Resumo
A insuficiência cardíaca com fração de ejeção preservada (ICFEP) é hoje uma epidemia cardiovascular emergente, sendo identificada como o principal fenótipo observado na prática clínica. Está mais associado ao sexo feminino, idade avançada e a comorbidades como hipertensão arterial, diabetes, obesidade e doença renal crônica. A amiloidose é uma desordem clínica caracterizada pelo depósito de agregados de fibrilas insolúveis originadas de proteínas que apresentam dobramento anômalo. Recentemente, têm sido descritos quadros de amiloidose senil em pacientes com ICFEP, demonstrando a necessidade de os cardiologistas clínicos investigarem esta etiologia em casos suspeitos. Deve-se aumentar a suspeição clínica de amiloidose diante dos casos de ICFEP onde os métodos de cardioimagem sejam compatíveis com o quadro de cardiomiopatia infiltrativa. Os avanços nos métodos de cardioimagem aliados à possibilidade de realização de testes genéticos e identificação do tipo do material amiloide permitem a realização do diagnóstico. O manejo dos pacientes diagnosticados pode ser feito em parceria com centros especializados no estudo de amiloidose, que, aliados às novas tecnologias, investigam a possibilidade de transplante de órgãos ou medula óssea e também o envolvimento dos pacientes em estudos clínicos que avaliam a ação das novas drogas emergentes.
Palavras-chave:
Amiloidose; Insuficiência Cardíaca Diastólica; Volume Sistólico; Cardiomiopatia Restritiva; Fatores de Risco
Abstract
Heart failure with preserved ejection fraction (HFpEF) is now an emerging cardiovascular epidemic, being identified as the main phenotype observed in clinical practice. It is more associated with female gender, advanced age and comorbidities such as hypertension, diabetes, obesity and chronic kidney disease. Amyloidosis is a clinical disorder characterized by the deposition of aggregates of insoluble fibrils originating from proteins that exhibit anomalous folding. Recently, pictures of senile amyloidosis have been described in patients with HFpEF, demonstrating the need for clinical cardiologists to investigate this etiology in suspect cases. The clinical suspicion of amyloidosis should be increased in cases of HFPS where the cardio imaging methods are compatible with infiltrative cardiomyopathy. Advances in cardio imaging methods combined with the possibility of performing genetic tests and identification of the type of amyloid material allow the diagnosis to be made. The management of the diagnosed patients can be done in partnership with centers specialized in the study of amyloidosis, which, together with the new technologies, investigate the possibility of organ or bone marrow transplantation and also the involvement of patients in clinical studies that evaluate the action of the new emerging drugs.
Keywords:
Amyloidosis; Heart Failure, Diastolic; Stroke Volume; Cardiomyopathy, Restrictive; Risk Factors
Introdução
A insuficiência cardíaca (IC) com fração de ejeção preservada (ICFEP) é hoje uma epidemia cardiovascular emergente, sendo identificada como o principal fenótipo observado na prática clínica em diferentes países, como Estados Unidos, Reino Unido, Portugal e Brasil. Está mais associado ao sexo feminino, idade avançada e a comorbidades como hipertensão arterial, diabetes, obesidade e doença renal crônica.11 Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355(3):251-9.
2 Morgan S, Smith H, Simpson I, Liddiard GS, Raphael H, Pickering RM, et al. Prevalence and clinical characteristics of left ventricular dysfunction among elderly patients in general practice setting: cross sectional survey. BMJ. 1999;318(7180):368-72.
3 Ceia F, Fonseca C, Mota T, Morais H, Matias F, De Sousa A, et al; EPICA Investigators. Prevalence of chronic heart failure in Southwestern Europe: The EPICA study. Eur J Heart Fail. 2002;4(4):531-9.
4 Mesquita ET, Jorge AJ. Heart failure with normal ejection fraction: new diagnostic criteria and pathophysiological advances. Arq Bras Cardiol. 2009;93(2):180-7.
5 Jorge AL, Rosa ML, Martins WA, Correia DM, Fernandes LC, Costa JA, et al. The prevalence of stages of heart failure in primary care: a population-based study. J Card Fail. 2016;22(2):153-7.-66 van Heerebeek L, Paulus WJ. Understanding heart failure with preserved ejection fraction: where are we today? Neth Heart J. 2016;24(4):227-36. Recentemente, tem sido descrito um quadro de amiloidose senil em pacientes com ICFEP, demonstrando a necessidade dos cardiologistas clínicos de investigarem esta etiologia. Os avanços nos métodos de cardioimagem aliados à possibilidade de realização de testes genéticos e identificação do tipo do material amiloide, permitem maior facilidade no processo diagnóstico diante da suspeita clínica da doença.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.
8 Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113-22.
9 Palka P, Lange A, Donnelly JE, Scalia G, Burstow DJ, Nihoyannopoulos P. Doppler tissue echocardiographic features of cardiac amyloidosis. J Am Soc Echocardiogr. 2002;15(11):1353-60.
10 Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546-54.
11 Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076-84.
12 Rapezzi C, Quarta CC, Guidalotti PL, Pettinato C, Fanti S, Leone O, et al. Role of 99mTc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging. 2011;4(6):659-70.
13 Arbustini E, Verga L, Concardi M, Palladini G, Obici L, Merlini G. Electron and immuno-electron microscopy of abdominal fat identifies and characterizes amyloid fibrils in suspected cardiac amyloidosis. Amyloid. 2002;9(2):108-14.
14 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.-1515 González-López E, Gallego-Delgado M, Guzzo-Merello G, De Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94.
A amiloidose é uma desordem clínica decorrente do depósito de agregados de fibrilas insolúveis originadas de proteínas que apresentam dobramento anômalo. Essas proteínas, em sua maioria, inicialmente solúveis e com configuração em alfa hélice, assumem a forma beta pregueada através do fenômeno de dobradura incorreta, com precipitação nos tecidos na forma de agregados fibrilares amiloides. Estes agregados apresentam a característica de se corarem pelo vermelho-congo, adquirindo uma tonalidade descrita como "maçã-verde" à luz polarizada. Através da alteração do órgão comprometido, determina inúmeras disfunções de curso irreversível, progressivo e indolente, como observado na amiloidose cardíaca.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.,1010 Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546-54.,1616 Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol. 2012;302(3):H544-52.
17 Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
18 Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.
19 Lawler PR, Bergmark BA, Laubach JP, Lakdawala NK. Having a heavy heart: approaches to infiltrative cardiomyopathy. Circulation. 2014;129(16):1703-11.
20 Rapezzi C, Lorenzini M, Longhi S, Milandri A, Gagliardi C, Bartolomei I, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117-24.
21 Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.
22 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.-2323 Roof L, Coker WJ, Lazarchick J, Kang Y. Senile transthyretin cardiac amyloidosis in patients with plasma cell dyscrasias: importance of cardiac biopsy for making the correct diagnosis. Aperito J Cell Mol Biol. 2014;1(1). pii. 102.
O acometimento cardíaco pode levar ao desenvolvimento de um modelo de IC restritiva. Os depósitos no miocárdio e nos vasos sanguíneos ocasionam disfunção diastólica, sistólica, isquemia e arritmias, sendo o retardo do diagnóstico a principal causa da redução da sobrevida destes pacientes.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.,1010 Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546-54.,1616 Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol. 2012;302(3):H544-52.
