Abstracts
Glycoconjugate vaccines, in which a cell surface carbohydrate from a micro-organism is covalently attached to an appropriate carrier protein are proving to be the most effective means to generate protective immune responses to prevent a wide range of diseases. The technology appears to be generic and applicable to a wide range of pathogens, as long as antibodies against surface carbohydrates help protect against infection. Three such vaccines, against Haemophilus influenzae type b, Neisseria meningitidis Group C and seven serotypes of Streptococcus pneumoniae, have already been licensed and many others are in development. This article discusses the rationale for the development and use of glycoconjugate vaccines, the mechanisms by which they elicit T cell-dependent immune responses and the implications of this for vaccine development, the role of physicochemical methods in the characterisation and quality control of these vaccines, and the novel products which are under development.
Glycoconjugate Vaccines; Capsular polysaccharides; lipopolysaccharides
Vacinas glicoconjugadas, cujo carboidrato da superfície de um microrganismo está covalentemente ligado a uma proteína carreadora, vêm sendo consideradas como efetivas para gerar respostas imunes que previnem um grande número de doenças. A tecnologia é genérica e aplicável a vários patógenos, se os anticorpos contra os carboidratos de superfície forem capazes de proteger contra a infecção. Três vacinas contra Haemophilus influenzae tipo b, Neissseria meningitidis Grupo C e sete sorotipos de Streptococcus pneumoniae já foram licenciadas e muitas outras estão em desenvolvimento. Este artigo discute o racional para o desenvolvimento e uso de vacinas glicoconjugadas; os mecanismos pelos quais elas induzem respostas imune dependentes de célula T e suas implicações para o seu desenvolvimento; o papel dos métodos físico-químicos na caracterização e no controle de qualidade dessas vacinas; e os produtos novos que estão em desenvolvimento.
vacinas glicoconjugadas; polissacarídeos capsulares; lipopolissacarídeos
BIOMEDICAL AND MEDICAL SCIENCES
Vaccines based on the cell surface carbohydrates of pathogenic bacteria
Christopher Jones
Laboratory for Molecular Structure, National Institute for Biological Standards and Control, Blanche Lane, South Mimms, Herts EN6 3QG. UK
ABSTRACT
Glycoconjugate vaccines, in which a cell surface carbohydrate from a micro-organism is covalently attached to an appropriate carrier protein are proving to be the most effective means to generate protective immune responses to prevent a wide range of diseases. The technology appears to be generic and applicable to a wide range of pathogens, as long as antibodies against surface carbohydrates help protect against infection. Three such vaccines, against Haemophilus influenzae type b, Neisseria meningitidis Group C and seven serotypes of Streptococcus pneumoniae, have already been licensed and many others are in development.
This article discusses the rationale for the development and use of glycoconjugate vaccines, the mechanisms by which they elicit T cell-dependent immune responses and the implications of this for vaccine development, the role of physicochemical methods in the characterisation and quality control of these vaccines, and the novel products which are under development.
Key words: Glycoconjugate Vaccines, Capsular polysaccharides, lipopolysaccharides.
RESUMO
Vacinas glicoconjugadas, cujo carboidrato da superfície de um microrganismo está covalentemente ligado a uma proteína carreadora, vêm sendo consideradas como efetivas para gerar respostas imunes que previnem um grande número de doenças. A tecnologia é genérica e aplicável a vários patógenos, se os anticorpos contra os carboidratos de superfície forem capazes de proteger contra a infecção. Três vacinas contra Haemophilus influenzae tipo b, Neissseria meningitidis Grupo C e sete sorotipos de Streptococcus pneumoniae já foram licenciadas e muitas outras estão em desenvolvimento.
Este artigo discute o racional para o desenvolvimento e uso de vacinas glicoconjugadas; os mecanismos pelos quais elas induzem respostas imune dependentes de célula T e suas implicações para o seu desenvolvimento; o papel dos métodos físico-químicos na caracterização e no controle de qualidade dessas vacinas; e os produtos novos que estão em desenvolvimento.
Palavras-chave: vacinas glicoconjugadas, polissacarídeos capsulares, lipopolissacarídeos.
INTRODUCTION
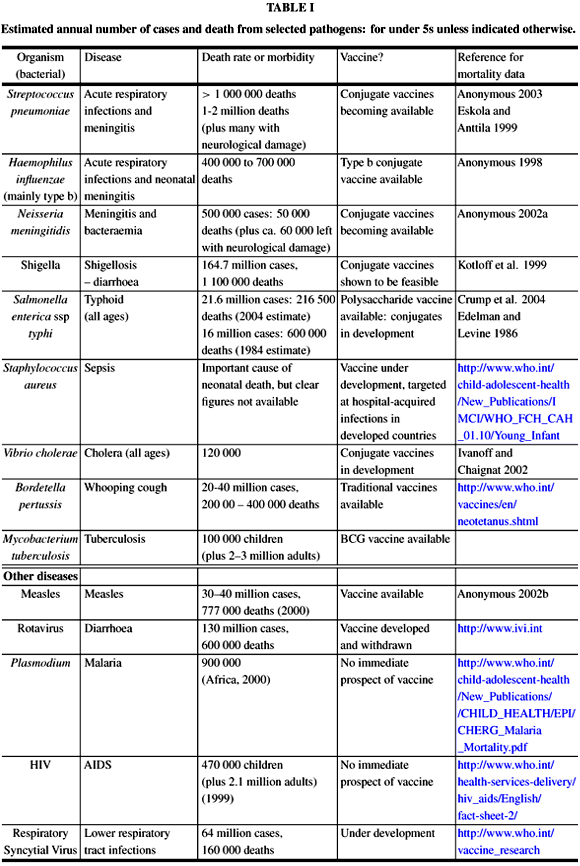
Bacterial infections remain major killers of infants and children, particularly in developing countries. Several million children die each year due to such infections (Table I). The most important pathogens are Streptococcus pneumoniae, Haemophilius influenzae type b, Neisseria meningitidis, Salmonella entericus subspecies typhi, Staphylococcus aureus, and diarrhoea-causing organisms such as Shigella, Salmonella and Vibrio cholerae. The disease profile can be endemic or epidemic, as occurs with Group A meningococcal disease in sub-Saharan Africa. Disease and mortality are concentrated amongst children in developing countries. Each of these pathogens possesses a cell surface capsular polysaccharide (CPS) or lipopolysaccharide (LPS) shell, or both, which helps the pathogen to establish an infection. The CPS hides cell surface components of the bacterium from the immune system of the host, preventing complement activation by cell surface proteins (Roitt 1997) and inhibiting phagocytosis. If the bacterium is phagocytosed, the CPS helps prevent bacterial killing. The role of LPS as a virulence factor is less well defined. In some cases it has been demonstrated, and in other cases it is suspected, that antibodies against a CPS or LPS O-chain will protect against infection (Lindberg et al. 1991, Popoff 1991, Waldor et al. 1994, Fulop et al. 2001, Coughlin and Bogard 1987, Brahmbhatt et al. 1992, Bowden et al. 1995).

For most of these pathogens, different strains express CPSs or LPSs of different structures, resulting in a number of different serotypes or serogroups. Virulence and pathogenicity may be serotype or serogroup dependent, or there may be geographic differences in the clinically relevant serotypes. Whilst Haemophilus influenzae disease is caused overwhelmingly by a single serotype, the type b, pneumococcal disease is caused by a very large number of the ninety known serotypes. Meningococcal disease in developed countries is principally Groups B and C, although Groups W135 and Y are becoming increasingly important, and Group A disease is virtually unknown (Racoosin et al. 1998, Mayer et al. 2002). Meningococcal Group A strains are, however, responsible for the regular meningitis epidemics which plague sub-Saharan Africa. Practically therefore, most of the saccharide-derived vaccines must contain multiple carbohydrates structures to provide adequate coverage against the disease-causing strains. In some cases there is immunological cross reactivity between related structures which can provide partial protection. The optimal choice of polysaccharide to include in the vaccine is therefore a complex epidemiological problem (Robbins et al. 1983). Pneumococcal Types 1 and 5, for example, are important pathogens in South America, but much less important in North America or Europe. For this reason, these serotypes were not included in the first 7-valent glycoconjugate vaccines licensed (Wyeth's Prevenar®), but have been included in subsequent 9- and 11-valent glycoconjugate vaccines under development. A number of these pathogens, including Neisseria meningitidis and Streptococcus pneumoniae, can leave surviving infants with severe neurological damage. This may affect as many infants as actually die from the infection. This has profound social and economic impacts.
Once it became clear that antibodies against CPSs protect against infection, it was a logical step to attempt to use these polysaccharides as immunogens. The first attempts were made in the late 1940s (McLeod et al. 1945), but the seemingly miraculous potential of antibiotics to control disease postponed development of this field until the late 1960s, when antibiotic resistance and the potential for neurological damage in "antibiotic-cured" infants became apparent (Cochi et al. 1985). CPS vaccines clearly work, and vaccines of this type are licensed and used in many countries, but this approach has several severe limitations. Repeating polysaccharides are T cell-independent type 2 (TI-2) immunogens: without the involvement of T cells they do not induce immunological memory, avidity maturation and isotype switching do not occur, and the antibodies induced, largely IgM and IgG2 (Musher et al. 1990, Lortan et al. 1993), are not good activators of complement. Crucially, vaccines of this type fail to induce immune responses in infants below the age of about two years, who are the major group at risk for these infections, because this aspect of the immune system develops relatively late. Repeat vaccination does not lead to increased antibody levels, so only one dose is given, but without immunological memory re-vaccination is required at regular intervals as antibody levels decline. This is typically every five years.
Whilst the specificity of the immune response depends upon the structure of the CPS, the magnitude of the response depends critically upon its molecular weight. Only very high molecular weight polysaccharides are immunogenic and product development focussed at first on the isolation of material of sufficiently high molecular weight. For this reason, LPS O-chains and low molecular weight CPSs, such as those from Staphylococcus aureus, are not effective as vaccines.
POLYSACCHARIDE STRUCTURE
CPSs and LPS O-chains have strict repeating structure, which may consist of either a single sugar unit or oligosaccharide units, containing as many as seven or eight sugar residues (Kamerling 2000). The repeat units can either be linear or branched and contain non-carbohydrate substituents such as O-acetyl, glycerol phosphate, or pyruvate ketals. Structural heterogeneity may occur as a result of the loss of or migration of labile O-acetyl groups between sites. Bacterial polysaccharides may contain unusual sugar residues including diamino-, deoxy- and branched chain sugars (Lindberg 1990a). As a general rule, CPSs tend to be anionic in character whilst LPS O-chains are neutral. The structures of some the repeat units of the capsular polysaccharides of clinically important bacteria are shown in Table II. Whilst these vaccines elicit a strong antibody response, it is likely that protection depends upon a relatively small proportion of high avidityantibodies, with those directed against the saccharide backbone perhaps most important. Whilst antibodies against substituents such as O-acetyl groups may predominate, they may be of relatively low avidity and not clinically important (Michon et al. 2000).
