Abstract
The effectiveness of methylene blue (MB) combined with pyrimethamine (PYR), chloroquine (CQ) or quinine (Q) was examined in a classical four-day suppressive test against a causative agent of rodent malaria, Plasmodium berghei. A marked potentiation was observed when MB was administered at a non-curative dose of 15 mg/kg/day in combination with PYR (0.19 mg/kg/day) or Q (25 mg/kg/day). No synergy was found between MB (15 mg/Kg) and CQ (0.75 mg/Kg). Our results suggest that the combination of MB with PYR or Q may improve the efficacy of these currently used antimalarial drugs.
antimalarials; methylene blue; drug combination
SHORT COMMUNICATION
The in vivo antimalarial activity of methylene blue combined with pyrimethamine, chloroquine and quinine
Giovanny GaravitoI, + + Corresponding author: ggaravitoc@unal.edu.co ; Stéphane BertaniII, III; Miguel QuilianoIV; Alexis ValentinII; Ignacio AldanaIV; Eric DeharoII, V
IDepartamento de Farmacia, Facultad de Ciencias, Universidad Nacional de Colombia, Bogotá, Colombia
IIPharma-Dev UMR 152, Faculté de Pharmacie, Université de Toulouse, Université Paul Sabatier, Toulouse, France
IIIPharma-Dev UMR 152, Institut de Recherche pour le Développement, Lima, Peru
IVNeglected Diseases Section, Drug R&D Unit, Center for Applied Pharmacobiology Research, University of Navarra, Pamplona, Spain
VPharma-Dev UMR 152, Faculté de Pharmacie, Institut de Recherche pour le Développement, Toulouse, France
ABSTRACT
The effectiveness of methylene blue (MB) combined with pyrimethamine (PYR), chloroquine (CQ) or quinine (Q) was examined in a classical four-day suppressive test against a causative agent of rodent malaria, Plasmodium berghei. A marked potentiation was observed when MB was administered at a non-curative dose of 15 mg/kg/day in combination with PYR (0.19 mg/kg/day) or Q (25 mg/kg/day). No synergy was found between MB (15 mg/Kg) and CQ (0.75 mg/Kg). Our results suggest that the combination of MB with PYR or Q may improve the efficacy of these currently used antimalarial drugs.
Key words: antimalarials - methylene blue - drug combination
With almost 3.3 billion people living in endemic areas, malaria remains a major global health issue (WHO 2011). In response to the emergence of resistance strains of malaria, new antimalarials that are combinable with classical treatments are urgently needed (WHO 2001, 2010). Methylene blue (MB) is the oldest and least expensive drug ever used in antimalarial therapy and has been proven safe and effective in patients (Guttmann & Ehrlich 1891, Ferreira 1893, Meissner et al. 2005, 2006). In vitro studies showed that the antiplasmodial activity of MB could be enhanced when this drug was combined with additional antimalarials (Vennerstrom et al. 1995, Akoachere et al. 2005, Garavito et al. 2007a). In many countries, MB has already been approved by regulatory agencies for the treatment of methemoglobinaemia and for ifosfamide-induced encephalopathy in cancer treatment (Schirmer et al. 2003, Mandi et al. 2005). Thus, MB could easily be reintroduced into antimalarial therapeutic schemes.
The purpose of our study was to determine whether MB in combination with the low-cost drug pyrimethamine (PYR) is able to inhibit parasitaemia in rodent malaria models and to compare the effectiveness of this treatment with that of combinations of MB with quinine (Q) or chloroquine (CQ) diphosphate.
