of the disease are signs and symptoms related to portal hypertension, often with gastrointestinal bleeding55. Rajekar H, Vasishta R, Chawla YK, Dhiman RK. Noncirrhotic Portal Hypertension. J Clin Exp Hepatol. 2011;1:94-108.. Hemathemesis and melena are initial symptoms in 30%-70% of cases99. Poala SB, Bisogon G, Colombatti R. Thrombocytopenia and splenomegaly: an unusual presentation of Congenital Hepatic Fibrosis. Orphanet J Rare Dis. 2010;5:4.. More rarely, they may present episodes of cholangitis. Depending on the associated diseases, patients will have signs and symptoms related to other organs such as kidneys and central nervous system (CNS)11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.. These patients classically do not have cirrhosis, maintain normal hepatic lobular architecture and liver function2121. Ghadir MR, Bagheri M, Ghanoon AH. Congenital Hepatic Fibrosis leading to cirrhosis and hepatocelular carcinoma: a case report. J Med Case Reports. 2011;5:160..
CHF is classified into 4 types55. Rajekar H, Vasishta R, Chawla YK, Dhiman RK. Noncirrhotic Portal Hypertension. J Clin Exp Hepatol. 2011;1:94-108.,77. Kwon JH, Kim MJ, Kim YH, Kang KJ, Kang YN, Kwon SY. Monosegmental Hepatobiliary Fibropolycystic Disease Mimicking a Mass: Report of Three Cases. Korean J Radiol 2014;15:54-60.,99. Poala SB, Bisogon G, Colombatti R. Thrombocytopenia and splenomegaly: an unusual presentation of Congenital Hepatic Fibrosis. Orphanet J Rare Dis. 2010;5:4.:
-
Portal hypertension
-
Cholestatic / Cholangitis
-
Mixed
-
Latent
The reported case probably has the latent form. The portal hypertension type of CHF is the most common and becomes more severe in the presence of portal vein abnormality. Latent type presents in old age or is an incidental finding, as do our case. On physical examination the patient may present with hepatomegaly, especially of the left lobe, splenomegaly and nephromegaly. The liver is firm in consistency and has a slightly nodular surface. In laboratory tests, there is a slight elevation of liver enzymes, where in cholestatic conditions predominate elevations in alkaline phosphatase, gamma-glutamyltransferase and bilirubin. Cytopenias may occur as a consequence of splenomegaly and this is probably the cause of low levels of platelets in our patient. Changes in renal function tests occur when there is CHF associated with extensive cystic kidney disease, which may progress to end-stage renal failure. When kidney and liver disease are combined, the severity of each is independent33. Gunay-Aygun M, Font-Montegomery E, Lukose L, Gerstein MT, Piwnica-Worms K, Choyke P, et al. Characteristics of congenital Hepatic Fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. 2013;144:112-21..
Diagnosis
Ultrasound is usually the first method employed due to availability, low cost and harmlessness. It is able to detect alterations in the bile ducts and hepatic parenchyma, besides renal alterations. The most common findings are hypertrophy of the left lateral and caudate hepatic segments, right segment atrophy, splenomegaly, intra- and extrahepatic biliary dilation with concomitant cystic and solid lesions (regeneration nodules), periportal thickening, hepatic and renal cysts, and CTPV.
Computed tomography (CT) has advantages over ultrasound by better morphological representation, with accurate volumetric measurements, as well as by adequately showing the vessels and biliary tree. However, nuclear magnetic resonance imaging (MRI) with cholangioresonance is an important diagnostic alternative because it does not expose the patient to radiation and allows a very detailed evaluation of the biliary tree.
Anatomopathological examination, interpreted in the clinical-radiological context, is of great importance for a diagnostic definition of CHF. It is possible various degrees of fibrosis with nodular formations, and when diagnosed late, clinical and even histological findings may be confused with liver cirrhosis11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.. In addition, large fibrous bands are seen in the portal tract, with an increased number of proliferated irregular bile ducts with normal cubic epithelium. These findings were seen in our patient’s liver biopsy, supporting the diagnosis. Hepatic lobes are normal with hepatocytes of usual morphology, especially in the early stages of the disease.