THE MOLECULAR MECHANISMS OF THE GENERATION OF IMMUNE RESPONSES AGAINST POLYSACCHARIDES
The molecular mechanisms by which TI-2 immunogens with repeating structures, such as bacterial polysaccharides, stimulate an antibody response have been revealed by the work of Snapper and coworkers (Mond et al. 1995, Snapper and Mond 1996, Snapper et al. 1997). In brief, the polysaccharide crosslinks approximately 15-20 surface immunoglobulin molecules (sIg) molecules present on a B cell of appropriate specificity, leading through a series of intermediate protein phosphorylation steps to an increase in free intracellular calcium. Such a cell is primed to secrete antibody, but a second signal is also required. The nature of this second signal has not been well defined, and may be different in the case of a natural infection than when a vaccine is used. When this second signal is received, the B cells mature into plasma cells and secrete antibodies. There appears to be no direct interaction between B cells and T cells. The necessity to crosslink many sIg molecules would seem to be the reason why only high mass CPSs are immunogenic. The mechanism by which glycoconjugate vaccines elicit an immune response is significantly different and is discussed in more detail below, but it is this difference which explains why glycoconjugate are so much more effective as vaccines, and why they can be used to stimulate an immune response against a much wider variety of carbohydrate immunogens. Processing of zwitterionic capsular polysaccharides by an MHC II pathway has very recently been suggested (Cobb et al. 2004).
POLYSACCHARIDE VACCINES IN CLINICAL USE TODAY
Three families of CPS vaccines are in widespread clinical use at present, whilst a fourth against Haemophilus influenzae type b (Hib) infection was used as a short term measure before the introduction of Hib conjugates. The simplest CPS vaccine, against typhoid, contains the so-called Vi (for virulence) antigen as its sole component, but with, typically, lactose present as a stabiliser. Clinical trials of the Salmonella typhi Vi polysaccharide in Nepal indicated an efficacy of approximately 70%, which is similar to older whole cell vaccines against typhoid but the side effects of the polysaccharide vaccine are much less severe (Acharya et al. 1987, Yang et al. 2001). Vaccines containing two (Groups A and C), three (Groups A, C and W135) or four meningococcal (Groups A, C, Y and W135) CPSs are licensed. In developed countries they are currently used for control of outbreaks, but vaccination is required by Muslims undertaking the Hajj pilgrimage to Mecca. These vaccines are also used to control epidemic Group A meningitis in sub-Saharan Africa (Anonymous 2002a).
The pneumococcal polysaccharide vaccine is a blend of 23 serotype-specific polysaccharides, and is used in developed countries to protect the elderly from pneumonia. There is active discussion about exactly how effective these vaccines are for that purpose: some recent metastudies have cast doubt upon its efficacy (Melegaro and Edmunds 2004, Jackson et al. 2003). It is known that, for genetic reasons, some vaccinees are incapable of generating an immune response against some of the serotypes (Musher et al. 1998). This appears to be linked to the very limited genetic diversity of the immune response to polysaccharides. The elderly population appear to produce antibodies of lower avidity (Romero-Steiner et al. 1999).
Despite their limitations, polysaccharide vaccines are available, have moderate efficacy in appropriate populations, are generally cheap (Fleck 2003, Plans 2002) and have an excellent safety record. Glycoconjugate vaccines against meningococcal Group C and against seven pneumococcal serotypes have been licensed, whilst glycoconjugate vaccines against other meningococcal CPSs, more pneumococcal serotypes and typhoid are in development (see below).
GLYCOCONJUGATE VACCINES
The means to increase the immunogenicity of polysaccharides was first discovered by Avery and Goebel in 1931 (Avery and Goebel 1931) covalent attachment of the polysaccharide to an appropriate protein carrier, to form a conjugate. Such conjugates provide T cell-dependent immunogenicity against the saccharide hapten. With the involvement of T cells, immunological memory is invoked, avidity maturation and isotypes switching occurs, to generate complement-activating antibody isotypes such as IgG1 (Wuorimaa et al. 2001). The avidity of the antibodies elicited is much higher than those from polysaccharide vaccines. Crucially, since a different arm of the immune system is involved, that used to process protein immunogens, glycoconjugate vaccines are effective in young infants. Multiple immunisations are necessary to provide the required immune response, but not regular revaccination. In the UK the vaccination regime for Hib conjugate vaccines is at two, three and four months, and a booster at 18 months has recently been introduced to ensure long term protection.
The mechanism by which glycoconjugates stimulate an immune response involves an initial binding of the conjugate to the surface immunoglobulin (sIg) of B cell with appropriate specificity for the saccharide hapten (Siber 1994). This complex is internalised and the carrier protein degraded by proteolytic enzymes. Suitable peptides are transported to and displayed by MHC II complexes. The peptide-loaded MHC II complex is recognised by T cells, which then provide appropriate signals through direct interactions of cell surface proteins and through cytokine signalling processes, to induce maturation of the B cell into an antibody secreting plasma cell. The role of dendritic cells in the process is not yet defined, and the process is probably different in adults who have already been exposed to the saccharide immunogens glycoconjugate vaccines typically invoke an antibody response in adults after a single dose (Baxendale et al. 2000, Hougs et al. 1999). Since crosslinking of surface immunoglobulin molecules is not required, glycoconjugate vaccines can be produced from small saccharide chains. In many cases, the glycans attached in the conjugate are oligosaccharides prepared by degradation of the original polysaccharide (Anderson et al. 1986, Costantino et al. 1999). In addition, glycoconjugates can be produced from relatively low molecular weight oligosaccharides related to the repeating polymers (Mawas et al. 2002, Benaissa-Trouw et al. 2001, Jansen et al. 2001, Jansen and Snippe 2004), or the short glycans of LPS O-chain (Gupta et al. 1998), or low molecular weight capsular polysaccharides such as those expressed by Staphylococcus aureus (Fattom et al. 2004). It has been shown possible to make effective glycoconjugate immunogens from low molecular weight oligosaccharides such as those present on the lipo-oligosaccharides of pathogens such as a Neisseria meningitidis (Mieszala et al. 2003). The same glycoconjugate technology has been used to prepare immunotherapeutics to slow the redevelopment of cancer following chemotherapy, prepared from the glycan chains of glycolipids overexpressed by tumour cells (Musselli et al. 2001). Further discussion of cancer immunotherapeutics is outside the remit of this review.
The first glycoconjugate vaccines against Haemophilus influenzae type b were licensed in the late 1980s. They arose from the academic work of Porter Anderson and of others (Anderson et al. 1981, 1985, Insel and Anderson 1986, Jennings and Lugowski 1981, Schneerson et al. 1980). The Anderson approach involved reductive amination of periodate-generated aldehyde-terminated oligosaccharides to a carrier protein. In modern preparations, CRM197, a genetically toxoided variant of diphtheria toxin is used. The resulting glycoconjugate is approximately 90 kDa in size, is approximately 30% carbohydrate and contains an average of six glycan chains per carrier protein. It is similar in size and saccharide content to many serum proteins, and can be termed a "neo-glycoconjugate" vaccine. Another approach, originally developed by Hilleman (Tai et al. 1987) and commercialised by Aventis Pasteur and GSK, involves random activation of the polysaccharide with cyanogen bromide, addition of linker such as 6-aminocaproic acid or adipic acid dihydrazide linker, and attachment to an appropriate carrier protein typically tetanus toxoid. As there are multiple activation points within each polysaccharide and multiple linkage points on each carrier protein, the resulting conjugate is a crosslinked network of polysaccharide and protein with a molecular weight of, on average, 5 ×106 Da. Such a vaccine can be described as a "crosslinked network". The third approach uses conjugation of size-reduced polysaccharide to LPS-depleted vesicles of outer membrane proteins a "vesicle vaccine" (Donnelly et al. 1990). Thus there are three fundamentally different structures for these conjugates, which are illustrated as cartoons in Figure 2.
The immune responses elicited by these different structural variants are generally similar all are T cell-dependent immunogens, although the vesicle-based vaccines seem to be characterised by a stronger antibody response following the first immunisation, a less pronounced booster effect on subsequent immunisations, and that the antibodies produced tend to be of lower avidity to than those produced by the other two structural types (Schlesinger and Granoff 1992), and different light chain V regions are used (Granoff et al. 1993). The time course for the development of an antibody response following administration of different Hib conjugate vaccines is shown in Figure 3. Usually only two doses of the vesicle vaccines and a booster dose are given.
These conjugate vaccines proved extremely effective at preventing disease in those countries which have adopted them as part of mass vaccination programmes, so that Hib meningitis, which had been the most common form of neonatal meningitis in developed countries, has been almost completely eradicated. The startling effectiveness of these vaccines stimulated a demand that their usage be expanded to other countries, with a WHO target of global coverage (Anonymous 1998). Trial introduction of Hib vaccination into some developing countries highlighted the fact that the disease burden due to this organism had been seriously underestimated. It had also become clear that glycoconjugate vaccines were an effective generic technology which could be used to protect against a wide variety of other pathogens, if the conjugates were made.
CHOICE OF CARRIER PROTEIN AND CONJUGATION CHEMISTRIES
Two types of carrier protein are most commonly used. The earliest and most widely used group have been bacterial toxoids, including tetanus and diphtheria toxoid. These proteins were already licensed for human use. Another popular carrier is CRM197, a genetically toxoided variant of diphtheria toxin, as chemical toxoiding is not required and the vaccines are simpler to characterise and control, and there is less variability possible in carrier protein production. The other approach, which has not yet been used in commercial vaccines, is to choose a carrier protein the immune response against which will complement the role of anti-saccharide antibodies. For example, a developmental vaccine using the pneumococcal toxin pneumolysin, in a genetically toxoided form, has been reported (Michon et al. 1998), and the use of Streptococcal proteins (Paoletti et al. 1999) or Moraxella OMPs as carriers (Hu et al. 2000). The key factor is that the carrier protein delivers a strong T cell involvement, and carrier proteins to which the infant has had prior exposure, such as tetanus toxoid, are particularly valuable in this respect.
Polysaccharides require activation before attachment to the carrier protein. Sometimes it is necessary to activate to the carrier protein as well. The optimum means to activate the polysaccharide depends upon its structure. This is particularly true if oligosaccharide haptens are to be used. For example, periodate oxidation of the Hib polysaccharide and meningococccal group C polysaccharide results in oligosaccharides with a reactive aldehyde group at either end (Anderson et al. 1986). These can be attached to the free amino groups of lysine residues in the carrier protein by reductive amination. Both the Hib and the meningococccal group C polysaccharides are susceptible to mild acid hydrolysis, generating a protected aldehyde group at the reducing end. This reducing end can be activated, for example by reductive amination, and a linker attached which allows covalently attachment to a carrier protein this approach has been adopted by Chiron Vaccines (Ravenscroft 2000). The polysaccharides from Salmonella typhi and Staphylococcus aureus, in contrast, have few free hydroxyl groups and are extremely resistant to hydrolysis. These polysaccharides have been activated by the addition of a bifunctional reagent to the uronic acid carboxyl group, which allows conjugation to a suitably activated carrier protein (Szu et al. 1987, Fattom et al. 1988, 1995). Treatment of a polysaccharide with cyanogen bromide, or a cyanogen bromide analogue such as CDAP (Shafer et al. 2000), results in random activation of hydroxyl groups to which a bifunctional linker, such as adipic acid dihydrazide can be attached (Shafer et al. 2000). This linker is then attached to Asp or Glu carboxyl groups on the carrier by treatment with a water-soluble carbodiimide.
Another conjugation chemistry that has been used involves partial de-N-acetylation of an N-acetylglucosamine residue in the repeat unit of the capsular polysaccharide from Streptococcus pneumoniae Type 14. Treatment of material with nitrous acid leads to the formation of an anhydromannose residue with a free aldehyde group, suitable for conjugation by reductive amination to the free Ne group of a lysine residue in an appropriate carrier protein (Laferrière et al. 1997, 1998). The effectiveness of this strategy depends on the nature and substitution pattern of the acetamido sugar.