The four-day suppressive test for the quantitative assessment of blood schizontocidal activity was performed according to the method of Peters and Robinson (1999). Male Swiss albino mice (Bioterio DFUNC) were inoculated with 107Plasmodium berghei ANKA-parasitised red blood cells (RBCs). Batches of 10 animals per group were then randomly distributed after infection and orally treated (0.1 mL) at the same time daily for four consecutive days beginning on the day of infection. A control group received 0.9% saline solution and a reference group was administered CQ orally at 3 mg/kg/day. The percentage of parasitised erythrocytes was determined in Giemsa-stained blood smears made from peripheral blood on day four post-infection. The percent inhibition of parasitaemia was calculated by the following formula:
(parasitaemia in control - parasitaemia in drug-treated) x 100
parasitaemia in control
The ED50 and ED90 values were estimated with a nonlinear regression logistic dose-response model. For the interaction study, batches of five animals (infected as described above) were orally treated for four consecutive days with the individual drugs at two different doses and with the combination of MB (15 mg/Kg) and PYR (0.19 mg/Kg), Q (25 mg/Kg) or CQ (0.75 mg/Kg). Statistical analysis was performed with GraphPad Prism 5.01 software (GraphPad Software, San Diego).
All drugs were obtained from Sigma-Aldrich (L'Isle d'Abeau Chesnes, France) and dissolved in water. The animals were treated according to the French and Colombian legislation on laboratory animal use and care.
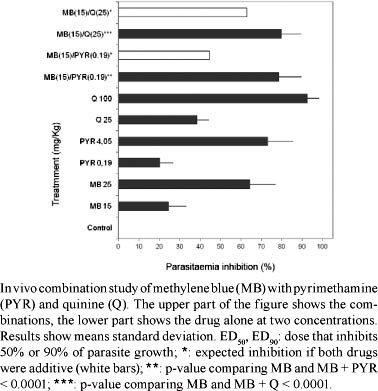
The results are reported in Table and Figure. PYR was the most active compound, with an ED50 less than half that of CQ and almost 150 times lower than that of Q. Under these conditions, Q showed an ED90 close to the 116 mg/Kg found by Peters (1987). In a previous study, we obtained an equivalent ED50 for CQ against P. berghei ANKA of 0.53 mg/Kg (Garavito et al. 2008). The ED90 of CQ reported in Table was similar to the ED90 of 3.3 mg/Kg previously demonstrated (Peters 1987). The combination of 15 mg/Kg of MB with 0.75 mg/Kg of CQ did not reveal any interaction (data not shown), in accordance with the in vitro data recorded previously (Garavito et al. 2007a).
The combination of 15 mg/Kg of MB with 0.19 mg/Kg of PYR resulted in an inhibition of parasitaemia of 78.7% ± -11.1%, displaying an activity superior to that expected if the effect of the two drugs was additive (44.1% inhibition). In vitro drug interaction experiments with the FcM29 strain of Plasmodium falciparum showed an antagonist effect when MB was combined with PYR and an antagonistic interaction was also described using the K1 strain of P. falciparum (Akoachere et al. 2005, Garavito et al. 2007a, Garavito 2007).
Combining 15 mg/Kg of MB with 25 mg/Kg of Q led to an inhibition of 78.8% ± 9.7%. This activity was greater than expected if the effect of both drugs was additive (63% inhibition). We reported a synergistic effect between MB and Q used against the CQ-resistant P. falciparum strain FcM29 in vitro (Garavito et al. 2007a, Garavito 2007), whereas Akoachere et al. (2005) found additivity in a CQ-resistant P. falciparum K1 strain.
The antimalarial activity of MB has been attributed to antioxidant defences that specifically target glutathione reductase (GR), a crucial enzyme for P. falciparum (Ginsburg & Atamna 1994, Atamna et al. 1996, Atamna & Ginsburg 1997, Färber et al. 1998), or inhibit the glutathione-dependent degradation of haeme (Garavito et al. 2007b). In a previous study, we demonstrated that MB inhibited almost 60% of parasite growth during the late ring/early trophozoite stages, when intense digestion of haemoglobin occurs; this inhibition of parasite growth correlates with a peak expression of plasmodial GR at the 24th h of the intraerythrocytic cycle (PlasmoDB 2005, Garavito et al. 2007a). MB reaches maximum plasma concentrations 1-2 h following oral administration at a dose of 100 mg in humans. Its plasmatic half-life is approximately 5-6.5 h and renal excretion is low, at approximately 20-30%. The rapid antimalarial effect of MB, combined with its pharmacokinetic properties, including a short half-life, might promote the slow development of resistance because exposure of parasites to sub-therapeutic levels of the drug would be very brief; this effect has been shown to occur with artemisinin.