17 Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
18 Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.
19 Lawler PR, Bergmark BA, Laubach JP, Lakdawala NK. Having a heavy heart: approaches to infiltrative cardiomyopathy. Circulation. 2014;129(16):1703-11.
20 Rapezzi C, Lorenzini M, Longhi S, Milandri A, Gagliardi C, Bartolomei I, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117-24.
21 Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.
22 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.-2323 Roof L, Coker WJ, Lazarchick J, Kang Y. Senile transthyretin cardiac amyloidosis in patients with plasma cell dyscrasias: importance of cardiac biopsy for making the correct diagnosis. Aperito J Cell Mol Biol. 2014;1(1). pii. 102.
O diagnóstico da amiloidose apresenta importantes avanços não invasivos para caracterizar a sua presença e o seu tipo. No passado, o diagnóstico estava centrado na biópsia endomiocárdica corada pelo vermelho-congo. Mais recentemente, novas técnicas como o ecodopplercardiograma (ECO) com análise do strain miocárdico, cintilografia miocárdica com radioisotopos como o Tc 99m ligado ao pirofosfato ou ao 2,3-dicarboxipropano-1,1-difosfonato (DPD) e a ressonância magnética, além de testes sanguíneos para avaliação da genotipagem têm promovido importantes avanços nessa área.1111 Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076-84.,1212 Rapezzi C, Quarta CC, Guidalotti PL, Pettinato C, Fanti S, Leone O, et al. Role of 99mTc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging. 2011;4(6):659-70.
Novos tratamentos direcionados para alvos específicos da doença já foram incorporados à prática clínica e outros ainda estão sendo testados, gradativamente melhorando a sobrevida e a qualidade de vida dos pacientes.2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.,2525 Marcoux J, Mangione PP, Porcari R, Degiacomi MT, Verona G, Taylor GW, et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015;7(10):1337-49.
Segundo dados obtidos no MedLine, publicações sobre o quadro de amiloidose cardíaca datam desde 1948, totalizando mais de 1000 artigos indexados em diversos idiomas. Observa-se, sobretudo nos últimos cinco anos, um crescente aumento no número de estudos que avaliam os diversos aspectos da doença, em especial no que se refere às inovações quanto aos métodos diagnósticos e às novas terapias (Figura 1). Essa tendência é ratificada pelo fato de o material produzido nestes últimos cinco anos representar um terço do total publicado até o momento.2626 Wessler S, Freedberg A. Cardiac amyloidosis; electrocardiographic and pathologic observations. Arch Intern Med (Chic). 1948;82(1):63-74.
Busca com o termo Cardiac Amyloidosis no período de 1996 a 2016 mostrando o crescimento de artigos relacionados ao tema nos últimos dez anos. (Medline)
O aumento no número de estudos acerca da doença conferem evidência suficiente para que os médicos aumentem o seu grau de suspeita clínica de amiloidose cardíaca, em especial do tipo senil, nos casos de ICFEP, sendo recomendado o encaminhamento desses pacientes para centros especializados. Nestes locais, métodos diagnósticos invasivos e não-invasivos permitem uma ampla avaliação, inclusive dispondo de testes genéticos, time multidisciplinar e acesso aos novos medicamentos.
Nessa presente revisão abordaremos os recentes avanços da etiofisiopatologia da amiloidose cardiaca, em especial a forma senil, de uma forma sistematizada para avaliação de indivíduos com suspeita de amiloidose no contexto da ICFEP e as terapias emergentes atualmente disponíveis na prática clínica.
Classificação e etiopatogenia da amiloidose
Diante da complexidade associada à doença e sua multiplicidade de apresentações, foram estabelecidas nomenclaturas e classificações específicas relacionadas à condição que a predispõe e ao tipo de fibrila amiloide depositada no tecido. De modo geral, a amiloidose pode ser classificada como primária, secundária, relacionada à diálise e associada à transtirretina.2727 Dember LM, Madias NE, Harrington JT, Perrone RD, Levey AS, Kausz A. Emerging treatment approaches for the systemic amyloidoses. Kidney Int. 2005;68(3):1377-90.
28 Loo D, Mollee PN, Renaut P, Hill MM. Proteomics in molecular diagnosis: Typing of amyloidosis. J Biomed Biotechnol. 2011;2011:754109.
29 Mollee P, Renaut P, Gottlieb D, Goodman H. How to diagnose amyloidosis. Intern Med J. 2014;44(1):7-17.-3030 Perfetto F, Moggi-Pignone A, Livi R, Tempestini A, Bergesio F, Matucci-Cerinic M. Systemic amyloidosis: a challenge for the rheumatologist. Nat Rev Rheumatol. 2010;6(7):417-29.
Classifica-se como amiloidose primária (AL) quando é definida pela produção de proteína amiloide composta por imunoglobulinas de cadeia leve (kappa e lambda), sintetizadas em condições clínicas que apresentam discrasias plasmocitárias, como o mieloma múltiplo e, menos frequentemente, a macroglobulinemia de Waldenström e o linfoma não-Hodgkin. Classicamente, a AL é uma doença sistêmica que predomina em uma população mais idosa e em indivíduos do sexo masculino.2727 Dember LM, Madias NE, Harrington JT, Perrone RD, Levey AS, Kausz A. Emerging treatment approaches for the systemic amyloidoses. Kidney Int. 2005;68(3):1377-90.,3131 Muchtar E, Buadi FK, Dispenzieri A, Gertz MA. Immunoglobulin light-chain amyloidosis: From basics to new developments in diagnosis, prognosis and therapy. Acta Haematol. 2016;135(3):172-90.
O quadro clínico varia diretamente com o órgão de predomínio de deposição do material amiloide e seu grau de comprometimento funcional. Os dois órgãos mais comumente afetados são o rim e o coração, correspondendo a 60-80% dos pacientes na maioria dos estudos. O implicação renal manifesta-se com síndrome nefrótica ou proteinúria assintomática. O envolvimento cardíaco está atrelado ao desenvolvimento de um quadro de ICFEP, além do possível acometimento do sistema de condução do coração e suas correspondentes complicações. Neuropatia autonômica, ou neuropatia periférica sensitivomotora, podem estar presentes em até 20% dos pacientes.2727 Dember LM, Madias NE, Harrington JT, Perrone RD, Levey AS, Kausz A. Emerging treatment approaches for the systemic amyloidoses. Kidney Int. 2005;68(3):1377-90.,3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.
33 Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1(6):1331-41.-3434 Desport E, Bridoux F, Sirac C, Delbes S, Bender S, Fernandez B, et al. Al amyloidosis. Orphanet J Rare Dis. 2012;7:54.
O acúmulo no fígado do material amiloide é achado frequente e cursa com hepatomegalia isolada ou ainda uma hepatoesplenomegalia, podendo apresentar padrão de elevação de enzimas hepáticas, compatível com quadro de colestase. Infiltração muscular pode ocorrer, cursando com pseudohipertrofia, como na clássica macroglossia, assim como quadro de artropatia devido a depósitos nas articulações. A púrpura periorbitária (sinal do guaxinim), a despeito de ser um achado pouco frequente, é fortemente característica da forma AL. Diátese hemorrágica é uma importante condição que pode estar presente e apresenta como nexos causais possíveis a ligação do material amiloide com o fator X da coagulação, a reduzida síntese de fatores de coagulação diante de um fígado comprometido e uma possível doença de von Willebrand adquirida.2727 Dember LM, Madias NE, Harrington JT, Perrone RD, Levey AS, Kausz A. Emerging treatment approaches for the systemic amyloidoses. Kidney Int. 2005;68(3):1377-90.,3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.