Traditionally, the source of the glycan used for conjugate production has been natural polysaccharide isolated from the pathogen. Whilst the Hib and meningococcal CPSs have been relatively easy to activate, by periodate oxidation or after dilute acid hydrolysis, this becomes more problematic for the pneumococcal CPSs, where the greater structural diversity between serotypes means that a single method may not be suitable. The CPSs from S. typhi and S. aureus Types 5 and 8 require activation through the carboxylate groups. Total synthesis of oligosaccharides therefore has considerable attractions, as these can be produced with a ready-made activated linker attached, so that a single conjugation chemistry can be used for a wide range of products. This approach has been developed by the Cuban group for Hib conjugate vaccines (Verez-Bencomo et al. 2004). The amounts of saccharide needed are accessible, if the synthesis is optimised. For example, South America would require ca. 500 g of saccharide for its birth cohort of ca. 14 million, at 10 micrograms of saccharide per dose and a three or four dose regime. These conjugation chemistries are illustrated in Figure 4.
PHYSICOCHEMICAL CHARACTERISATION AND QUALITY CONTROL OF GLYCOCONJUGATE VACCINES
For many of the diseases which glycoconjugate vaccines prevent, there is no useful animal model which correlates with the efficacy in man. For this reason, quality control of these vaccines is heavily dependent upon physicochemical methods, and this, in turn, required careful characterisation of the final vaccine and its individual components. In addition, these relatively simple vaccines are capable of being well characterised by a combination of physical and spectroscopic methods.
Work initiated to characterise CPSs used as vaccines has carried over to the saccharide components of glycoconjugate vaccines. In our laboratory we have assigned the NMR spectra of all the CPSs used in glycoconjugate production (Jones and Lemercinier 1999). NMR spectroscopy is the sole method used by us to establish the identity and purity of the CPSs, with de-O-acetylation in situ to determine the degree of O-acetylation (Jones and Lemercinier 2002). We also have extensive NMR data to define characteristic resonances arising from CPS degradation (Jones et al. 2000). This in turn defines the predominant degradation pathway. NMR spectra can also be obtained on the activated polysaccharide, which provides information on the average size (where oligosaccharides are used), the average degree of activation and addition of a linker when intact CPSs are used, and whether there is selectivity in the activation of specific hydroxyl groups in the repeat unit (Jones 2005a; Xu et al. 2005). Molecular weight profiles of activated oligosaccharides have been obtained using size exclusion chromatography (SEC) linked with multiple angle laser-light scattering (MALLS) (D'Ambra et al. 1997) or anion exchange chromatography (Ravenscroft et al. 1999). In most cases it is possible to obtain high quality NMR spectra of the final bulk conjugate, and this is a means to ensure the integrity (identity, degree of O-acetylation, absence of degradation) of the saccharide in the final vaccine (Jones 2005a).
When toxoids used as components of other vaccines are employed as carriers in conjugate vaccines they should meet the appropriate pharmacopoeial specifications. In some cases it has proven worthwhile to use more highly purified toxoids for conjugate production. Carrier proteins not been previously used in vaccine manufacture have been subjected to more extensive characterisation, including mass spectrometry, confirmation of the amino acid sequence, size exclusion chromatography, and analysis of secondary structure and stability by optical spectroscopy (Jones et al. 2000). The applicability of different physicochemical methods to the characterisation of glycoconjugate vaccines is summarised in Figure 5.
The extent to which the three major structural types have been characterised differs. The most extensive characterisation has been performed on the neo-glycoconjugate vaccines which use CRM197 as the carrier protein, as the heterogeneity introduced by chemical toxoiding is absent. These studies have included NMR spectroscopy (which reports principally on the glycan chains), optical spectroscopy to assess the integrity and stability of the carrier protein, and molecular sizing to determine the proportion of protein molecules carrying different numbers of glycan chains. In all of the current neo-glycoprotein conjugates except the Cuban synthetic vaccine (Verez-Bencomo et al. 2004), the glycan chains are attached to amino groups, either the Ne of lysine residues or the amino group at the N-terminus. CRM197 contains 39 lysine residues and one N-terminus, of which six are substituted in the average protein. Different conjugation chemistries are likely to have different specificities for the available amines, depending on differences in pKa and steric accessibility. Attempts have been made to define this pattern for vaccines produce using active ester chemistry (the Chiron process) (Jones et al. 2000). Studies of the crosslinked conjugate vaccines have focused principally on the molecular size, which is a simple but valuable means to assess stability, and the integrity of carrier protein (Parisi and von Hunolstein 1999). Toxoids, such as tetanus toxoid, tend to be very robust.
In some respects these vaccines are no longer considered as traditional biological products. For example, it is generally accepted that a change in the manufacturing process for the polysaccharide component can be validated by detailed physicochemical characterisation of the polysaccharide, and does not require a clinical bridging trial to demonstrate equal immunogenicity with the original product for which clinical trial data was obtained. However, this probably would not be true for a change in the conjugation process (Holliday and Jones 1999). The Guidelines published by the World Health Organisation are a valuable starting point to understand the control tests and characterisation necessary for production of a new vaccine (Anonymous 2000, 2004b, c).
IMPORTANT FACTORS DEFINING THE EFFICACY AND STABILITY OF CONJUGATE VACCINES
The general principles for the quality control of vaccines are well established but there is a growing emphasis on the application of sophisticated physicochemical approaches (Dellepiane et al. 2000). Experience has shown that a very wide range of structural types can function as effective vaccines. In this section I will discuss those factors which are known to be important, or not important, in the production of an effective and stable glycoconjugate vaccine. In most cases, there is no direct link between an individual assay and the expected efficacy of the final vaccine: the intention is to demonstrate consistency in the manufacturing process and that successive production batches are essentially the same as the material used in the clinical trial.
-
The identity of the polysaccharide used to make the vaccine and confirmation of its structure in the final vaccine. This can be achieved using immunological methods or NMR spectroscopy of the bulk conjugate. Experience with the meningococcal Groups C and Y and Staphylococcus aureus conjugate vaccines seems to indicate that the absence of O-acetyl groups on the glycan chain does not result in an ineffective vaccine, consistent with the idea that an antibody population recognising the saccharide backbone is sufficient for protection (Michon et al. 2000, Doares and Cowell 2001, Fattom et al. 1998).
-
The identity of the carrier protein used to manufacture the vaccine and confirmation of its structure in the final vaccine. If the carrier protein is chemically toxoided, confirmation is required that reversion to toxicity has not occurred.
-
The polysaccharide-protein ratio. The desired immune response is against the saccharide, and low polysaccharide-protein ratios require immunisation of the infant with larger than necessary amounts of protein. On the other hand, proteolysis of the carrier protein is required to generate peptides for display by MHC II, and high polysaccharide-protein ratios can interfere with this process. Such vaccines have reduced immunogenicity. The optimal ratio has to be determined by experiment in preclinical studies or clinical trials. The mean polysaccharide-protein ratio may not be sufficient to fully define the vaccine if a single population is not present (Egan et al. 1995). A variety of procedures have been adopted to determine this parameter, principally separate determination of the polysaccharide and protein contents. These have been conjugate specific, reflecting the differences in the chemistry of the glycan chains. NMR spectroscopy provides a more generic approach (Figure 6).
-
The presence of free polysaccharide. Free (unconjugated) polysaccharide reduces the immunogenicity of the vaccine, possibly by competing for the sIg molecules. The amount of free saccharide (and unconjugated carrier protein) in the vaccine are key quality control specifications (Peeters et al. 1992).
-
Degradation and depolymerisation of the saccharide chain is the principal mechanism by which some glycoconjugate vaccines lose immunogenicity. Coincidentally, the first vaccines against Hib and MenC employed some of the least stable CPSs. Degradation of the saccharide chains can be monitored by an increase in free saccharide, by changes in molecular size, or by the appearance of characteristic resonances in the NMR spectrum of bulk conjugate (Jones et al. 2000: Figure 7). With a neoglycoconjugate vaccine, cleavage of the glycan chain inevitably gives rise to an increase in free saccharide, whilst in a crosslinked network vaccine two cleavages in close proximity are required. Consequently there is a lag phase between the beginning of glycan depolymerisation and the appearance of free saccharide. Aluminium hydroxide adjuvants have been shown to catalyse the depolymerisation of the Hib CPS (Sturgess et al. 1999).
-
The size of the glycan chains. Small glycan chains can be used (Jansen and Snippe 2004), but all manufacturers maintain specifications of the glycans conjugated as an indication that they are producing a consistent product (Costantino et al. 1999).
-
Molecular sizing of the conjugates is a simple and effective means to ensure consistency of the final conjugate (Parisi and von Hunolstein 1999).
-
Mild denaturation of the carrier protein almost certainly does not reduce the immunogenicity of the conjugate, since the role of the carrier is to be degraded and to provide peptides for display by the MHC II (Siber 1994). More extensive denaturation may result in aggregation, which may influence availability of the conjugate, although such conditions are also likely to cause depolymerisation of the glycan chains.
-
The stability of the linkage between the polysaccharide and the carrier protein.
-
Limit specifications exist for reagents used in the conjugation process and for uncapped activation sites (Anonymous 2002a, 2004b, c).
CLINICAL EFFICACY OF GLYCOCONJUGATE VACCINES
The clinical trials of 4 different Hib conjugate vaccines (called PRP-D, PRP-T, PRP-OMP and HbOC) manufactured using different sized glycan chains, different carrier proteins and different conjugation chemistries showed high efficacy, a reduction in the incidence of disease, in each case (Heath 1998, Booy et al. 1994, Black et al. 1991, Santosham et al. 1991, Eskola et al. 1987). Of these vaccines, PRP-D, in which the carrier protein is diphtheria toxoid, was not demonstrated to be effective in populations with high levels of endemic disease. On the other hand, PRP-OMP (which used a complex of meningococcal outer membrane proteins as the carrier) proved particularly effective in this population, perhaps because a protective antibody response is elicited after the first dose (Figure 3). Whilst different target populations can react in quantitatively different ways (Santosham et al. 1992), the clear conclusion was that the Hib conjugate vaccines produced by a variety of routes are efficacious in the prevention of disease. Because of the low incidence of meningococccal disease in the UK and the well-established correlation between antibody level and protection (Goldschneider et al. 1969a, b), licensing of meningococccal group C conjugate vaccines in the UK was on basis of immunogenicity, and particularly functional antibody levels as measured by a bacteriocidal assay, rather than protection against disease. Subsequent surveillance of disease incidence in the UK has indicated an efficacy for these vaccines of approximately 93% (Balmer et al. 2002). Clinical trials of the heptavalent pneumococcal conjugate vaccine in California were so clear cut that the trial was stopped at an early stage (Shinefield et al. 1999, Black et al. 2001). However, clinical trials in Soweto, South Africa (Huebner et al. 2002), and in a Navajo Indian population in the US have indicated lower clinical efficacy (O'Brien et al. 2003). It is not clear what factors influence this poorer nutrition, or environmental or genetic factors.