Among the major complications, signs of cholestasis and even recurrent cholangitis can be seen. The development of cholangiocarcinoma is another possible complication. When CHF is associated with Caroli’s disease, cholangiocarcinoma occurs in 2.5%-16% of cases11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.. On the other hand, in cases of isolated CHF, there are few reports of cholangiocarcinoma2222. Daroca PJ Jr, Tuthill R, Reed RJ. Cholangiocarcinoma arising in congenital hepatic fibrosis. A case report. Arch Pathol. 1975;99:592-5.,2323. Yamato T, Sasaki M, Hoso M, Sakai J, Ohta H, Watanabe Y, Nakanuma Y. Intrahepatic cholangiocarcinoma arising in congenital hepatic fibrosis: report of an autopsy case. J Hepatol . 1998;28:717-22.. It is suggested that malignant transformation via dysplasia occurs in abnormal intrahepatic bile trees in older patients with CHF2323. Yamato T, Sasaki M, Hoso M, Sakai J, Ohta H, Watanabe Y, Nakanuma Y. Intrahepatic cholangiocarcinoma arising in congenital hepatic fibrosis: report of an autopsy case. J Hepatol . 1998;28:717-22.. There are also some cases of hepatocellular carcinoma associated with CHF reported in the literature2121. Ghadir MR, Bagheri M, Ghanoon AH. Congenital Hepatic Fibrosis leading to cirrhosis and hepatocelular carcinoma: a case report. J Med Case Reports. 2011;5:160..
Treatment
To date, there is no specific therapy capable of stopping or reversing the pathological process in CHF11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.,1818. Rockey DC. Current and future anti-fibrotic therapies for chronic liver disease. Clin Liver Dis. 2008;12:939-62, xi.. When considering the antifibrotic treatment, it is necessary to remember that fibrosis is a dynamic process in which the degradation of the extracellular matrix, and not only the matrix formation, is important1818. Rockey DC. Current and future anti-fibrotic therapies for chronic liver disease. Clin Liver Dis. 2008;12:939-62, xi.. Moreover, many studies in the literature emphasize that liver fibrosis is a reversible process2424. Debernardi-Venon W, Martini S, Biasi F, Vizio B, Termine A, Poli G, Brunello F, Alessandria C, Bonardi R, Saracco G, Rizzetto M, Marzano A. AT1 receptor antagonist Candesartan in selected cirrhotic patients: effect on portal pressure and liver fibrosis markers. J Hepatol . 2007;46:1026-33.. Different researchers try to find a drug that can control the progression of the disease, stopping and even reversing fibrosis. Some drugs have shown success in animal studies1818. Rockey DC. Current and future anti-fibrotic therapies for chronic liver disease. Clin Liver Dis. 2008;12:939-62, xi., but have failed to present benefits in humans, including colchicine, angiotensin II blocker, interferon gamma and pirfenidone11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.,1818. Rockey DC. Current and future anti-fibrotic therapies for chronic liver disease. Clin Liver Dis. 2008;12:939-62, xi.,2424. Debernardi-Venon W, Martini S, Biasi F, Vizio B, Termine A, Poli G, Brunello F, Alessandria C, Bonardi R, Saracco G, Rizzetto M, Marzano A. AT1 receptor antagonist Candesartan in selected cirrhotic patients: effect on portal pressure and liver fibrosis markers. J Hepatol . 2007;46:1026-33.,2525. Armendáriz-Borunda J, Islas-Carbajal MC, Meza-García E, Rincón AR, Lucano S, Sandoval AS, Salazar A, Berumen J, Alvarez A, Covarrubias A, Aréchiga G, García L. A pilot study in patients with established advanced liver fibrosis using pirfenidone. Gut. 2006;55:1663-5.. Therefore, the therapeutic strategy for CHF, so far, is to treat the complications of the disease55. Rajekar H, Vasishta R, Chawla YK, Dhiman RK. Noncirrhotic Portal Hypertension. J Clin Exp Hepatol. 2011;1:94-108..
Endoscopy is an important option for primary and secondary prophylaxis of bleeding esophageal and gastric varices, as well as for treatment of acute bleeding2626. de Francis R, Baveno VI Faculty. Expanding consensus in portal hypertension - Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J Hepatol . 2015;63:743-52.. Likewise, it plays an important role in the management of recurrent cholangitis (especially in Caroli syndrome)11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.,55. Rajekar H, Vasishta R, Chawla YK, Dhiman RK. Noncirrhotic Portal Hypertension. J Clin Exp Hepatol. 2011;1:94-108..