Q is relatively inexpensive and remains the preferred anti-malarial drug for the treatment of severe and complicated malaria in most African countries. Although Q resistance is not yet a significant problem, drug failure may occur as a result of poor compliance with the prolonged treatment course and problems with tolerability. An alternative is the development of shorter-course treatments in combination with therapies that have the potential to improve compliance and treatment outcomes (Achan et al. 2011). To treat malaria in the first trimester of pregnancy, the WHO recommends the use of Q plus clindamycin; unfortunately, this combination is too expensive for most affected people (Achan et al. 2011). Q is quickly absorbed after oral or parenteral administration, readily crossing the placental barrier and detectable in cerebral spinal fluid. Q has a half-life of between 9.7-18 h (3.2 h in healthy paediatric subjects and 12.1 h in patients infected with malaria). We have previously shown that Q targeted the late ring/early trophozoite stages, when digestion of haemoglobin occurs, but had a greater impact on the haeme-glutathione interaction than on haemozoin formation (Garavito 2007, Garavito et al. 2007b).
Although the combination of Q with MB was effective in our experiment, as expected, this combination has some drawbacks that preclude its therapeutic use, such as the fact that both drugs target similar developmental stages of the parasite with similar mechanisms of action. Further, both drugs exhibit similarly short half-lives, requiring multiple administrations, which can lead to poor compliance. Finally, Q is approximately 10 times more expensive than PYR or MB.
PYR was discovered more than 60 years ago as a result of an intense antimalarial research program conducted by the United States army to find an alternative to Q during World War II (Plowe 2001). PYR has been used for decades because of its high therapeutic index and very low cost. Unfortunately, used alone, PYR generates a high rate of resistance and must therefore be combined with another antimalarial. It has been hypothesised that PYR's mechanism of action mainly involves its interference in folate metabolism through the inhibition of dihydrofolate reductase (DHFR) (Peterson & Wellems 1988). Recently, Legorreta-Herrera et al. (2010) provided new details on the effect of PYR by highlighting its production of free radicals in rodent models of malaria. These authors hypothesised that PYR induces oxidative stress in Plasmodium yoelii-infected RBCs; in support of this proposed mechanism, increased redox enzyme activities (superoxide dismutase and glutathione peroxidase) were demonstrated in both erythrocytes and the liver. PYR also increases nitric oxide levels in the serum and upregulates mRNA concentrations of superoxide dismutase, glutathione peroxidase, catalase and iNOS in the spleen. PYR is also well absorbed when administered orally, reaching maximum plasma concentrations 2-6 h after administration and exhibiting a plasma half-life of 80-96.5 h.
A rapid analysis of the structure-activity relationship between PYR and MB emphasises the complementary and synergic mechanisms of action of these drugs. PYR is a pyrimidine compound that interacts with the active site of the DHFR protein, establishing hydrogen bond interactions and the number of flexible bonds plays a crucial role in drug resistance (Yuvaniyama et al. 2003). MB is a phenothiazine compound that exhibits a planar geometry in its "blue form" and a partial planar geometry, uncharged and colourless, in its "Leuco form". To date, crystallographic data for the MB-GR complex have not been reported, although due to a high number of aromatic and non-polar residues in the region, stacking-π and van der Waals interactions occur in the interdimeric cavity of GR (Sarma et al. 2003, Schirmer et al. 2011). From a chemical research standpoint, fragment-base discovery is an exciting and novel method of lead generation (Warr 2009). Fragments are small, low-molecular-weight molecules that usually form part of a drug compound. Based on fragment-base discovery, the pyrimidine-diamine system of PYR may be found to serve as an effective scaffold upon which to build further antimalarial drug development. In addition, both phenothiazine and quinoline templates have been promoted as "privileged structures" that can help in the modification and improvement of existing active molecules (Wermuth 2004).