Licensing of new pneumococcal conjugate vaccines may prove complicated due to a requirement from the FDA that a new vaccine is "non-inferior" to existing vaccines (Jodar et al. 2003, Lee et al. 2003). For example, is a vaccine which induces a slightly lower immune response to one serotype but contains several additional serotype immunogens "inferior" to an existing vaccine? Production and quality control factors for the development of pneumococcal conjugate vaccines have been codified by WHO (Anonymous 2004c).
GLYCOCONJUGATE VACCINES UNDER DEVELOPMENT
There are a large number of novel glycoconjugate vaccines under development by academic laboratories, by established vaccine manufacturers and smaller start-up biotechnology companies and by public non-governmental institutions. Table III contained details of the novel glycoconjugate vaccines reported to be in development or in clinical trials. Whilst most of these vaccines continue to use high molecular weight repeating polysaccharides as the saccharide hapten, in some lower molecular weight glycans such as the short saccharide chains present on lipo-oligosaccharides are used. In addition, the developmental conjugate vaccine against the encapsulated fungal pathogen Cryptococcus neoformans shows that this technology is not limited to protection against bacterial infection (Devi 1996).
In developed countries, the costs of vaccine delivery to the target population usually exceed the costs of the vaccine, and there is considerable pressure to reduce the number of injections that a baby receives. These pressures, and commercial concerns, favour the development of combination vaccines, such a hexavalent DTP-Hib-HB-IPV combinations. However, the immune response to one or more of the components in such combination vaccines is frequently reduced (Granoff 2001, Redhead et al. 1994), although this is not generally considered to be clinically significant. Clinical assessment and quality control of these complex combination vaccines can be extremely difficult, especially in countries where mass immunisation programmes are already in place and disease incidence may be extremely low.
VACCINE DEMAND AND VACCINE PRICES
Most vaccines are commercial products, and to be widely used demands that they be available to purchasers at prices that they can afford. The major burden of bacterial disease is in amongst the young in developing countries. Whilst novel vaccines may be expensive when initially introduced, the expectation is that mass production, amortisation of capital investment and the emergence of multiple producers will drive prices down, expanding vaccine usage into less wealthy countries, leading to further reduction in prices. A number of non-governmental organisations and charities are working to speed the introduction of novel vaccines into developing countries. The Hib glycoconjugate vaccine, global use of which is a WHO aim, is a example. The global birth cohort is nearly 130 million (http://www.census.gov/ipc/prod/wp02/tabA-03.pdf). Vaccination of these infants with three or four dose regimes will require between 400 million and 500 million doses of vaccine per year. Production in developed countries is currently of the order of 100 million doses per year. According to published figures, the US government pays approximately $8 per dose for Hib conjugate vaccines in single dose vials (Jacobson et al. 2003). Hib conjugate vaccine is available to South American governments through the PAHO Revolving Fund for $3.50 per dose (http://www.paho.org/English/AD/FCH/IM/sne2601.pdf). The World Health Organisation believes that global uptake of Hib conjugate vaccine requires a target price of approximately $1 per dose. A number of manufacturers in developing countries are attempting to fill the production gap using a variety of different techniques. The Cuban government have recently licensed a Hib conjugate vaccines produced from synthetic oligosaccharides linked to tetanus toxoid (Verez-Bencomo et al. 2004). Manufacturers in Indonesia, Brazil, India and Mexico are developing crosslinked network vaccines, sometimes in collaboration with other organisations.
Introduction of the other conjugate vaccines has been more recent. Meningococccal Group C conjugates were first introduced into the UK in 1998 (Miller et al. 2001) whilst pneumococcal conjugate vaccines are even more recent. Current prices for these vaccines are higher than for the Hib conjugate vaccine. A recent study assessing cost-effectiveness of Men C conjugate in Canada was based on a price of CDN$50 per dose (De Wals et al. 2004). The prices of complex and multivalent conjugates, such as a pneumococcal conjugates, are particularly high, with the heptavalent pneumococcal conjugate being more than $50 per dose, and a four dose regime is required (Pai et al. 2002). Thus the conjugate vaccines are relatively expensive products. Prices for polysaccharide vaccines are of the order of a few cents per dose for S. typhi Vi polysaccharide, while trivalent meningococccal A,C,W135 polysaccharide vaccine cost 1 euro per dose (Anonymous 2004a). Other Revolving Fund prices for other vaccines are often of the order of cents per dose or a few US$s per dose (http://www.paho.org/English/AD/FCH/IM/sne2601.pdf). There is an acknowledged fragmentation of the global vaccine market, with the use of complex, expensive combination vaccine in industrialised countries, whilst developing countries require simpler monovalent vaccines. A key factor in this is the introduction of expensive acellular pertussis and inactivated polio components into combinations for industrialised countries, replacing the cheaper whole cell pertussis vaccines. This provides opportunities for manufacturers in developing countries/emerging economies such as Brazil, Indonesia or India to supply the mass markets through contracts with PAHO and UNICEF (http://www.who.int/vaccines-access/supply/Divergence_vaccines.pdf). In other cases conjugate vaccines are not the object of commercial development due to low incidence of disease in rich developed countries and the low income of a target population in developing countries. The Programme for Appropriate Technologies in Health (PATH) are, for example, co-ordinating a project funded by the Gates Foundation, the Rockefeller Foundation, and the World Bank to produce a cheap US$0.40 per dose target price meningococcal Group A conjugate vaccine appropriate for use in sub-Saharan Africa (http://www.meningvax.org/timeline.htm). The International Vaccine Institute in Korea, through its Diseases of the Most Impoverished (DOMI) programme is working to introduce affordable vaccines against typhoid, shigellosis, and cholera (http://www.ivi.int).
THE FUTURE
Existing glycoconjugate vaccines represent the first examples of the use of a generic technology which offers the possibility to reduce the death toll taken on young infants by many other infectious diseases. Unlike, for example, DNA vaccines, they have been proven to work in mass paediatric vaccine campaigns and there would seem to be no major technological hurdles to the development of many other highly effective vaccines of this type. There is a now an extensive body of academic work demonstrating proof-of-principle for a wide range of vaccines. However, conjugates are expensive and the limitation to the development of this technology is likely to be commercial. To some extent, this is being addressed by the involvement of philanthropic foundations and non-governmental organisations, but there is clearly much more to be done in this field.
Manuscript received on February 14, 2005; accepted for publication on February 16, 2005; presented by LUCIA MENDONÇA PREVIATO
E-mail: cjones@nibsc.ac.uk
- ACHARYA IL, LOWE CU, THAPA R, GURUBACHARYA VL, SHRESTHA MB, CADOZ M, SCHULZ D, ARMAND J, BRYLA DA, TROLLFORS B, CRAMTON T, SCHNEERSON R AND ROBBINS JB. 1987. Prevention of typhoid fever in Nepal with the Vi capsular polysaccharide of Salmonella typhi A preliminary report. N Eng J Med 317: 1101-1104.
- AKKOYUNLU M, MELHUS A, CAPIAU C, VAN OPSTAL O AND FORSGREN A. 1997. The acylated form of protein D of Haemophilus influenzae is more immunogenic than the nonacylated form and elicits an adjuvant effect when it is used as a carrier conjugated to polyribosyl ribitol phosphate. Infect Immun 65: 5010-5016.
- ALEXANDER J, DEL GUERCIO MF, MAEWAL A, QIAO L, FIKES J, CHESNUT RW, PAULSON J, BUNDLE DR, DEFREES S AND SETTE A. 2000. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J Immunol 164: 1625-1633.
- ANDERSON P, INSEL RA, SMITH DH, CATE TR, COUCH RB AND GLEZEN WP. 1981. A polysaccharide-protein complex from Haemophilus influenzae type b. 3. Vaccine trial in human adults. J Infect Dis 144: 530-538.
- ANDERSON P, PICHICHERO ME AND INSEL RA. 1985. Immunogens consisting of oligosaccharides from the capsule of Haemophilus influenzae type b coupled to diphtheria toxoid or the toxin protein CRM197. J Clin Invest 76: 52-59.
- ANDERSON PW, PICHICHERO ME, INSEL RA, BETTS R, Eby R AND SMITH DH. 1986. Vaccines consisting of periodate-cleaved oligosaccharides from the capsule of Haemophilus influenzae type b coupled to a protein carrier: structural and temporal requirements for priming in the human infant. J Immunol 137: 1181-1186.
- ANONYMOUS. 1998. Global Programme for Vaccinesand Immunisation: the WHO position paper on Haemophilus influenzae type b conjugate vaccines. Weekly Epidemiological Record 73: 64-68. Available at: www.who.int/vaccines-documents/PP-WER/wer7310.pdf
- ANONYMOUS. 2000. Recommendations for the production and control of Haemophilus influenzae type b conjugate vaccines. WHO Technical Report Series 897: 27-59. Available at: http://whqlibdoc.who.int/trs/WHO_TRS_897.pdf
- ANONYMOUS. 2002a. Meningococcal vaccines: polysaccharide and polysaccharide conjugate vaccines: WHO position paper. Weekly Epidemiological Record 77: 331-339. Available at: www.who.int/vaccines-documents/PP-WER/wer7740.pdf
- ANONYMOUS. 2002b. WHO-UNICEF joint statement of strategies to reduce measles mortality wordwide. Weekly Epidemiological Record 77: 224-228.
- ANONYMOUS. 2003. Pneumococcal vaccines: WHO position paper. Weekly Epidemiological Record 78: 110-119. Available at: www.who.int/vaccines-documents/PP-WER/wer7423.pdf
- ANONYMOUS. 2004a. Potential meningitis tragedy averted. Bull WHO 82: 398.
- ANONYMOUS. 2004b. Recommendations for the production and quality control of group C meningococcal conjugate vaccines. WHO Technical Report Series 924: 102-128.
- ANONYMOUS. 2004c. Recommendations for the production and quality control of pneumococcal conjugate vaccines. WHO Technical Report Series in press.
- ASHKENAZI S, PASSWELL JH, HARLEY E, MIRON D, DAGAN R, FARZAN N, RAMON R, MAJADLY F, BRYLA DA, KARPAS AB, ROBBINS JB, SCHNEERSON R AND THE ISRAEL PEDIATRIC SHIGELLA STUDY GROUP. 1999. Safety and immunogenicity of Shigella sonnei and Shigella flexneri 2a O-specific polysaccharide conjugates in children. J Infect Dis 179: 1565-1568.
- AVERY OT AND GOEBEL WF. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins: V. The immunological specificity of an antigen prepared by combining the capsular polysaccharide of Type III pneumococcus with foreign protein. J Exp Med 54: 437-447.
- BAKER CJ, PAOLETTI LC, WESSELS MR, GUTTORMSEN HK, RENCH MA, HICKMAN ME AND KASPER DL. 1999. Safety and immunogenicity of capsular polysaccharide-tetanus toxoid conjugate vaccines for group B streptococcal types Ia and Ib. J Infect Dis 179: 142-50.
- BAKER CJ, PAOLETTI LC, RENCH MA, GUTTORMSEN HK, CAREY VJ, HICKMAN ME AND KASPER DL. 2000. Use of capsular polysaccharide-tetanus toxoid conjugate vaccine for type II group B Streptococcus in healthy women. J Infect Dis 182: 1129-1138.
- BALMER P, BORROW R AND MILLER E. 2002. Impact of meningococcal C conjugate vaccine in the UK. J Med Microbiol 51: 717-722.
- BAXENDALE HE, DAVIS Z, WHITE HN, SPELLERBERG MB, STEVENSON FK AND GOLDBLATT D. 2000. Immunogenetic analysis of the immune response to pneumococcal polysaccharide. Eur J Immunol 30: 1214-1223.