33 Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1(6):1331-41.-3434 Desport E, Bridoux F, Sirac C, Delbes S, Bender S, Fernandez B, et al. Al amyloidosis. Orphanet J Rare Dis. 2012;7:54.
A amiloidose secundária (AA) é identificada em quadros clínicos de inflamação crônica, como artrite reumatoide, psoríase e, mais recentemente, as denominadas doenças autoinflamatórias, como a doença inflamatória intestinal, febre familiar do mediterrâneo e a síndrome de Muckle-Wells. As fibrilas são compostas pela proteína amiloide A e são produzidas pelo fígado durante a fase aguda de doenças inflamatórias. Esta proteína apresenta originalmente a função de aumentar a afinidade das lipoproteínas de alta densidade (HDL) por macrófagos e adipócitos, assim como mediar a quimioatração e indução da síntese de citocinas pró-inflamatórias. O quadro inflamatório crônico aumenta a sua síntese e, diante de um processamento incorreto com clivagem e dobramento errôneo, resulta na sua forma patogênica. Os rins são comprometidos em aproximadamente 80% dos pacientes, sendo o órgão mais afetado pela AA. No entanto, também há relatos de envolvimento cardíaco.2727 Dember LM, Madias NE, Harrington JT, Perrone RD, Levey AS, Kausz A. Emerging treatment approaches for the systemic amyloidoses. Kidney Int. 2005;68(3):1377-90.,3535 Real de Asúa D, Costa R, Galván JM, Filigheddu MT, Trujillo D, Cadiñanos J. Systemic AA amyloidosis: epidemiology, diagnosis, and management. Clin Epidemiol. 2014;6:369-77.
36 Scarpioni R, Ricardi M, Albertazzi V. Secondary amyloidosis in autoinflammatory diseases and the role of inflammation in renal damage. World J Nephrol. 2016;5(1):66-75.-3737 Gallimore JR, Sc B, Sabin CA, Ph D, Gillmore JD, Ph D, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356(23):2361-71.
A amiloidose relacionada à diálise ocorre em função do depósito de fibrilas originadas da proteínas beta-2 microglobulina, que se acumulam em níveis crescentes em pacientes com doença renal em estágio avançado e que são submetidos a longo tempo em diálise. Nesta forma, em especial, o quadro predominante é de acometimento osteoarticular, como a síndrome do túnel do carpo e envolvimento do manguito rotador.3838 Danesh F, Ho LT. Dialysis-related amyloidosis: history and clinical manifestations. Semin Dial. 2001;14(2):80-5.,3939 Stoppini M, Bellotti V. Systemic amyloidosis: Lessons from ?2-microglobulin. J Biol Chem. 2015;290(16):9951-8.
A amiloidose cardíaca associada à transtirretina (TTR) é a segunda forma de amiloidose com maior prevalência de acometimento cardíaco, podendo ser dividida nas formas hereditária e senil. A proteína precursora é sintetizada predominantemente no fígado e tem papel de transportadora de retinol e tiroxina.1010 Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546-54.,1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.
15 González-López E, Gallego-Delgado M, Guzzo-Merello G, De Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94.
16 Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol. 2012;302(3):H544-52.
17 Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.-1818 Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.
Na forma senil, identificamos o depósito tecidual da forma selvagem da TTR, sobretudo no miocárdio, sendo observado um quadro clinico de IC. A associação com síndrome do túnel do carpo tem sido descrita, enquanto o acometimento renal é um achado raro. Observou-se em estudos com necrópsias que o depósito deste material amiloide no coração é um achado frequente, em especial nos pacientes previamente assintomáticos.2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66. Dados do grupo da Mayo Clinic indicam que a prevalência desta forma entre os pacientes portadores de amiloidose é de aproximadamente 8,5%, com idade média de 77 anos e o sexo masculino representando 82% dos indivíduos acometidos. Identificou-se que raramente ocorre em pacientes abaixo dos 70 anos. Estudo observacional, observou que os pacientes costumam ter uma progressão lenta e uma sobrevida, após o diagnóstico, de aproximadamete 43 meses, em comparação com os 26,6 meses da forma mutante.1818 Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.,4040 Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2(2):1-11.
41 Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1(2):e000364.-4242 Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164(2):222-8.
A forma hereditária, ao contrário da senil, acomete pacientes em diferentes faixas etárias, mas predominando uma idade média inferior à encontrada nos pacientes portadores da forma senil. A codificação da TTR ocorre no cromossomo 18 e já foram identificadas mais de 70 mutações associadas a esta proteína. Diante da suspeita de um quadro de amiloidose por TTR, o sequenciamento desta proteína a partir de amostra de tecido ou sangue deve ser realizado para o diagnóstico e identificação de uma possível mutação específica que nos permite definir o curso prognóstico do paciente e orientar a investigação dos familiares. A mutação Val122Ile está mais associada a idosos e predomínio no sexo masculino, apresentando, em 90% dos casos, manifestação clínica de uma cardiomiopatia.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.,1010 Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546-54.,1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.,1515 González-López E, Gallego-Delgado M, Guzzo-Merello G, De Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94.
A mutação mais prevalente na população mundial é a Val30Met, que apresenta marcado envolvimento neurológico, aliado a um acometimento cardíaco tardio e é relacionada a doença de Corino de Andrade, também conhecida como doença dos pezinhos, que cursa com polineuropatia periférica sensitivo-motora que se manifesta, especialmente, aos 20 anos de idade, caracterizando-se por parestesias, distúrbios motores e autonômicos, além de cursar com comprometimento cardíaco e renal na fase tardia da doença. Essa condição tem sido identificada como uma doença genética associada à mutação TTR.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.,1010 Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546-54.,1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.
Apresentações clínicas
A cardiomiopatia da amiloidose é classicamente descrita como um modelo de IC direita, cursando muitas das vezes com ascite, antecedendo o edema de membros inferiores e aliado a hepatomegalia ao exame físico. Diferentemente da disfunção cardíaca com aumento das pressões de enchimento, o edema pulmonar é um quadro pouco frequente na cardiomiopatia da amiloidose.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.,88 Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113-22.,1010 Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546-54.,1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.
Um fenótipo mais raro é o envolvimento do septo interventricular com depósito do material amiloide promovendo o espessamento desproporcional da região, mimetizando um quadro de cardiomiopatia hipertrófica. Esta apresentação configura o que se denomina uma fenocópia, ou seja, uma condição clínica que se apresenta através de manifestações típicas de uma doença de origem genética bem definida.2121 Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.,2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.,4141 Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1(2):e000364.
O relato de síncope, devido ao acometimento do sistema nervoso autônomo pela amiloidose, é um achado comum nestes pacientes e sua presença diante de esforço físico está ligada a um pior prognóstico, apresentando elevada mortalidade em três meses, muitas vezes devido à morte súbita.2121 Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.,4141 Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1(2):e000364.