MB has been reported not only to exhibit non-competitive inhibitor, but also as a substrate reduced by flavoenzymes that produce reactive oxygen species (Buchholz et al. 2008). The complementary mechanisms of PYR and MB seem clear; both compounds generate oxidative stress, but in different ways, whereas MB also induces indirect structural changes in the interdimeric region of GR. Both MB and PYR target the crucial redox machinery of Plasmodium, but at two different stages of the intraerythrocytic development: PYR acts against young schizonts (Maerki et al. 2006), whereas MB is effective against the late ring/early trophozoite stages (Garavito 2007). Therefore, in theory, the effects of these drugs should be synergistic. This premise is corroborated by the experimental data obtained herein. Moreover, the effective therapeutic half-life of MB is much shorter than that of PYR and, consequently, selection for resistance should be less intense. Finally, both compounds are affordable for afflicted populations, with the combination approximately 30 times less expensive than the combination of artesunate and PYR (Baird 2005). These results confirm that the combination of MB and PYR, two established, reliable and effective molecules, should be considered with renewed interest as a new, low-cost antimalarial therapy.
ACKNOWLEDGEMENTS
To Pr Irene Landau (MNHN, Paris), for providing P. berghei ANKA rodent malaria parasite, and to Joshua L Halford and David Parker Jr, for their critical discussions.
Received 25 January 2012
Accepted 29 May 2012
Financial support: DIB/UNC, IRD (France)
- Achan J, Talisuna AO, Erhart A, Yeka A, Tibenderana JK, Baliraine FN, Rosenthal PJ, D'Alessandro U 2011. Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malar J 10: 144.
- Akoachere M, Buchholz K, Fischer E, Burhenne J, Haefeli WE, Schirmer RH, Becker K 2005. In vitro assessment of methylene blue on CQ-sensitive and resistant Plasmodium falciparum strains reveals synergistic action with artemisinins. Antimicrob Agents Chemother 49: 4592-4597.
- Atamna H, Ginsburg H 1997. The malaria parasite supplies glutathione to its host cell. Investigation of glutathione transport and metabolism in human erythrocytes infected with Plasmodium falciparum Eur J Biochem 250: 670-679.
- Atamna H, Krugliak M, Shalmiev G, Deharo E, Pescarmona G, Ginsburg H 1996. Mode of antimalarial effect of methylene blue and some of its analogues on Plasmodium falciparum in culture and their inhibition of P. vinckei petteri and P. yoelii nigeriensis in vivo Biochem Pharmacol 51: 693-700.
- Baird JK 2005. Effectiveness of antimalarial drugs. N Engl J Med 352: 1565-1577.
- Buchholz K, Schirmer RH, Eubel JK, Akoachere MB, Dandekar T, Becker K, Gromer S 2008. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum Antimicrob Agents Chemother 52: 183-191.
- Färber PM, Arscott LD, Williams CH, Becker K, Schirmer RH 1998. Recombinant Plasmodium falciparum glutathione reductase is inhibited by the antimalarial dye methylene blue. FEBS Lett 422: 311-314.
- Ferreira 1893. Treatment of malaria with methylene blue. Lancet: 821-822.
- Garavito G 2007. Etude pharmacologique expérimentale de l'activité antipaludique d'un composé de synthèse: le chlorure de methyl-thioninium, PhD Thesis, Université Toulouse III/Paul Sabatier École Doctorale BSB, IOP Publishing Physics, Toulouse, 155 pp.
- Garavito G, Bertani S, Deharo E 2008. Complementary findings on the antimalarial activity and toxicity of methylene blue. Vitae 15: 91-95.
- Garavito G, Bertani S, Rincon J, Maurel S, Monje MC, Landau I 2007a. Blood schizontocidal activity of methylene blue in combination with antimalarials against Plasmodium falciparum Parasite 14: 135-140.
- Garavito G, Monje MC, Maurel S, Valentin A, Nepveu F, Deharo E 2007b. A non-radiolabeled heme-GSH interaction test for the screening of antimalarial compounds. Exp Parasitol 116: 311-313.
- Ginsburg H, Atamna H 1994. The redox status of malaria-infected erythrocytes: an overview with an emphasis on unresolved problems. Parasite 1: 5-13.
- Guttmann P, Ehrlich P 1891. Guttmann ueber die wirkung des methylen-blau bei malaria. Berlin Klin Wochenschr 28: 953-956.