- BENAISSA-TROUW B, LEFEBER DJ, KAMERLING JP, VLIEGENTHART JFG, KRAAIJEVELD K AND SNIPPE H. 2001. Synthetic polysaccharide type 3-related di-, tri-, and tetrasaccharide-CRM197 conjugates induce protection against Streptococcus pneumoniae type 3 in mice. Infect Immun 69: 4698-4701.
- BHATTACHARJEE AK, JENNING HJ, KENNY CP, MARTIN A AND BUNDLE DR. 1975. Structural determination of the sialic acid polysaccharide antigens of Neisseria meningitidis serogroups B and C with carbon 13 nuclear magnetic resonance. J Biol Chem 250: 1926-1932.
- BHATTACHARJEE AK, JENNING HJ, KENNY CP, MARTIN A AND BUNDLE DR. 1976. Structural determination of the polysaccharide antigens of Neisseria meningitidis serogroups Y, W-135, and BO1. Can J Biochem 54: 1-8.
- BLACK SB, SHINEFIELD HR, FIREMAN B, HIATT R, POLEN M AND VITTINGHOFF E. 1991. Efficacy in infancy of oligosaccharide conjugate Haemophilus influenzae type b (HbOC) vaccine in a United States population of 61,080 children. The Northern California Kaiser Permanente Vaccine Study Center Pediatrics Group. Pediatr Infect Dis J 10: 97-104.
- BLACK SB, SHINEFIELD HR, HANSEN J, ELVIN L, LAUFER D AND MALINOSKI F. 2001. Postlicensure evaluation of the effectiveness of seven valent pneumococcal conjugate vaccine. Ped Infect Dis J 20: 1105-1107.
- BLUM MD, DAGAN R, MENDELMAN PM, PINSK V, GIORDANI M, LI S, BOHIDAR N AND MCNEELY TB. 2000. A comparison of multiple regimens of pneumococcal polysaccharide-meningococcal outer membrane protein complex conjugate vaccine and pneumococcal polysaccharide vaccine in toddlers. Vaccine 18: 2359-2367.
- BOOY R, HODGSON S, CARPENTER L, MAYON-WHITE RT, SLACK MP, MACFARLANE JA, HAWORTH EA, KIDDLE M, SHRIBMAN S AND ROBERTS JS. 1994. Efficacy of Haemophilus influenzae type b conjugate vaccine PRP-T. Lancet 344: 362-366.
- BOUTONNIER A, VILLENEUVE S, NATO F, DASSY B AND FOURNIER J-M. 2001. Preparation, immunogenicity, and protective efficacy, in a murine model, of a conjugate vaccine composed of the polysaccharide moiety of the lipopolysaccharide of Vibrio cholerae O139 bound to tetanus toxoid. Infect Immun 69: 3488-3493.
- BOWDEN RA, CLOECKAERT A, ZYGMUNT MS AND DUBRAY G. 1995. Outer-membrane protein- and rough lipopolysaccharide-specific monoclonal antibodies protect mice against Brucella ovis J Med Microbiol 43: 344-347.
- BRAHMBHATT HN, LINDBERG AA AND TIMMIS KN. 1992. Shigella lipopolysaccharide: structure, genetics, and vaccine development. Curr Topics Microbiol Immunol 180: 45-64.
- BRANEFORS-HELANDER P, ERBING C, KENNE L AND LINDBERG B. 1976. Structural studies of the antigen from Haemophilus influenzae type b. Acta Chem Scand B 30: 276-277.
- BRUGE J, BOUVERET-LE CAM N, DANVE B, ROUGON G AND SCHULZ D. 2004. Clinical evaluation of a group B meningococcal N-propionylated polysaccharide conjugate vaccine in adult, male volunteers. Vaccine 22: 1087-1096.
- BUNDLE DR, SMITH ICP AND JENNING HJ. 1974. Determination of the structure and conformation of bacterial polysaccharides by carbon 13 nuclear magnetic resonance. Studies on the group-specific antigens of Neisseria meningitidis serogroups A and X. J Biol Chem 249: 2275-2281.
- BYSTRICKY S, PAULOVICOVA E AND MACHOVA E. 2003. Candida albicans mannan-protein conjugate as vaccine candidate. Immunol Lett 85: 251-255.
- CASADEVALL A, MUKHERJEE J, DEVI SJN, SCHNEERSON R, ROBBINS JB AND SCHARFF MD. 1992. Antibodies elicited by a Cryptococcus neoformans-tetanus toxoid conjugate vaccine have the same specificity as those elicited in infection. J Infect Dis 165: 1086-1093.
- CHERNYAK A, KONDO S, WADE TK, MEEKS MD, ALZARI PM, FOURNIER JM, TAYLOR RK, KOVACS P AND WADE WF. 2002. Induction of protective immunity by synthetic Vibrio cholerae hexasaccharide derived from V. cholerae O1 Ogawa lipopolysaccharide bound to a carrier protein. J Infect Dis 185: 950-952.
- CHOI JI, SCHIFFERLE RE, YOSHIMURA F AND KIM BW. 1998. Capsular polysaccharide-fimbrial protein conjugate vaccine protects against Porphyromonas gingivalis infection in SCID mice reconstituted with human peripheral blood lymphocytes. Infect Immun 66: 391-393.
- CHU CY, LIU BK, WATSON D, SZU SC, BRYLA D, SHILOACH J, SCHNEERSON R AND ROBBINS JB. 1991. Preparation, characterization, and immunogenicity of conjugates composed of the O-specific polysaccharide of Shigella dysenteriae type 1 (Shiga's bacillus) bound to tetanus toxoid. Infect Immun 59: 4450-4458.
- COBB BA, WANG Q, TZIANABOS AO AND KASPER DL. 2004. Polysaccharide processing and presentation by the MHCII pathway. Cell 117: 677-687.
- COCHI SL, BROOME CV AND HIGHTOWER AW. 1985. Immunisation of US children with Haemophilus influenzae type b polysaccharide vaccine. JAMA 253: 521-527.
- COHEN D, ASHKENAZI S, GREEN MS, GDALEVICH M, ROBIN G, SLEPON R, YAVZORI M, ORR N, BLOCK C, ASHKENAZI I, SHEMER J, TAYLOR DN, HALE TL, SADOFF JC, PAVLIAKOVA D, SCHNEERSON R AND ROBBINS JB. 1997. Double-blind vaccine-controlled randomised efficacy trial of an investigational Shigella sonnei conjugate vaccine in young adults. Lancet 349: 155-159.
- CONLAN JW, COX AD, KUOLEE R, WEBB A AND PERRY MB. 1999. Parenteral immunization with a glycoconjugate vaccine containing the O157 antigen of Escherichia coli O157:H7 elicits a systemic humoral immune response in mice, but fails to prevent colonization by the pathogen. Can J Microbiol 45: 279-286.
- CONLAN JW, SHEN H, WEBB A AND PERRY MB. 2002. Mice vaccinated with the O-antigen of Francisellatularensis LVS lipopolysaccharide conjugated to bovine serum albumin develop varying degrees of protective immunity against systemic or aerosol challenge with virulent type A and type B strains of the pathogen. Vaccine 20: 3465-3471.
- COSTANTINO P, NORELLI F, GIANNOZZI A, D'ASCENZI S, BARTOLINI A, KAUR S, TANG D, SEID R, VITI S, PAFFETTI R, BIGIO M, PENNATINI C, AVERANI G, GUARNIERI V, GALLO E, RAVENSCROFT N, LAZZERONI C, RAPPUOLI R AND CECCARINI C. 1999. Size fractionation of bacterial capsular polysaccharides for their use in conjugate vaccines. Vaccine 17: 1251-1263.
- COUGHLIN RT AND BOGARD JR WC. 1987. Immunoprotective murine monoclonal antibodies specific for the outer-core polysaccharide and for the O-antigen of Escherichia coli 0111:B4 lipopolysaccharide (LPS). J Immunol 139: 557-561.
- CRISEL RM, BAKER RS AND DORMAN DE. 1975. Capsular polysaccharide of Haemophilus influenzae, type b: 1 Structural characterisation of the capsular polymer of strain Eagan. J Biol Chem 250: 4926-4930.
- CROSS AS, SADOFF JC, FURER E AND CRYZ JR SJ. 1990. Escherichia coli and Klebsiella vaccines and immunotherapy. Infect Dis Clin North Am 4: 271-282.
- CROSS AS, OPAL SM, PALARDY JE, DRABICK JJ, WARREN HS, HUBER C, COOK P AND BHATTACHARJEE AK. 2003. Phase I study of detoxified Escherichia coli J5 lipopolysaccharide (J5dLPS)/group B meningococcal outer membrane protein (OMP) complex vaccine in human subjects. Vaccine 21: 4576-4587.
- CRUMP JA, LUBY SP AND MINTZ ED. 2004. The global burden of typhoid fever. Bull WHO 82: 346-353.
- CRYZ JR SJ, CROSS AS, SADOFF JC, WEGMANN A, QUE JU AND FURER E. 1991. Safety and immunogenicity of Escherichia coli O18 O-specific polysaccharide (O-PS)-toxin A and O-PS-cholera toxin conjugate vaccines in humans. J Infect Dis 163: 1040-1045.
- CRYZ JR SJ, LANG A, RUDEBERG A, WEDGWOOD J, QUE JU, FURER E AND SCHAAD U. 1997. Immunization of cystic fibrosis patients with a Pseudomonas aeruginosa O-polysaccharide-toxin A conjugate vaccine. Behring Inst Mittl, p. 345-349.
- D'AMBRA A, BAUGHER JE, CONCAMMON PE, PON RA AND MICHON F. 1997. Direct and indirect methods for molar-mass analysis of fragments of the capsular polysaccharide of Haemophilus influenzae type b. Analyt Biochem 250: 228-236.
- DE WALS P, NGUYEN VH, ERICKSON LJ, GUAY M, DRAPEAU J AND ST-LAURENT J. 2004. Cost-effectiveness of immunization strategies for the control of serogroup C meningococcal disease. Vaccine 22: 1233-1240.
- DECKER MD, EDWARDS KM, BRADLEY R AND PALMER P. 1992. Comparative trial in infants of four conjugate Haemophilus influenzae type b vaccines. J Pediatrics 120: 184-189.
- DELLEPIANE N, GRIFFITHS E AND MILSTEIN JB. 2000. New challenges in assuring vaccine quality. Bull WHO 78: 155-162.
- DEVI SJ, ZOLLINGER WD, SNOY PJ, TAI JY, COSTANTINI P, NORELLI F, RAPPUOLI R AND FRASCH CE. 1997. Preclinical evaluation of group B Neisseria meningitidis and Escherichia coli K92 capsular polysaccharide-protein conjugate vaccines in juvenile rhesus monkeys. Infect Immun 65: 1045-1052.
- DEVI SJN. 1996. Preclinical efficacy of a glucuronoxylomannan-tetanus toxoid conjugate vaccine of Cryptococcus neoformans in a murine model. Vaccine 14: 841-844.
- DEVI SJN, SCHNEERSON R, EGAN W, ULRICH TJ, BRYLA DA, ROBBINS JB AND BENNETT JA. 1991. Cryptococcus neoformans serotype A glucuronoxylomannan-protein conjugate vaccines: synthesis, characterization, and immunogenicity. Infect Immun 59: 3700-3707.