Transjugular portosystemic intrahepatic shunt (TIPS) is indicated in cases not eligible for endoscopic sclerotherapy and also in cases of refractory upper gastrointestinal bleeding, such as a bridge to liver transplantation.
Surgical shunts for portal decompression are indicated in cases of upper gastrointestinal bleeding not amenable to upper endoscopy. In cases of CHF associated with Caroli’s disease with recurrent cholangitis, partial hepatic resection may be indicated in cases of extensive heterogeneous involvement11. Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.,55. Rajekar H, Vasishta R, Chawla YK, Dhiman RK. Noncirrhotic Portal Hypertension. J Clin Exp Hepatol. 2011;1:94-108..
Hepatic transplantation is the only curative treatment for CHF. It is indicated in the advanced stages of the disease, with development of hepatic insufficiency, or in cases of recurrent cholangitis or malignant transformation2727. De Kerckhove L, De Meyer M, Verbaandert C, Mourad M, Sokal E, Goffette P, et al. The place of liver transplantation in Caroli’s disease and syndrome. Transpl Int. 2006;19:381-8.. The vast majority of cases of CHF who underwent liver transplantation, described in the literature, had Caroli’s disease associated with recurrent cholangitis. On the other hand, Geramizadeh et al., in 2010, reported two cases of CHF patients who required hepatic transplantation due to hepatic insufficiency, without previous or current history of cholangitis44. Geramizadeh B, Bahador PK, Salahi H, Nikeghbalaian S, Dehghani SM, Malek-Hosseini SA. Congenital Hepatic Fibrosis and need for liver transplantation. Int J Organ Transplant Med. 2010;1:98-100.. In cases of associated renal and hepatic diseases, combined kidney and liver transplantation may be necessary. In addition, one study showed that in patients with liver and kidney disease who underwent only liver transplantation, there was an improvement in renal function2828. Arikan C, Ozgenc F, Akman SA, Kilic M, Tokat Y, Yagci RV, Aydogdu S. Impact of liver transplantation on renal function of patients with congenital hepatic fibrosis associated with autosomal recessive polycystic kidney disease. Pediatr Transplantation. 2004;8:558-60..
CONCLUSION
The case reported herein corresponds to a form that is practically asymptomatic, but has a well-defined histological pattern of Congenital Hepatic Fibrosis (CHF), a rare or rarely diagnosed disease. The clinical spectrum of the CHF is very broad, and it can affect several organs, being a disease to be considered associated with other conditions, especially the kidney fibropolycystic disease.
REFERENCES
-
1Shorbagi A, Bayraktar Y. Experience of a single center with congenital hepatic fibrosis: A review of literature. World J Gastroenterol 2010;16:683-90.
-
2Locatelli L, Cadamuro M, Spirli C, Fiorotto R, Lecchi S, Morell CM, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in Congenital Hepatic Fibrosis. Hepatology. 2016;63(3).
-
3Gunay-Aygun M, Font-Montegomery E, Lukose L, Gerstein MT, Piwnica-Worms K, Choyke P, et al. Characteristics of congenital Hepatic Fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. 2013;144:112-21.
-
4Geramizadeh B, Bahador PK, Salahi H, Nikeghbalaian S, Dehghani SM, Malek-Hosseini SA. Congenital Hepatic Fibrosis and need for liver transplantation. Int J Organ Transplant Med. 2010;1:98-100.
-
5Rajekar H, Vasishta R, Chawla YK, Dhiman RK. Noncirrhotic Portal Hypertension. J Clin Exp Hepatol. 2011;1:94-108.
-
6Wen J. Congenital Hepatic fibrosis in autosomal recessive polycystic kidney diesease. Clin Transl Sci. 2011;4:460-5.
-
7Kwon JH, Kim MJ, Kim YH, Kang KJ, Kang YN, Kwon SY. Monosegmental Hepatobiliary Fibropolycystic Disease Mimicking a Mass: Report of Three Cases. Korean J Radiol 2014;15:54-60.
-
8Pawar S, Zanwar V, Mohite A, Surude R, Rathi P, Balasubramani M. A family of congenital hepatic fibrosis and atypical retinitis pigmentosa. Clinics and Pratice. 2015;5:792.
-
9Poala SB, Bisogon G, Colombatti R. Thrombocytopenia and splenomegaly: an unusual presentation of Congenital Hepatic Fibrosis. Orphanet J Rare Dis. 2010;5:4.