As arritmias ventriculares são causas pouco frequentes de síncope nessa população. Isto se justifica pelo fato de o miocárdio infiltrado pelo material amiloide apresentar-se mais suscetível a episódios de hipoperfusão. As doenças do sistema de condução podem estar presentes nas diferentes formas de amiloidose. No entanto, são encontradas mais frequentemente na forma associada à TTR, tanto a senil quanto a hereditária. A sincope em pacientes portadores de amiloidose cardíaca está associada principalmente a fenômenos de hipotensão arterial devido a disautonomia e bradiarritmias e menos relacionada a arritmias ventriculares. A arritmia ventricular maligna, quando presente, é uma causa comum de morte no portador de amiloidose cardíaca e esses pacientes são fortes candidatos ao implante de cardiodesfibriladores.2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.
Envolvimento do pericárdio pode ocorrer em alguns casos, resultando em derrame pericárdico que, na maioria das vezes, não desenvolve tamponamento cardíaco. Em decorrência das alterações próprias da amiloidose cardíaca, este quadro pode ser mascarado e não dispor de sinais ecocardiográficos como a compressão atrial e ventricular direita.2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.,4141 Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1(2):e000364.
O acúmulo do material no átrio promove sua disfunção eletromecânica e, consequentemente, aumenta de maneira considerável o risco de formação de trombo intracavitário. Esse processo é evidente, sobretudo nos portadores de amiloidose do tipo AL e é um fator independente da fibrilação atrial, e quando ambos os fatores estão presentes, o risco de tromboembolismo é extremamente elevado. Dessa forma, deve-se considerar o emprego de anticoagulantes nesses indivíduos.2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.,4141 Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1(2):e000364.
Amiloidose cardíaca e seu novo fenótipo clínico
Uma nova visão sobre a ICFEP torna-se fundamental, tendo em vista sua crescente relevância como fenótipo clínico mais prevalente de IC na população mundial. Esse dado é presente em nosso meio, como evidenciado através do estudo DIGITALIS, que investigou a prevalência da IC e seus fenótipos na atenção básica na cidade de Niterói. De acordo com esse estudo, dentre a população que apresentava IC, 59% eram portadores do fenótipo ICFEP.55 Jorge AL, Rosa ML, Martins WA, Correia DM, Fernandes LC, Costa JA, et al. The prevalence of stages of heart failure in primary care: a population-based study. J Card Fail. 2016;22(2):153-7.
Dados provenientes de um centro especializado em amiloidose no Brasil apontam para uma elevada prevalência de acometimento miocárdico em portadores de polineuropatia amiloidótica a partir de anormalidades no eletrocardiograma (ECG).4343 Queiroz MC, Pedrosa RC, Berensztejn AC, Pereira Bde B, Nascimento EM, Duarte MM, et al. Frequency of cardiovascular involvement in familial amyloidotic polyneuropathy in Brazilian patients. Arq Bras Cardiol. 2015;105(5):503-9.
Apesar de inúmeros estudos acerca desta condição clínica, muito se desconhece sobre sua etiofisiopatogenia, o que tem causado resultados negativos nos estudos de tratamento da ICFEP. Isso é dificultado, em especial, diante das inúmeras fenocópias que mimetizam sua apresentação.66 van Heerebeek L, Paulus WJ. Understanding heart failure with preserved ejection fraction: where are we today? Neth Heart J. 2016;24(4):227-36.,4444 Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263-71.
Diante desse cenário, destaca-se a possibilidade de uma parcela dos pacientes com ICFEP apresentarem, na realidade, um quadro de amiloidose cardíaca. Isso é ratificado através de estudos recentes com pacientes com diagnóstico de ICFEP, na ausência de hipertensão arterial ou diabetes, apresentarem, através das novas modalidades de cardioimagem, acúmulo do material amiloide no miocárdio. Ao lado disso, encontra-se o fato de o material amiloide ter sido identificado em estudos com necrópsias de pacientes com quadro de IC.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.,88 Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113-22.,1515 González-López E, Gallego-Delgado M, Guzzo-Merello G, De Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94.,4545 Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232-9.
De forma a facilitar a identificação de um quadro suspeito para amiloidose cardíaca, independente do tipo de fibrila depositada, devemos atentar a algumas evidências clínicas e de exames complementares conforme apresentamos na Tabela 1.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.
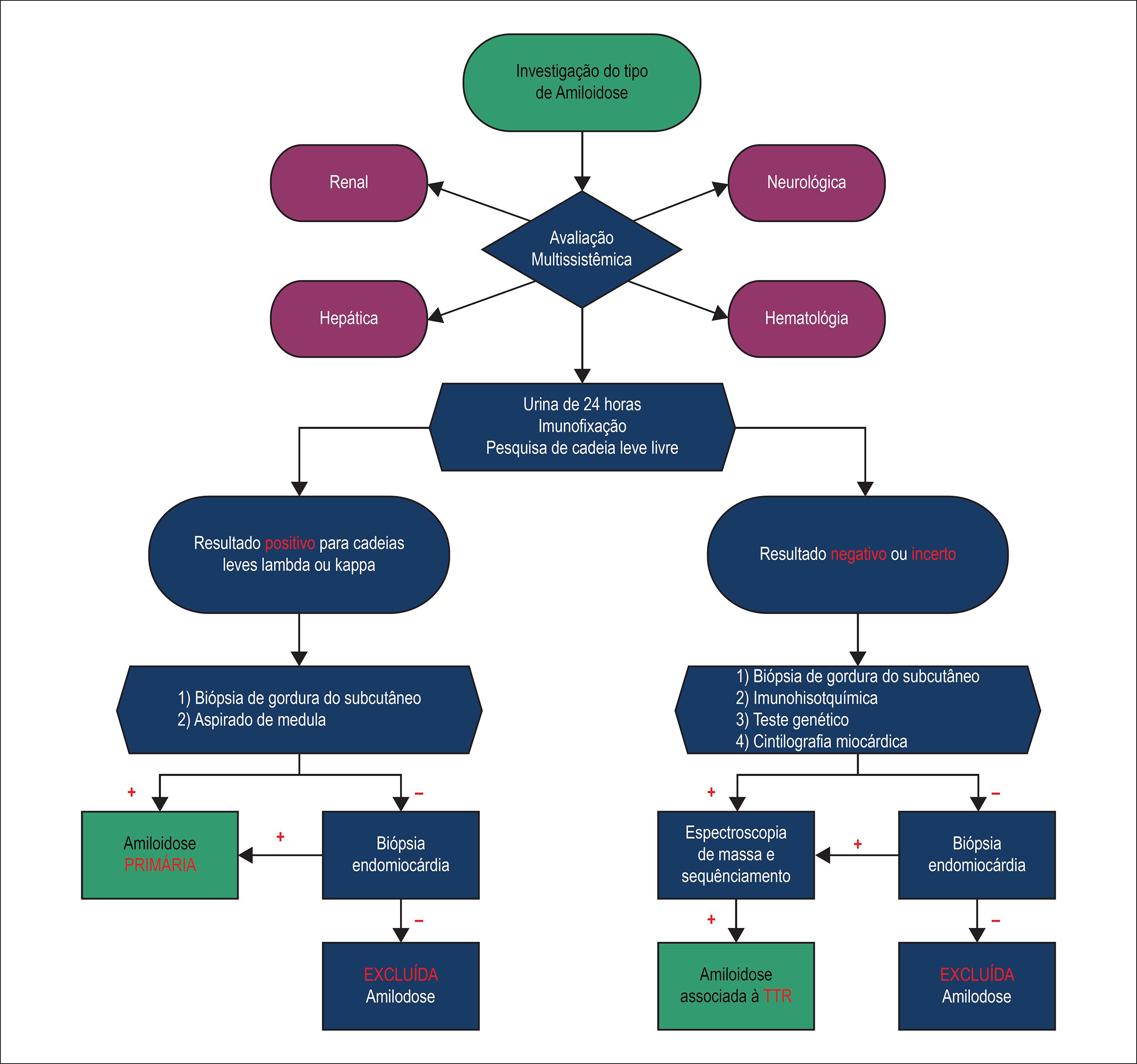
Neste cenário em que não são confirmadas as etiologias mais comums aplicáveis ao quadro de ICFEP, devemos suspeitar de um quadro de amiloidose cardíaca. Diante disso propomos um fluxograma norteador para o manejo destes pacientes (Figuras 2 e 3).1818 Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.