- DOARES SH AND COWELL JL. 2001. Role of O-acetyl groups in saccharide-protein conjugate vaccines for the production of functional antibodies. Abstracts of papers of Am Chem Soc 222: 49-CARB Part 1.
- DONNELLY JJ, DECK RR AND LIU MA. 1990. Immunogenicity of a Haemophilus influenzae polysaccharide-Neisseria meningitidis outer membrane protein complex conjugate vaccine. J Immunol 145: 3071-3079.
- EDELMAN R AND LEVINE MM. 1986. Summary of an international workshop on typhoid fever. Rev Infect Dis 8: 329-349.
- EGAN W, FRASCH CE AND ANTONY BF. 1995. Lot-release criteria, post-licensure quality control and the Haemophilus influenzae type b conjugate vaccines. J Am med Assoc 273: 888-889.
- ESKOLA J AND ANTTILA M. 1999. Pneumococcal conjugate vaccines. Pediatr Infect Dis J 18: 543-551.
- ESKOLA J, PELTOLA H, TAKADA AK, KAYTHY H, HAKULINEN M, KARANKO V, KELA E, REKOLA P, RONNBERG PR AND SAMUELSON JS. 1987. Efficacy of Haemophilus influenzae type b polysaccharide-diphtheria toxoid conjugate vaccine in infancy. N Engl J Med 317: 717-722.
- FATTOM A, VANN WF, SZU SC, SUTTON A, LI X, BRYLA D, SCHIFFMAN G, ROBBINS JB AND SCHNEERSON R. 1988. Synthesis and physicochemical and immunological characterization of pneumococcus type 12F polysaccharide-diphtheria toxoid conjugates. Infect Immun 56: 2292-2298.
- FATTOM A, SCHNEERSON R, SZU SC, VANN WF, SHILOACH J, KARAKAWA WW AND ROBBINS JB. 1990. Synthesis and immunologic properties in mice of vaccines composed of Staphylococcus aureus type 5 and type 8 capsular polysaccharides conjugated to Pseudomonas aeruginosa exotoxin A. Infect Immun 58: 2367-2374.
- FATTOM A, SCHNEERSON R, WATSON DC, KARAKAWA WW, FITZGERALD D, PASTAN I, LI X, SHILOACH J, BRYLA DA AND ROBBINS JB. 1993. Laboratory and clinical evaluation of conjugate vaccines composed of Staphylococcus aureus type 5 and type 8 capsular polysaccharides bound to Pseudomonas aeruginosa recombinant exoprotein A. Infect Immun 61: 1023-1032.
- FATTOM A, LI X, CHO YH, BURNS A, HAWWARI A, SHEPHERD SE, COUGHLIN R, WINSTON S AND NASO R. 1995. Effect of conjugation methodology, carrier protein, and adjuvants on the immune response to Staphylococcus aureus capsular polysaccharides. Vaccine 13: 1288-1293.
- FATTOM AI, SARWAR J, BASHAM L, ENNIFAR S AND NASO R. 1998. Antigenic determinants of Staphylococcus aureus type 5 and type 8 capsular polysaccharide vaccines. Infect Immun 66: 4588-4592.
- FATTOM AI, HORWITH G, FULLER S, PROPST M AND NASO R. 2004. Development of StaphVAX, a polysaccharide conjugate vaccine against S. aureus infection: from the lab bench to phase III clinical trials. Vaccine 22: 880-887.
- FLECK F. 2003. WHO and MSF appeal for funds for new meningitis vaccine. Brit Med J 372: 769.
- FULOP M, MASTROENI M, GREEN M AND TITBALL RW. 2001. Role of antibody to lipopolysaccharide in protection against low- and high-virulence strains of Francisella tularensis Vaccine 19: 4465-4472.
- FUSCO PC, MICHON F, TAI JY AND BLAKE MS. 1997. Preclinical evaluation of a novel group B meningococcal conjugate vaccine that elicits bactericidal activity in both mice and nonhuman primates. J Infect Dis 175: 364-372.
- GILBERT FB, POUTREL B AND SUTRA L. 1994. Immunogenicity in cows of Staphylococcus aureus type 5 capsular polysaccharide-ovalbumin conjugate. Vaccine 12: 369-374.
- GOLDSCHNEIDER I, GOTSHLICH EC AND ARTENSTEIN MS. 1969a. Human immunity to the meningococcus: I. The role of humoral antibodies. J Exp Med 129: 1307-1326.
- GOLDSCHNEIDER I, GOTSHLICH EC AND ARTENSTEIN MS. 1969b. Human immunity to the meningococcus: II. Development of natural immunity. J Exp Med 129: 1327-1348.
- GRANOFF DM. 2001. Assessing efficacy of Haemophilus influenzae type b combination vaccines. Clin Infect Dis 33 (Suppl 4): S278-287.
- GRANOFF DM, SHACKELFORD PG, HOLMES SJ AND LUCAS AH. 1993. Variable expression in the antibody response of infants vaccinated with Haemophilus influenzae type b polysaccharide-protein conjugates: description of a new l light chain-associated idiotype and the relationship between idiotype expression, avidity and vaccine formulation. J Clin Invest 91: 788-796.
- GRAVEKAMP C, KASPER DL, PAOLETTI LC AND MADOFF LC. 1999. Alpha C protein as a carrier for type III capsular polysaccharide and as a protective protein in group B streptococcal vaccines. Infect Immun 67: 2491-2499.
- GU XX AND TSAI CM. 1993. Preparation, characterization, and immunogenicity of meningococcal lipo-oligosaccharide-derived oligosaccharide-protein conjugates. Infect Immun 61: 1873-1880.
- GU XX, TSAI CM, UEYAMA T, BARENKAMP SJ, ROBBINS JB AND LIM DJ. 1996. Synthesis, characterization, and immunologic properties of detoxified lipooligosaccharide from nontypeable Haemophilus influenzae conjugated to proteins. Infect Immun 64: 4047-4053.
- GU XX, SUN J, JIN S, BARENKAMP SJ, LIM DJ, ROBBINS JB AND BATTEY J. 1997. Detoxified lipooligosaccharide from nontypeable Haemophilus influenzae conjugated to proteins confers protection against otitis media in chinchillas. Infect Immun 65: 4488-4493.
- GU XX, CHEN J, BARENKAMP SJ, ROBBINS JB, TSAI CM, LIM DJ AND BATTEY J. 1998. Synthesis and characterization of lipooligosaccharide-based conjugates as vaccine candidates for Moraxella (Branhamella) catarrhalis Infect Immun 66: 1891-1897.
- GUPTA RK, EGAN W, BRYLA DA, ROBBINS JB AND SZU SC. 1995. Comparative immunogenicity of conjugates composed of Escherichia coli O111 O-specific polysaccharide, prepared by treatment with acetic acid or hydrazine, bound to tetanus toxoid by two synthetic schemes. Infect Immun 63: 2805-2810.
- GUPTA RK, TAYLOR DN, BRYLA DA, ROBBINS JB AND SZU SC. 1998. Phase 1 evaluation of Vibrio cholerae O1, serotype Inaba, polysaccharide-cholera toxin conjugates in adult volunteers. Infect Immun 66: 3095-3099.
- HAMASUR B, HAILE M, PAWLOWSKI A, SCHRODER U, WILLIAMS A, HATCH G, HALL G, MARSH P, KALLENIUS G AND SVENSON SB. 2003. Mycobacterium tuberculosis arabinomannan-protein conjugates protect against tuberculosis. Vaccine 21: 4081-4093.
- HAN Y, ULRICH MA AND CUTLER JE. 1999. Candida albicans mannan extract-protein conjugates induce a protective immune response against experimental candidiasis. J Infect Dis 179: 1477-1484.
- HATANO K, BOISOT S, DESJARDINS D, WRIGHT DC, BRISKER J AND PIER GB. 1994. Immunogenic and antigenic properties of a heptavalent high-molecular-weight O-polysaccharide vaccine derived from Pseudomonas aeruginosa Infect Immun 62: 3608-3616.
- HEATH PT. 1998. Haemophilus influenzae type b conjugate vaccines: a review of efficacy data. Pediatr Infect Dis J 17: S117-122.
- HEYNS K AND KIESSLING G. 1967. Struckturaufklarung das Vi-Antigens aus Citrobacter freundii (E. coli) 5396/38. Carbohydr Res 3: 340-353.
- HOLLIDAY MR AND JONES C. 1999. Meeting Report: WHO-Co-sponsored informal Workshop on the use of physicochemical methods for the characterization of Haemophilus influenzae type b conjugate vaccines. Biologicals 27: 51-53.
- HOUGS L, JUUL L, DITZEL HJ, HEILMANN C, SVEJGAARD A AND BARINGTON T. 1999. The first dose of a Haemophilus influenzae type b conjugate vaccine reactivates memory B cells: evidence for extensive clonal selection, intraclonal affinity maturation, and multiple isotype switches to IgA2. J Immunol 162: 224-237. Erratum published in J Immunol 2001 166: 2147.
- HU WG, CHEN J, BATTEY JF AND GU XX. 2000. Enhancement of clearance of bacteria from murine lungs by immunization with detoxified lipooligosaccharide from Moraxella catarrhalis conjugated to proteins. Infect Immun 68: 4980-4985.
- HUEBNER J, QUAAS A, KRUEGER WA, GOLDMANN DA AND PIER GB. 2000. Prophylactic and therapeutic efficacy of antibodies to a capsular polysaccharide shared among vancomycin-sensitive and -resistant enterococci. Infect Immun 68: 4631-4636.
- HUEBNER RE, MBELLE N, FORREST B, MADORE DV AND KLUGMAN KP. 2002. Immunogenicity after one, two or three doses and impact on the antibody response to coadministered antigens of a nonavalent pneumococcal conjugate vaccine in infants of Soweto, South Africa. Ped Infect Dis J 21: 1004-1007.
- INSEL RA AND ANDERSON PW. 1986. Oligosaccharide-protein conjugate vaccines induce and prime for oligoclonal IgG antibody responses to the Haemophilus influenzae type b capsular polysaccharide in human infants. J Exp Med 163: 262-269.
- IVANOFF B AND CHAIGNAT C. 2002. Anticholera vaccines and vaccination. Bull Societe Pathologie Exotique (1990) 95: 355-358.
- JACKSON LA, NEUZIL KM, YU O, BENSON P, BARLOW WE, ADAMS AL, HANSON CA, MAHONEY LD, SHAY DK, THOMPSON ET AL. 2003. Effectiveness of pneumococcal polysaccharide vaccine in older adults. New Eng J Med 348: 1747-1755.
- JACOBSON SH, KARNANI T AND SEWELL EC. 2003. Analyzing the economic value of the hepatitis B-Haemophilus influenzae type B combination vaccine by reverse engineering a formulary selection algorithm. Vaccine 21: 2169-2177.
- JANSEN WT AND SNIPPE H. 2004. Short-chain oligosaccharide protein conjugates as experimental pneumococcal vaccines. Indian J Med Res 119 (Suppl): 7-12.
- JANSEN WT, HOGENBOOM S, THIJSSEN MJ, KAMERLING JP, VLIEGENTHART JFG, VERHOEF J, SNIPPE H AND VERHEUL AF. 2001. Synthetic 6B di-, tri-, and tetrasaccharide-protein conjugates contain pneumococcal type 6A and 6B common and 6B-specific epitopes that elicit protective antibodies in mice. Infect Immun 69: 787-793.