-
10Yönem O, Ozkayar N, Balkanci F, Harmanci O, Sökmensüer C, Ersoy O, Bayraktar Y. Is congenital hepatic fibrosis a pure liver disease? Am J Gastroenterol. 2006;101:1253-9.
-
11Masyuk T, Larusso N. Policistic liver disease: New insights into disease pathogenesis. Hepatology. 2006;43(5).
-
12Strazzabosco M, Fabris L. Development of the bile ducts: Essentials for the clinical hepatologist. J Hepatol. 2012;56:1159-70.
-
13Yusuf B. Portal ductopathy: clinical importance and nomenclature. World J Gastroenterol . 2011;17:1410-5.
-
14Yonem O, Bayraktar Y. Is portal vein cavernous transformation a component of congenital hepatic fibrosis? World J Gastroenterol . 2007;13:1928-9.
-
15Bayraktar Y, Balkanci F, Kayhan B, Uzunalimoglu B, Ozenc A, Ozdemir A, Dündar S, Arslan S, Sivri B, Telatar H. Congenital hepatic fibrosis associated with cavernous transformation of the portal vein. Hepatogastroenterology. 1997;44:1588-94.
-
16Spirli C, Locatelli L, Morell C, Fiorotto R, Morton SD, Cadamuro M, Fabris L, Strazzabosco M. Protein Kinase A-dependent pSer 675 -Beta-catenin, a Novel Signaling in a mouse model of Congenital Hepatic Fibrosis. Hepatology 2013;58:1713-23.
-
17Alcolado R, Arthur MJ, Iredale JP. Pathogenesis of liver fibrosis. Clin Sci (Lond). 1997;92:103-12.
-
18Rockey DC. Current and future anti-fibrotic therapies for chronic liver disease. Clin Liver Dis. 2008;12:939-62, xi.
-
19Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med 1994;331:1286-92.
-
20Chu AS, Friedman JR. A role for microRNA in cystic liver and kidney diseases. J Clin Invest. 2008;118:3585-7.
-
21Ghadir MR, Bagheri M, Ghanoon AH. Congenital Hepatic Fibrosis leading to cirrhosis and hepatocelular carcinoma: a case report. J Med Case Reports. 2011;5:160.
-
22Daroca PJ Jr, Tuthill R, Reed RJ. Cholangiocarcinoma arising in congenital hepatic fibrosis. A case report. Arch Pathol. 1975;99:592-5.
-
23Yamato T, Sasaki M, Hoso M, Sakai J, Ohta H, Watanabe Y, Nakanuma Y. Intrahepatic cholangiocarcinoma arising in congenital hepatic fibrosis: report of an autopsy case. J Hepatol . 1998;28:717-22.
-
24Debernardi-Venon W, Martini S, Biasi F, Vizio B, Termine A, Poli G, Brunello F, Alessandria C, Bonardi R, Saracco G, Rizzetto M, Marzano A. AT1 receptor antagonist Candesartan in selected cirrhotic patients: effect on portal pressure and liver fibrosis markers. J Hepatol . 2007;46:1026-33.
-
25Armendáriz-Borunda J, Islas-Carbajal MC, Meza-García E, Rincón AR, Lucano S, Sandoval AS, Salazar A, Berumen J, Alvarez A, Covarrubias A, Aréchiga G, García L. A pilot study in patients with established advanced liver fibrosis using pirfenidone. Gut. 2006;55:1663-5.
-
26de Francis R, Baveno VI Faculty. Expanding consensus in portal hypertension - Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J Hepatol . 2015;63:743-52.
-
27De Kerckhove L, De Meyer M, Verbaandert C, Mourad M, Sokal E, Goffette P, et al. The place of liver transplantation in Caroli’s disease and syndrome. Transpl Int. 2006;19:381-8.
-
28Arikan C, Ozgenc F, Akman SA, Kilic M, Tokat Y, Yagci RV, Aydogdu S. Impact of liver transplantation on renal function of patients with congenital hepatic fibrosis associated with autosomal recessive polycystic kidney disease. Pediatr Transplantation. 2004;8:558-60.
-
Disclosure of funding: no funding received
Publication Dates
-
Publication in this collection
Oct-Dec 2018
History
-
Received
14 Mar 2018 -
Accepted
08 Sept 2018