Fluxograma para avaliação de pacientes com ICFEP e suspeita de amilodose. ICFEP: insuficiência cardíaca com fração de ejeção preservada; ECO: ecodopplercardiograma; ECG: eletrocardiograma; DPD: 2,3-dicarboxipropano-1,1-difosfonato.
Abordagem diagnóstica
A amiloidose cardíaca apresenta um curso indolente e o diagnóstico é feito, muitas vezes tardiamente, dessa forma contribuindo para um pior prognóstico. Em diversas situações o acometimento cardíaco acarreta grande morbidade ao paciente, entretanto o diagnóstico não é, com frequência, suspeitado, nem mesmo em condições onde existe a caracterização de cardiopatia restritiva.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.
Na história clínica podemos suspeitar de amiloidose cardíaca em pacientes com idade igual ou superior a 50 anos, que apresentam sinais e sintomas de IC, com fração de ejeção do VE maior ou igual a 50% e que com o tratamento não apresentam melhora dos sintomas. Deve-se considerar manifestações extra cardíacas na história do paciente como norteadoras do diagnóstico da IC pela amiloidose, tais como neuropatia periférica e síndrome do túnel do carpo, principalmente as recorrentes e bilaterais. Podem estar presentes também sinais e sintomas como hipotensão ortostática, macroglossia, consumo muscular da região tenar e hipotenar, hematomas de origem desconhecida e distúrbios de condução elétrica cardíaca.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.,2929 Mollee P, Renaut P, Gottlieb D, Goodman H. How to diagnose amyloidosis. Intern Med J. 2014;44(1):7-17.
O ECG é um exame de fácil acesso que pode oferecer alterações que levantam a suspeita de amiloidose na presença de um quadro de IC. O achado de um complexo QRS de baixa voltagem, desvios do eixo elétrico e bloqueios de ramo podem ser encontrados. Arritmias são frequentes, em especial a fibrilação atrial, que está relacionada com infiltração amiloide do átrio, e arritmias ventriculares complexas.1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.
O ECO é um importante método de investigação diagnóstica e a presença de hipertrofia significativa do ventrículo esquerdo associada ao ECG com QRS de baixa voltagem pode levar a suspeição de amiloidose cardíaca. Outro critério para a suspeita de amiloidose cardíaca é o espessamento da parede ventricular esquerda acima de 12 mm na ausência de história de hipertensão arterial sistêmica. Outros achados que podem estar presentes no ECO são o aumento biatrial com ventrículos de tamanho normal, derrame pericárdico e evidências de disfunção diastólica devido ao padrão de cardiomiopatia resritiva.99 Palka P, Lange A, Donnelly JE, Scalia G, Burstow DJ, Nihoyannopoulos P. Doppler tissue echocardiographic features of cardiac amyloidosis. J Am Soc Echocardiogr. 2002;15(11):1353-60.,1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.
A medida da espessura do septo interventricular pode sugerir o tipo de amiloidose presente no paciente, sendo frequentemente maior nos casos de amiloidose por TTR do que a forma AL, podendo em muitos casos ser superior a 20 mm. No entanto, a separação entre essas duas formas em bases clínicas nem sempre é possível.99 Palka P, Lange A, Donnelly JE, Scalia G, Burstow DJ, Nihoyannopoulos P. Doppler tissue echocardiographic features of cardiac amyloidosis. J Am Soc Echocardiogr. 2002;15(11):1353-60.,1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.
Em alguns pacientes com amiloidose cardíaca podemos observar o fenótipo clínico semelhante a miocardiopatia hipertrófica obstrutiva devido a presença do gradiente dinâmico de pressão que está relacionado a um "estreitamento" adicional da via de saída do VE. Mais recentemente, o strain sistólico longitudinal tem sido utilizado para o diagnóstico da disfunção sistólica em pacientes com amiloidose cardíaca, podendo mostrar a preservação da ponta do coração em relação as demais paredes. Na presença de alterações do ECG e do ECO devemos recorrer a exames complementares. Em uma primeira etapa na elucidação de um caso de amiloidose cardíaca, investigamos o status da função renal deste paciente através das escórias nitrogenadas e, em especial, a quantificação de perda protéica e o tipo desta, feitas através da coleta de urina de 24h com dosagem da proteinúria e da eletroforese de proteína urinária. Esta etapa na investigação permite a identificação da forma primária de amiloidose e consiste na identificação das cadeias leves que estão em altos títulos nestes pacientes. A imunofixação, quando associada, possibilita um aumento na acurácia diagnóstica deste tipo de apresentação.99 Palka P, Lange A, Donnelly JE, Scalia G, Burstow DJ, Nihoyannopoulos P. Doppler tissue echocardiographic features of cardiac amyloidosis. J Am Soc Echocardiogr. 2002;15(11):1353-60.,1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.,3131 Muchtar E, Buadi FK, Dispenzieri A, Gertz MA. Immunoglobulin light-chain amyloidosis: From basics to new developments in diagnosis, prognosis and therapy. Acta Haematol. 2016;135(3):172-90.,3434 Desport E, Bridoux F, Sirac C, Delbes S, Bender S, Fernandez B, et al. Al amyloidosis. Orphanet J Rare Dis. 2012;7:54.
O aspirado de gordura abdominal para estudo histopatológico é uma alternativa mais acessível, por ser um procedimento diagnóstico simples, de fácil execução, seguro e que apresenta boa sensibilidade, porém tendo menor acurácia na forma associada à TTR. Contudo, ao se obter resultado negativo com o aspirado de gordura abdominal, a biopsia endomiocárdica de ventrículo direito pode ser essencial para o diagnóstico de amiloidose cardíaca. Através deste método, a proteína amiloide é corada pelo vermelho-congo. Outros tecidos também podem ser avaliados como o reto, gengiva, medula óssea, rim, entre outros. O estudo histoquímico das amostras teciduais é importante a fim de se estabelecer uma distinção entre as formas hereditária, senil, sistêmica e primária, em virtude das diferenças de tratamento e prognóstico.1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.,2929 Mollee P, Renaut P, Gottlieb D, Goodman H. How to diagnose amyloidosis. Intern Med J. 2014;44(1):7-17.