- JANSSON PE, LINDBERG B AND LINDQUIST U. 1985. Structural studies of the capsular polysaccharide from Streptococcus pneumoniae type 5. Carbohydr Res 140: 101-110.
- JANSSON PE, LINDBERG B, ANDER SSON M, LINDQUIST U AND HENRICHSEN J. 1998. Structural studies of the capsular polysaccharide from Streptococcus pneumoniae type 2, a re-investigation. Carbohydr Res 182: 111-117.
- JENNINGS HJ AND LUGOWSKI C. 1981. Immunochemistry of Group-A, Group-B and Group-C meningococcal polysaccharide tetanus toxoid conjugates. J Immunol 127: 1011-1018.
- JENNINGS HJ, ROSELL K-G AND CARLO DJ. 1980. Structural determination of the capsular polysaccharide of Streptococcus pneumoniae type-19 (19F). Can J Chem 58: 1069-1074.
- JIAO X, HIRANO T, HOU Y AND GU XX. 2002. Specific immune responses and enhancement of murine pulmonary clearance of Moraxella catarrhalis by intranasal immunization with a detoxified lipooligosaccharide conjugate vaccine. Infect Immun 70: 5982-5989.
- JIN Z, CHU C, ROBBINS JB AND SCHNEERSON R. 2003. Preparation and characterization of group A meningococcal capsular polysaccharide conjugates and evaluation of their immunogenicity in mice. Infect Immun 71: 5115-5120.
- JODAR L, BUTLER J, CARLONE G, DAGAN R, GOLDBLATT D, KAYTHY H, KLUGMAN K, PLIKYATIS B, SIBER G, KOHBERGER R, CHANG I AND CHERIAN T. 2003. Serological criteria for evaluation and licensure of new pneumococcal conjugate vaccine formulations for use in infants. Vaccine 21: 3265-3272.
- JONES C. 2005a. NMR Assays for Carbohydrate-based Vaccines. J Pharm Biomed Anal in press.
- JONES C. 2005b. Revised structures for the capsular polysaccharides from Staphylococcus aureus Types 5 and 8, components of novel glycoconjugate vaccines. Carbohydr Res 340: 1097-1106.
- JONES C AND LEMERCINIER X. 1999. NMR identity tests for polysaccharides used in vaccines: methods and validation, in 'Biologicals' beyond 2000: challenges for quality standards in an evolving field. Pharmeuropa Special issue April 2000, p. 119-129.
- JONES C AND LEMERCINIER X. 2002. Use and Validation of an NMR Test for the Identity and O-Acetyl Content of Capsular Polysaccharides from Neisseria meningitidis used in vaccine manufacture. J Pharm Biomedical Anal 30: 1233-1247.
- JONES C, MULLOY B, WILSON A, DELL A AND OATES JE. 1985. Structure of the capsular polysaccharide from Streptococcus pneumoniae Type 9. J Chem Soc Perkin Trans 1: 1665-1673.
- JONES C, CURRIE F AND FORSTER MJ. 1991. NMR and conformational analysis of the capsular poly-saccharide from Streptococcus pneumoniae Type 4. Carbohydr Res 221: 95-121.
- JONES C, LEMERCINIER X, CRANE DT, GEE CK AND AUSTIN S. 2000. Spectroscopic studies of the structure and stability of glycoconjugate vaccines. Dev Biologicals 103: 121-136.
- JORBECK HJA, SVENSON SB AND LINDBERG AA. 1981. Artificial Salmonella vaccines Salmonella typhimurium O-antigen-specific oligosaccharide-protein conjugates elicit opsonising antibodies that enhance phagocytosis. Infect Immun 32: 497-502.
- KAMERLING JP. 2000. Pneumococcal polysaccharides: a chemical view. In: TOMASZ A (Ed.), Streptococcus pneumoniae, Larchmont: Mary Anne Liebert, p. 81-114.
- KASPER DL, PAOLETTI LC, WESSELS MR, GUTTORMSEN HK, CAREY VJ, JENNINGS HJ AND BAKER CJ. 1996. Immune response to type III group B streptococcal polysaccharide-tetanus toxoid conjugate vaccine. J Clin Invest 98: 2308-2314.
- KENNE L, LINDBERG B AND MADDEN JK. 1979. Structural studies of the capsular antigen from Streptococcus pneumoniae type 26. Carbohydr Res 73: 175-182.
- KOJIMA Y, TOJO M, GOLDMANN DA, TOSTESON TD AND PIER GB. 1990. Antibody to the capsular polysaccharide/adhesin protects rabbits against catheter-related bacteremia due to coagulase-negative staphylococci. J Infect Dis 162: 435-441.
- KONADU E, ROBBINS JB, SHILOACH J, BRYLA DA AND SZU SC. 1994. Preparation, characterization, and immunological properties in mice of Escherichia coli O157 O-specific polysaccharide-protein conjugate vaccines. Infect Immun 62: 5048-5054.
- KONADU E, SHILOACH J, BRYLA DA, ROBBINS JB AND SZU SC. 1996. Synthesis, characterization, and immunological properties in mice of conjugates composed of detoxified lipopolysaccharide of Salmonella paratyphi A bound to tetanus toxoid with emphasis on the role of O-acetyls. Infect Immun 64: 2709-2715.
- KONADU EY, PARKE JR JC, TRAN HT, BRYLA DA, ROBBINS JB AND SZU SC. 1998. Investigational vaccine for Escherichia coli O157: phase 1 study of O157 O-specific polysaccharide-Pseudomonas aeruginosa recombinant exoprotein A conjugates in adults. J Infect Dis 177: 383-387.
- KONADU E, DONOHUE-ROLFE A, CALDERWOOD SB, POZSGAY V, SHILOACH J, ROBBINS JB AND SZU SC. 1999. Syntheses and immunologic properties of Escherichia coli O157 O-specific polysaccharide and Shiga toxin 1 B subunit conjugates in mice. Infect Immun 67: 6191-6193.
- KONADU EY, LIN FY, HO VA, THUY NT, VAN BAY P, THANH TC, KHIEM HB, TRACH DD, KARPAS AB, LI ET AL. 2000. Phase 1 and phase 2 studies of Salmonella enterica serovar paratyphi A O-specific polysaccharide-tetanus toxoid conjugates in adults, teenagers, and 2- to 4-year-old children in Vietnam. Infect Immun 68: 1529-1534.
- KORKEILA M, LEHTONEN H, AHMAN H, LEROY O, ESKOLA J AND KAYTHY H. 2000. Salivary anti-capsular antibodies in infants and children immunised with Streptococcus pneumoniae capsular polysaccharides conjugated to diphtheria or tetanus toxoid. Vaccine 18: 1218-1226.
- KOSSACZKA Z, BYSTRICKY S, BRYLA DA, SHILOACH J, ROBBINS JB AND SZU SC. 1997. Synthesis and immunological properties of Vi and di-O-acetyl pectin protein conjugates with adipic acid dihydrazide as the linker. Infect Immun 65: 2088-2093.
- KOSSACZKA Z, LIN FY, HO VA, THUY NT, VAN BAY P, THANH TC, KHIEM HB, TRACH DD, KARPAS A, HUNT S, BRYLA DA, SCHNEERSON R, ROBBINS JB AND SZU SC. 1999. Safety and immunogenicity of Vi conjugate vaccines for typhoid fever in adults, teenagers, and 2- to 4-year-old children in Vietnam. Infect Immun 67: 5806-5810.
- KOSSACZKA Z, SHILOACH J, JOHNSON V, TAYLOR DN, FINKELSTEIN RA, ROBBINS JB AND SZU SC. 2000. Vibrio cholerae O139 conjugate vaccines: synthesis and immunogenicity of V. cholerae O139 capsular polysaccharide conjugates with recombinant diphtheria toxin mutant in mice. Infect Immun 68: 5037-5043.
- KOTLOFF KL, WINICKOFF JP, IVANOFF B, CLEMENS JD, SWERDLOW DL, SANSONETTI PJ, ADAK GK AND LEVINE MM. 1999. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Org 77: 651-666.
- KUO J, DOUGLAS M, REE HK AND LINDBERG AA. 1995. Characterization of a recombinant pneumolysin and its use as a protein carrier for pneumococcal type 18C conjugate vaccines. Infect Immun 63: 2706-2713.
- LAFERRIÈRE CA, SOOD RK, DE MUYS JM, MICHON F AND JENNINGS HJ. 1997. The synthesis of Streptococcus pneumoniae polysaccharide-tetanus toxoid conjugates and the effect of chain length on immunogenicity. Vaccine 15: 179-186.
- LAFERRIÈRE CA, SOOD RK, DE MUYS JM, MICHON F AND JENNINGS HJ. 1998. Streptococcus pneumoniae type 14 polysaccharide-conjugate vaccines: length stabilization of opsonophagocytic conformational polysaccharide epitopes. Infect Immun 66: 2441-2446.
- LAGERGARD T, SHILOACH J, ROBBINS JB AND SCHNEERSON R. 1990. Synthesis and immunological properties of conjugates composed of group B streptococcus type III capsular polysaccharide covalently bound to tetanus toxoid. Infect Immun 58: 687-694.
- LEE LH, FRASCH CE, FALK LA, KLEIN DL AND DEAL CD. 2003. Correlates of immunity for pneumococcal conjugate vaccines. Vaccine 21: 2190-2196.
- LEMERCINIER X AND JONES C. 1996. Full 1H NMR assignments and detailed O-acetylation patterns of capsular polysaccharides from Neisseria meningitidis used in vaccine production. Carbohydr Res 296: 83-96.
- LEONTEIN K, LINDBERG B AND LONNGREN J. 1981. Structural studies of the capsular polysaccharide from Streptococcus pneumoniae type 12F. Can J Chem 59: 2081-2085.
- LIN FY, HO VA, KHIEM HB, TRACH DD, BAY PV, THANH TC, KOSSACZKA Z, BRYLA DA, SHILOACH J, ROBBINS JB, SCHNEERSON R AND SZU SC. 2001. The efficacy of a Salmonella typhi Vi conjugate vaccine in two-to-five-year-old children. N Engl J Med 344: 1263-1269.
- LINDBERG AA, KARNELL A AND WEINTRAUB A. 1991. The lipopolysaccharide of Shigella bacteria as a virulence factor. Rev Infect Dis 13: S279-284.
- LINDBERG B. 1990a. Components of bacterial polysaccharides. Adv Carbohydr Chem Biochem 48: 279-318.
- LINDBERG B. 1990b. Structural studies of bacterial polysaccharides using NMR spectroscopy and mass spectrometry. Thesis Stockholm University.
- LINDBERG B, LONNGREN J AND POWELL DA. 1977. Structural studies on the specific type-14 pneumococcal polysaccharide. Carbohydr Res 58: 177-186.
- LORTAN JE, KANIUK AS AND MONTEIL MA. 1993. Relationship of in vitro phagocytosis of serotype 14 Streptococcus pneumoniae to specific class and IgG subclass antibody levels in healthy adults. Clin Exp Immunol 91: 54-57.
- MAWAS FJ, NIGGEMANN J, JONES C, CORBEL MJ, KAMERLING JP AND VLIEGENTHART JFG. 2002. A conjugate vaccine made with a synthetic single repeating unit of Type 14 pneumococcal polysaccharide coupled to CRM197 is immunogenic in a mouse model. Infect Immun 70: 5107-5114.