- Legorreta-Herrera M, Retana-Ugalde R, Ventura-Gallegos JL, Narváez V 2010. Pyrimethamine induces oxidative stress in Plasmodium yoelii 17XL-infected mice: a novel immunomodulatory mechanism of action for an old antimalarial drug? Exp Parasitol 126: 381-388.
- Maerki S, Brun R, Charman SA, Dorn A, Matile H, Wittlin S 2006. In vitro assessment of the pharmacodynamic properties and the partitioning of OZ277/RBx-11160 in cultures of Plasmodium falciparum J Antimicrob Chemother 58: 52-58.
- Mandi G, Witte S, Meissner P, Coulibaly B, Mansmann U, Rengelshausen J 2005. Safety of the combination of chloroquine and methylene blue in healthy adult men with G6PD deficiency from rural Burkina Faso. Trop Med Int Health 10: 32-38.
- Meissner PE, Mandi G, Coulibaly B, Witte S, Tapsoba T, Mansmann U 2006. Methylene blue for malaria in Africa: results from a dose-finding study in combination with chloroquine. Malar J 5: 84.
- Meissner PE, Mandi G, Witte S, Coulibaly B, Mansmann U, Rengelshausen J 2005. Safety of the methylene blue plus chloroquine combination in the treatment of uncomplicated falciparum malaria in young children of Burkina Faso. Malar J 4: 45-54.
- Peters W 1987. Chemotherapy and drug resistance in malaria, 2nd ed., Academic Press, London, 1100 pp.
- Peters W, Robinson BL 1999. Handbook of animal models of infection, Academic Press, New York, 1136 pp.
- Peterson DW, Wellems TE 1988. Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc Natl Acad Sci USA 85: 9114-9118.
- PlasmoDB 2005. [database on the Internet]. The plasmodium genome resource. Expression of plasmodial glutathione reductase. [cited 2005 Nov 17]. Available from: plasmodb.org/
- Plowe C 2001. Folate antagonists and mechanisms of resistance. In PJ Rosenthal, Antimalarial chemotherapy: mechanisms of action, resistance, and new directions in drug discovery, Humana Press Inc, New Jersey, p. 173.
- Sarma GN, Savvides SN, Becker K, Schirmer M, Schirmer RH, Karplus PA 2003. Glutathione reductase of the malarial parasite Plasmodium falciparum: crystal structure and inhibitor development. J Mol Biol 328: 893-907.
- Schirmer RH, Adler H, Pickhardt M, Mandelkow E 2011. "Lest we forget you - methylene blue...". Neurobiol Aging 32: 2325.
- Schirmer RH, Coulibaly B, Stich A, Scheiwein M, Merkle H, Eubel J 2003. Methylene blue as an antimalarial agent. Redox Rep 8: 272-275.
- Vennerstrom JL, Makler MT, Angerhofer CK, Williams JA 1995. Antimalarial dyes revisited: xanthenes, azines, oxazines and thiazines. Antimicrob Agents Chemother 39: 2671-2677.
- Warr WA 2009. Fragment-based drug discovery. J Comput Aided Mol Des 23: 453-458.
- Wermuth C 2004. Selective optimization of side effects: another way for drug discovery. J Med Chem 47: 1303-1314.
- WHO - World Health Organization 2001. Antimalarial drug combination therapy Report of a WHO technical consultation, WHO, Geneva, 36 pp.
- WHO - World Health Organization 2010. [homepage on the Internet]. WHO guidelines for the treatment of malaria [cited 2011 Jun 11]. Available from: who.int/malaria
- WHO - World Health Organization 2011. [homepage on the Internet]. Malaria. WHO-TDR Special Programme for Research and Training in Tropical Diseases. [cited 2011 March 10]. Available from: apps.who.int/tdr/svc/diseases/malaria
- Yuvaniyama J, Chitnumsub P, Kamchonwongpaisan S, Vanichtanankul J, Sirawaraporn W, Taylor P 2003. Insights into antifolate resistance from malarial DHFR-TS structures. Nat Struct Biol 10: 357-365.
Publication Dates
-
Publication in this collection
14 Sept 2012 -
Date of issue
Sept 2012
History
-
Received
25 Jan 2012 -
Accepted
29 May 2012