A ressonância magnética surge como mais uma alternativa para o diagnóstico da amiloidose cardíaca, com uma sensibilidade de 87% e especificidade de 96% para a forma associada à TTR. Através desse exame é possível identificar o espessamento miocárdico e do septo interatrial, sinais de disfunção diastólica, e o típico padrão de realce tardio subendocárdico no ventrículo esquerdo, que pode também acometer todas as câmaras cardíacas.1111 Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076-84.,1212 Rapezzi C, Quarta CC, Guidalotti PL, Pettinato C, Fanti S, Leone O, et al. Role of 99mTc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging. 2011;4(6):659-70.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.,2929 Mollee P, Renaut P, Gottlieb D, Goodman H. How to diagnose amyloidosis. Intern Med J. 2014;44(1):7-17.
A imagem molecular também tem revolucionado o diagnóstico. Pode ser utilizado o método não invasivo a partir do emprego do radiotraçador Tc99m, que se liga à TTR mas não aos derivados da cadeia leve, sendo um método eficaz de avaliação diante das formas mutante ou selvagem da amiloidose cardíaca associada à TTR. A tomografia por emissão de pósitrons em conjunção com o traçador C-RiB pode ser uma nova estratégia a ser empregada no diagnóstico desses pacientes.1414 Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.
A biópsia da medula óssea com análise de coloração imuno-histoquímica ou de citometria de fluxo é fundamental nos pacientes em que se identificou o quadro de amiloidose do tipo AL. Esta demonstrará uma população clonal de plasmócitos, que estão produzindo as cadeias leves defeituosas do anticorpo. Caso esses testes sejam negativos, devemos investigar as formas hereditárias da doença.2929 Mollee P, Renaut P, Gottlieb D, Goodman H. How to diagnose amyloidosis. Intern Med J. 2014;44(1):7-17.,3333 Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1(6):1331-41.
Existem novos estudos envolvendo ciências ômicas que pretendem aumentar a precisão diagnóstica da amiloidose. A proteômica envolve o estudo de toda a expressão protéica de uma célula em diferentes condições, sendo seu estudo complementar ao genoma, identificando qualquer proteína, com ou sem mutações genéticas. A principal técnica empregada é a microdissecção a laser seguido por espectrometria de massa (LMD-MS), através das quais amostras positivas na coloração vermelho-congo são dissecadas e decompostas em componentes menores denominados peptídeos.2525 Marcoux J, Mangione PP, Porcari R, Degiacomi MT, Verona G, Taylor GW, et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015;7(10):1337-49.,2828 Loo D, Mollee PN, Renaut P, Hill MM. Proteomics in molecular diagnosis: Typing of amyloidosis. J Biomed Biotechnol. 2011;2011:754109.
Tratamento da amiloidose cardíaca
O tratamento da amiloidose cardíaca é melhor realizado em centros especializados da doença. O tratamento requer duas abordagens: controle das complicações relacionadas ao coração devido ao depósito amiloide e tratamento da doença de base para prevenir novas formações amiloides.
O tratamento da amiloidose cardíaca tem como objetivo melhorar os sinais e sintomas da IC. O emprego de diuréticos em baixa dose melhora os sintomas relacionados a congestão, enquanto que o uso da combinação de betabloqueadores e inibidores da enzima conversora da angiotensina tem o seu beneficio ainda não definido na amiloidose.1616 Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol. 2012;302(3):H544-52.,1818 Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.,2121 Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.,2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.
O emprego de digitálicos não apresenta benefícios nesse grupo de pacientes, visto que o miocárdio em disfunção pelo material amiloide está mais suscetível aos efeitos tóxicos, o que predispõe a ocorrencia de arritmias.1616 Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol. 2012;302(3):H544-52.,1818 Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.,2121 Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.,2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.
Deve ser aventado o uso de anticoagulantes em caso de fibrilação atrial e na detecção de trombos intracavitários.1818 Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.,2121 Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.,4141 Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1(2):e000364.
Amiloidose da forma AL
A sobrevida global é de aproximadamente quatro anos após o diagnóstico e tem melhorado nas últimas três décadas. A amiloidose da forma AL é, frequentemente, resultado de um aumento clonal dos plasmócitos na medula óssea e dessa forma a terapia com quimioterápicos citotóxicos, pode ser eficaz. A atuação do hematologista no processo de estadiamento e definição da estratégia terapêutica neste cenário é fundamental.2121 Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.,3131 Muchtar E, Buadi FK, Dispenzieri A, Gertz MA. Immunoglobulin light-chain amyloidosis: From basics to new developments in diagnosis, prognosis and therapy. Acta Haematol. 2016;135(3):172-90.,3333 Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1(6):1331-41.,3434 Desport E, Bridoux F, Sirac C, Delbes S, Bender S, Fernandez B, et al. Al amyloidosis. Orphanet J Rare Dis. 2012;7:54.
Pacientes que apresentam resposta hematológica ao tratamento tem melhora sintomática e dos biomarcadores cardíacos, podendo cursar com regressão do depósito amiloide, o que já é evidente logo nos primeiros três meses, confirmando um melhor prognóstico nestes casos.1919 Lawler PR, Bergmark BA, Laubach JP, Lakdawala NK. Having a heavy heart: approaches to infiltrative cardiomyopathy. Circulation. 2014;129(16):1703-11.,2121 Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.,3131 Muchtar E, Buadi FK, Dispenzieri A, Gertz MA. Immunoglobulin light-chain amyloidosis: From basics to new developments in diagnosis, prognosis and therapy. Acta Haematol. 2016;135(3):172-90.,3333 Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1(6):1331-41.,3434 Desport E, Bridoux F, Sirac C, Delbes S, Bender S, Fernandez B, et al. Al amyloidosis. Orphanet J Rare Dis. 2012;7:54.
Terapia com melfalano associada a dexametasona (MelDex) em pacientes inelegiveis para transplante autólogo de células tronco apresentou uma taxa de resposta em torno de 70%, sendo essa pior nos casos com envolvimento cardíaco avançado. Em recente ensaio clínico randomizado comparando MelDex com altas doses de melfalano seguido pelo transplante de células tronco mostrou uma melhor taxa de sobrevida nos pacientes que fizeram uso do MelDex.3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.,4646 Palladini G, Russo P, Nuvolone M, Lavatelli F, Perfetti V, Obici L, et al. Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood. 2007;110(2):787-9.
47 Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997;336(17):1202-7.-4848 Mehta J. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2008;358(1):91.
O Bortezomib tem demonstrado ser uma droga eficaz quando associado à Ciclofosfamida e Dexametasona, com resposta hematológica importante (71%), após dois meses de utilização. Segundo grupo da Mayo Clinic, no caso de três pacientes inicialmente inelegíveis para terapia com célula-tronco, o emprego de Bortezomib tornou possível o procedimento. O transplante de células-tronco é utilizado em 25% dos pacientes com amiloidose cardíaca. Após o procedimento episódios de taquicardia supraventricular podem ocorrer, com mortalidade de 11% nesses indivíduos. A resposta hematológica positiva em pacientes submetidos a terapia com células-tronco é de aproximadamente 56%.1616 Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol. 2012;302(3):H544-52.,1919 Lawler PR, Bergmark BA, Laubach JP, Lakdawala NK. Having a heavy heart: approaches to infiltrative cardiomyopathy. Circulation. 2014;129(16):1703-11.,2727 Dember LM, Madias NE, Harrington JT, Perrone RD, Levey AS, Kausz A. Emerging treatment approaches for the systemic amyloidoses. Kidney Int. 2005;68(3):1377-90.,3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.,3333 Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1(6):1331-41.
Estudo que avaliou o emprego de Bortezomid, Dexametasona e agentes alquilantes (BDEX+AA) em 106 pacientes com IC sintomática devido amiloidose cardíaca AL mostrou uma melhora da sobrevida após ajuste das variáveis clínicas. (hr: 0,209; 95% IC: 0,069 a 0,636; p = 0,006).3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.,4949 Sperry BW, Ikram A, Hachamovitch R, Valent J, Vranian MN, Phelan D, et al. Efficacy of chemotherapy for light-chain amyloidosis in patients presenting with symptomatic heart failure. J Am Coll Cardiol. 2016;67(25):2941-8.
Forma associada à TTR
O Tafamidis surge como uma importante opção para o tratamento da amiloidose, atuando como um estabilizador cinético do tetrâmero da TTR. A interação de moléculas em determinados sítios de ligação da TTR promove a estabilidade à proteína em seu estado tetramérico, diminuindo acentuadamente sua dissociação e, consequentemente, a amiloidogênese. O tafamidis apresenta capacidade de se ligar seletivamente em um dos sítios de tiroxina na TTR, promovendo a estabilização cinética do tetrâmero.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.,2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.,3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.,5050 Waddington Cruz M, Benson MD. A Review of Tafamidis for the treatment of transthyretin-related amyloidosis. Neurol Ther. 2015;4(2):61-79.
51 Coelho T, Maia LF, Da Silva AM, Cruz MW, Planté-Bordeneuve V, Suhr OB, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802-14.
52 Coelho LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, Suhr OB, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785-92.
53 Merlini G, Planté-Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013;6(6):1011-20.-5454 Maurer MS, Grogan DR, Judge DP, Mundayat R, Packman J, Lombardo I, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Hear Fail. 2015;8(3):519-26.
Em estudo multicêntrico, randomizado, duplo-cego, placebo controlado, foram demonstradas a segurança e eficácia do Tafamidis oral em pacientes com amiloidose e envolvimento do sistema nervoso periférico. Os ensaios clínicos mostram que esta medicação retarda a progressão da doença, melhora a função de fibras nervosas de pequeno e grande calibre e, consequentemente, reduz a perda funcional dos sistemas acometidos, otimizando a qualidade de vida do paciente. Em outro estudo, o Tafamidis resultou em estabilização da transtirretina em 97% dos pacientes com quadro de IC leve a moderada devido à forma selvagem da amiloidose cardíaca.77 Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.,2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.,2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.,3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.,5050 Waddington Cruz M, Benson MD. A Review of Tafamidis for the treatment of transthyretin-related amyloidosis. Neurol Ther. 2015;4(2):61-79.
51 Coelho T, Maia LF, Da Silva AM, Cruz MW, Planté-Bordeneuve V, Suhr OB, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802-14.
52 Coelho LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, Suhr OB, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785-92.
53 Merlini G, Planté-Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013;6(6):1011-20.-5454 Maurer MS, Grogan DR, Judge DP, Mundayat R, Packman J, Lombardo I, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Hear Fail. 2015;8(3):519-26.
Outro fármaco em processo de avaliação para os quadros de amiloidose cardíaca associada à TTR é o Diflunisal, um antinflamatório não-esteróide que pode estabilizar o tetrâmero, evitando a amiloidogênese. Uma coorte avaliou a tolerância e os efeitos promovidos em 13 pacientes portadores de amiloidose cardíaca por TTR, tanto a forma mutante quanto a selvagem. Não foram observadas mudanças significativas na estrutura e função cardíaca, bem como dos biomarcadores.3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.,5555 Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda SI. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015;22(2):79-83.,5656 Castaño A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 2012;18(6):315-9.
O emprego de doxiciclina e do ácido tauroursodeoxicólico foi realizado em estudo com pequeno número de pacientes, sendo identificada uma possível melhora clínica. Uma nova terapia antisense de segunda geração, ISIS-TTRrx, atua reduzindo o nível sérico da proteína TTR ao suprimir a expressão gênica de sua síntese. Além destes, um total de 28 estudos estão cadastrados na base do Clinical Trials para intervenções em pacientes com amiloidose cardíaca.3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.,5757 Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819-29.,5858 Benson MD, Pandey S, Witchell D, Jazayeri A, Siwkowski A, Monia B, et al. Antisense oligonucleotide therapy for TTR amyloidosis. Amyloid. 2011;18 Suppl 1:60.
Uma alternativa de tratamento para alguns tipos de amiloidose seria o transplante de fígado com o objetivo de substituir o gene mutado TTR que produz a maioria da transtirretina circulante por um gene encontrado em um órgão do doador geneticamente normal. Desta forma, o transplante de fígado pode ser uma alternativa para retardar a progressão da doença e prolongar a sobrevida do paciente. Contudo, a imunossupressão crônica pertinente ao transplante leva a uma taxa de mortalidade alta no primeiro ano, cerca de 10%, e uma morbidade elevada. O transplante não impede a síntese extra-hepática de proteína amiloide e com isso não retarda a progressão da doença.2222 Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.,2424 Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.,3232 Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.
Conclusão
A amiloidose cardíaca inaugura uma nova era da cardiologia personalizada, onde o diagnóstico preciso através de técnicas que envolvem análise genética molecular, biomarcadores e métodos de cardioimagem possibilitam classificar a forma da amiloidose e definir seu curso clínico e prognóstico e, futuramente, guiar a terapêutica desses quadros.
Deve-se aumentar a suspeição clínica de amiloidose diante dos casos de ICFEP em que os métodos de cardioimagem sejam compatíveis com o quadro de cardiomiopatia restritiva ou com os sinais de dissociação entre os achados do ECO e do ECG. A parceria com centros especializados em amiloidose aliado às novas tecnologias são fundamentais no manejo desses pacientes através de tratamentos especializados, incluindo transplante de órgãos, ou, ainda, envolvendo os pacientes em estudos clínicos que avaliam a ação dos novos medicamentos emergentes.
-
Fontes de financiamentoO presente estudo não teve fontes de financiamento externas.
-
Vinculação acadêmicaNão há vinculação deste estudo a programas de pós-graduação.
References
-
1Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355(3):251-9.
-
2Morgan S, Smith H, Simpson I, Liddiard GS, Raphael H, Pickering RM, et al. Prevalence and clinical characteristics of left ventricular dysfunction among elderly patients in general practice setting: cross sectional survey. BMJ. 1999;318(7180):368-72.
-
3Ceia F, Fonseca C, Mota T, Morais H, Matias F, De Sousa A, et al; EPICA Investigators. Prevalence of chronic heart failure in Southwestern Europe: The EPICA study. Eur J Heart Fail. 2002;4(4):531-9.
-
4Mesquita ET, Jorge AJ. Heart failure with normal ejection fraction: new diagnostic criteria and pathophysiological advances. Arq Bras Cardiol. 2009;93(2):180-7.
-
5Jorge AL, Rosa ML, Martins WA, Correia DM, Fernandes LC, Costa JA, et al. The prevalence of stages of heart failure in primary care: a population-based study. J Card Fail. 2016;22(2):153-7.
-
6van Heerebeek L, Paulus WJ. Understanding heart failure with preserved ejection fraction: where are we today? Neth Heart J. 2016;24(4):227-36.
-
7Ton V, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. 2014;8(Suppl 1):39-44.
-
8Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113-22.
-
9Palka P, Lange A, Donnelly JE, Scalia G, Burstow DJ, Nihoyannopoulos P. Doppler tissue echocardiographic features of cardiac amyloidosis. J Am Soc Echocardiogr. 2002;15(11):1353-60.
-
10Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546-54.
-
11Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076-84.
-
12Rapezzi C, Quarta CC, Guidalotti PL, Pettinato C, Fanti S, Leone O, et al. Role of 99mTc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging. 2011;4(6):659-70.
-
13Arbustini E, Verga L, Concardi M, Palladini G, Obici L, Merlini G. Electron and immuno-electron microscopy of abdominal fat identifies and characterizes amyloid fibrils in suspected cardiac amyloidosis. Amyloid. 2002;9(2):108-14.
-
14Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.
-
15González-López E, Gallego-Delgado M, Guzzo-Merello G, De Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94.
-
16Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol. 2012;302(3):H544-52.
-
17Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
-
18Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.
-
19Lawler PR, Bergmark BA, Laubach JP, Lakdawala NK. Having a heavy heart: approaches to infiltrative cardiomyopathy. Circulation. 2014;129(16):1703-11.
-
20Rapezzi C, Lorenzini M, Longhi S, Milandri A, Gagliardi C, Bartolomei I, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117-24.
-
21Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease. New frontiers and insights in pathophysiology, diagnosis, and management. Texas Heart Inst J. 2005;32(2):178-84.
-
22Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.
-
23Roof L, Coker WJ, Lazarchick J, Kang Y. Senile transthyretin cardiac amyloidosis in patients with plasma cell dyscrasias: importance of cardiac biopsy for making the correct diagnosis. Aperito J Cell Mol Biol. 2014;1(1). pii. 102.
-
24Castaño A, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.
-
25Marcoux J, Mangione PP, Porcari R, Degiacomi MT, Verona G, Taylor GW, et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015;7(10):1337-49.
-
26Wessler S, Freedberg A. Cardiac amyloidosis; electrocardiographic and pathologic observations. Arch Intern Med (Chic). 1948;82(1):63-74.
-
27Dember LM, Madias NE, Harrington JT, Perrone RD, Levey AS, Kausz A. Emerging treatment approaches for the systemic amyloidoses. Kidney Int. 2005;68(3):1377-90.
-
28Loo D, Mollee PN, Renaut P, Hill MM. Proteomics in molecular diagnosis: Typing of amyloidosis. J Biomed Biotechnol. 2011;2011:754109.
-
29Mollee P, Renaut P, Gottlieb D, Goodman H. How to diagnose amyloidosis. Intern Med J. 2014;44(1):7-17.
-
30Perfetto F, Moggi-Pignone A, Livi R, Tempestini A, Bergesio F, Matucci-Cerinic M. Systemic amyloidosis: a challenge for the rheumatologist. Nat Rev Rheumatol. 2010;6(7):417-29.
-
31Muchtar E, Buadi FK, Dispenzieri A, Gertz MA. Immunoglobulin light-chain amyloidosis: From basics to new developments in diagnosis, prognosis and therapy. Acta Haematol. 2016;135(3):172-90.
-
32Chakraborty R, Muchtar E, Gertz MA. Newer therapies for amyloid cardiomyopathy. Curr Heart Fail Rep. 2016;13(5):237-46.
-
33Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1(6):1331-41.
-
34Desport E, Bridoux F, Sirac C, Delbes S, Bender S, Fernandez B, et al. Al amyloidosis. Orphanet J Rare Dis. 2012;7:54.
-
35Real de Asúa D, Costa R, Galván JM, Filigheddu MT, Trujillo D, Cadiñanos J. Systemic AA amyloidosis: epidemiology, diagnosis, and management. Clin Epidemiol. 2014;6:369-77.
-
36Scarpioni R, Ricardi M, Albertazzi V. Secondary amyloidosis in autoinflammatory diseases and the role of inflammation in renal damage. World J Nephrol. 2016;5(1):66-75.
-
37Gallimore JR, Sc B, Sabin CA, Ph D, Gillmore JD, Ph D, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356(23):2361-71.
-
38Danesh F, Ho LT. Dialysis-related amyloidosis: history and clinical manifestations. Semin Dial. 2001;14(2):80-5.
-
39Stoppini M, Bellotti V. Systemic amyloidosis: Lessons from ?2-microglobulin. J Biol Chem. 2015;290(16):9951-8.
-
40Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2(2):1-11.
-
41Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1(2):e000364.
-
42Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164(2):222-8.
-
43Queiroz MC, Pedrosa RC, Berensztejn AC, Pereira Bde B, Nascimento EM, Duarte MM, et al. Frequency of cardiovascular involvement in familial amyloidotic polyneuropathy in Brazilian patients. Arq Bras Cardiol. 2015;105(5):503-9.
-
44Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263-71.
-
45Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232-9.
-
46Palladini G, Russo P, Nuvolone M, Lavatelli F, Perfetti V, Obici L, et al. Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood. 2007;110(2):787-9.
-
47Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997;336(17):1202-7.
-
48Mehta J. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2008;358(1):91.
-
49Sperry BW, Ikram A, Hachamovitch R, Valent J, Vranian MN, Phelan D, et al. Efficacy of chemotherapy for light-chain amyloidosis in patients presenting with symptomatic heart failure. J Am Coll Cardiol. 2016;67(25):2941-8.
-
50Waddington Cruz M, Benson MD. A Review of Tafamidis for the treatment of transthyretin-related amyloidosis. Neurol Ther. 2015;4(2):61-79.
-
51Coelho T, Maia LF, Da Silva AM, Cruz MW, Planté-Bordeneuve V, Suhr OB, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802-14.
-
52Coelho LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, Suhr OB, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785-92.
-
53Merlini G, Planté-Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013;6(6):1011-20.
-
54Maurer MS, Grogan DR, Judge DP, Mundayat R, Packman J, Lombardo I, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Hear Fail. 2015;8(3):519-26.
-
55Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda SI. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015;22(2):79-83.
-
56Castaño A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 2012;18(6):315-9.
-
57Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819-29.
-
58Benson MD, Pandey S, Witchell D, Jazayeri A, Siwkowski A, Monia B, et al. Antisense oligonucleotide therapy for TTR amyloidosis. Amyloid. 2011;18 Suppl 1:60.
Datas de Publicação
-
Publicação nesta coleção
29 Jun 2017 -
Data do Fascículo
Jul 2017
Histórico
-
Recebido
15 Ago 2016 -
Revisado
30 Dez 2016 -
Aceito
09 Mar 2017