- MAYER LW, REEVES MW, AL-HAMDAN N, SACCHI CT, THA MK, AIELLO GW, SCHMINK SE, NOBLE CA, TONDELLA MLC, WHITNEY AM, AL-MAZROU Y, AL-JEFRI M, MISHKHIS A, SABBAN S, CAUGANT DA, LINGAPPA J, ROSENSTEIN NE AND POPOVIC T. 2002. Outbreak of W135 Meningococcal Disease in 2000: Not Emergence of a New W135 Strain but Clonal Expansion within the Electrophoretic Type 37 Complex. J Infect Dis 185: 1596-1605.
- MCKENNEY D, POULIOT K, WANG Y, MURTHY V, ULRICH M, DORING G, LEE JC, GOLDMANN DA AND PIER GB. 2000. Vaccine potential of poly-1-6 b-D-N-succinyl-glucosamine, an immunoprotective surface polysaccharide of Staphylococcus aureus and Staphylococcus epidermidis J Biotech 83: 37-44.
- MCLEOD CM, HODGES RC, HEIDELBERGER M AND BERNHARDT WG. 1945. Prevention of pneumococcal pneumonia by immunization with specific capsular polysaccharides. J Exp Med 82: 445-465.
- MELEGARO A AND EDMUNDS WJ. 2004. The 23-valent pneumococcal polysaccharide vaccine. Part I. Efficacy of PPV in the elderly: a comparison of meta-analyses. Eur J Epidemiology 19: 353-363.
- MICHON F, FUSCO PC, MINETTI CA, LAUDE-SHARP M, UITZ C, HUANG CH, D'AMBRA AJ, MOORE S, REMETA DP, HERON I AND BLAKE MS. 1998. Multivalent pneumococcal capsular polysaccharide conjugate vaccines employing genetically detoxified pneumolysin as a carrier protein. Vaccine 16: 1732-1741.
- MICHON F, HUANG CH, FARLEY EK, HRONOWSKI L, DI J AND FUSCO PC. 2000. Structure activity studies on group C meningococcal polysaccharide-protein conjugate vaccines: effect of O-acetylation on the nature of the protective epitope. Dev Biol (Basel) 103: 151-160.
- MIESZALA M, KOGAN G AND JENNINGS HJ. 2003. Conjugation of meningococcal lipooligosaccharides through their lipid A terminus conserves their inner epitopes and results in conjugate vaccines having improved immunological properties. Carbohyd Res 338: 167-175.
- MILLER E, SALISBURY D AND RAMSAY M. 2001. Planning, registration, and implementation of an immunisation campaign against meningococcal serogroup C disease in the UK: a success story. Vaccine 20: S58-67.
- MOND JJ, LEES A AND SNAPPER CM. 1995. T cell-independent antigens type 2. Ann Rev Immunol 13: 655-692.
- MOREAU M, RICHARDS JC, FOURNIER JM, BYRD RA, KARAKAWA WW AND VANN WF. 1990. Structure of the Type 5 capsular polysaccharide of Staphylococcus aureus Carbohydr Res 201: 285-297.
- MUSHER DM, LUCHI MJ, WATSON DA, HAMILTON R AND BAUGHN RE. 1990. Pneumococcal polysaccharide vaccine in young adults and older bronchitics: determination of IgG responses by ELISA and the effect of adsorption of serum with non-type-specific cell wall polysaccharide. J Infect Dis 161: 728-735.
- MUSHER DM, GROOVER JE, WATSON DA, RODRIGUEZ-BARRADAS MC AND BAUGHN RE. 1998. IgG responses to protein-conjugated pneumococcal capsular polysaccharides in persons who are genetically incapable of responding to unconjugated polysaccharides. Clin Infect Dis 27: 1487-1490.
- MUSSELLI C, LIVINGSTON PO AND RAGAPATHI G. 2001. Keyhole limpet hemocyanin conjugate vaccines against cancer: the Memorial Sloan Kettering experience. J Cancer Res Clin Oncology 127: R20-R26.
- NUSSBAUM G, ANANDASABAPATHY S, MUKHERJEE J, FAN M, CASADEVALL A AND SCHARFF MD. 1999. Molecular and idiotypic analyses of the antibody response to Cryptococcus neoformans glucuronoxylomannan-protein conjugate vaccine in autoimmune and nonautoimmune mice. Infect Immun 67: 4469-4476.
- O'BRIEN KL, MOULTON LH, REID R, WEATHERHOLTZ R, OSKI J, BROWN L, KUMAR G, PARKINSON A, HU D AND HACKELL ET AL. 2003. Efficacy and safety of seven-valent conjugate pneumococcal vaccine in American Indian children: group randomised trial. Lancet 362: 355-361.
- PAI VB, HEYNEMAN CA AND ERRAMOUSPE J. 2002. Conjugated heptavalent pneumococcal vaccine. Ann Pharmacotherapy 36: 1403-1413.
- PAOLETTI LC, KASPER DL, MICHON F, DIFABIO J, HOLME K, JENNINGS HJ AND WESSELS MR. 1990. An oligosaccharide-tetanus toxoid conjugate vaccine against type III group B Streptococcus. J Biol Chem 265: 18278-18283.
- PAOLETTI LC, KASPER DL AND MICHON F. 1992a. Effects of chain length on the immunogenicity in rabbits of group B Streptococcus type III oligosaccharide-tetanus toxoid conjugates. J Clin Invest 89: 203-209.
- PAOLETTI LC, KASPER DL, MICHON F, DIFABIO J, JENNINGS HJ, TOSTESON TD AND WESSELS MR. 1992b. Group B Streptococcus type II polysaccharide-tetanus toxoid conjugate vaccine. Infect Immun 60: 4009-4014.
- PAOLETTI LC, KENNEDY RC, CHANH TC AND KASPER DL. 1996. Immunogenicity of group B Streptococcus type III polysaccharide-tetanus toxoid vaccine in baboons. Infect Immun 64: 677-679.
- PAOLETTI LC, PINEL J, JOHNSON KD, REINAP B, ROSS RA AND KASPER DL. 1999. Synthesis and preclinical evaluation of glycoconjugate vaccines against group B Streptococcus types VI and VIII. J Infect Dis 180: 892-895.
- PARISI L AND VON HUNOLSTEIN C. 1999. Determination of the molecular size distribution of Haemophilus influenzae type b-tetanus toxoid conjugate vaccines by size-exclusion chromatography. J Chromatog A 847: 209-211.
- PASSWELL JH, HARLEY E, ASHKENAZI S, CHU C, MIRON D, RAMON R, FARZAN N, SHILOACH J, BRYLA DA, MAJADLY E, ROBERTSON R, ROBBINS JB AND SCHNEERSON R. 2001. Safety and immunogenicity of improved Shigella O-specific polysaccharide-protein conjugate vaccines in adults in Israel. Infect Immun 69: 1351-1357.
- PASSWELL JH, ASHKENAZI S, HARLEY E, MIRON D, RAMON R, FARZAM N, LERNER-GEVA L, LEVI Y, CHU C, SHILOACH ET AL. 2003. Safety and immunogenicity of Shigella sonnei-CRM9 and Shigella flexneri type 2a-rEPAsucc conjugate vaccines in one- to four-year-old children. Ped Infect. Dis J 22: 701-706.
- PEETERS CCM, TENBERGEN-MEEKES AM, POOLAMN JT, ZEGERS BJ AND RIJKERS GT. 1992. Immunogenicity of a Streptococcus pneumoniae type 4 polysaccharide-protein conjugate vaccine is decreased by admixture of high doses of free saccharide. Vaccine 10: 833-840.
- PIER GB. 2003. Promises and pitfalls of Pseudomonas aeruginosa lipopolysaccharide as a vaccine antigen. Carbohydr Res 338: 2549-2556.
- PIROFSKI L, LUI R, DESHAW M, KRESSEL AB AND ZHONG Z. 1995. Analysis of human monoclonal antibodies elicited by vaccination with a Cryptococcus neoformans glucuronoxylomannan capsular polysaccharide vaccine. Infect Immun 63: 3005-3014.
- PLANS P. 2002. Cost-effectiveness of 23-valent antipneumococcal vaccination in Catalonia (Spain). Gaceta Sanitaria / SESPAS 16: 392-400.
- POLOTSKY VY, SCHNEERSON R, BRYLA D AND ROBBINS JB. 1994a. Immunogenicity of two types of Shigella flexneri 2a O-specific polysaccharide-tetanus toxoid conjugates. Ann NY Acad Sci 730: 359-360.
- POLOTSKY VY, ROBBINS JB, BRYLA D AND SCHNEERSON R. 1994b. Comparison of conjugates composed of lipopolysaccharide from Shigella flexneri type 2a detoxified by two methods and bound to tetanus toxoid. Infect Immun 62: 210-214.
- POPOFF MY. 1991. Virulence factors of Salmonella: from molecular genetics to diagnostic applications. Bull l'Academie Nationale Medecine 175: 811-821.
- POZSGAY V, CHU C, PANNELL L, WOLFE J, ROBBINS JB AND SCHNEERSON R. 1999. Protein conjugates of synthetic saccharides elicit higher levels of serum IgG lipopolysaccharide antibodies in mice than do those of the O-specific polysaccharide from Shigella dysenteriae type 1. Proc Natl Acad Sci USA 96: 5194-5197.
- PUUMALAINEN T, ZETA-CAPEDING MR, KAYTHY H, LUCERO MG, AURAMEN K, LEROY O AND NOHYNEK H. 2002. Antibody response to an eleven valent diphtheria- and tetanus-conjugated pneumococcal conjugate vaccine in Filipino infants. Pediatr Inf Dis J 21: 309-314.
- RACOOSIN JA, WHITNEY CG, CONOVER C AND DIAZ PS. 1998. Serogroup Y meningococcal disease in Chicago, 1991-1997. JAMA 280: 2094-2098.
- RAVENSCROFT N. 2000. The application of NMR spectroscopy to track industrial preparation of polysaccharide and derived glycoconjugate vaccines, Pharmeuropa, 2000, Special issue "Biologicals beyond 2000: challenge for quality standards in an evolving field", p. 131-144.
- RAVENSCROFT N, AVERANI G, BARTOLINI A, BERTI S, BIGIO M, CARINCI V, COSTANTINO P, D'ASCENZI S, GIANNOZZI A, NORELLI F, PENNATINI C, PROIETTI D, CECCARINI C AND CESCUTTI P. 1999. Size determination of bacterial capsular oligosaccharides used to prepare conjugate vaccines. Vaccine 17: 2802-2816.
- REDHEAD K, SESARDIC D, YOST SE, ATTWELL AM, WATKINS J, HOY CS, PLUMB JE AND CORBEL MJ. 1994. Combination of DTP and Haemophilus influenzae type b conjugate vaccines can affect laboratory evaluation of potency and immunogenicity. Biologicals 22: 339-345.
- REEVES RE AND GOEBEL WF. 1941. Chemoimmunological studies on the soluble specific substance of pneumococcus. V. The structure of the Type III polysaccharide. J Biol Chem 139: 511-519.
- RICHARDS JC AND PERRY MB. 1988. Structural analysis of the specific capsular polysaccharide of Streptococcus pneumoniae 23F (American type 23). Biochem Cell Biol 66: 758-771.
- ROBBINS JB, AUSTRIAN R, LEE CJ, RASTOGI SC, SCHIFFMAN G, HENRICHSEN J, MAKELA PH, BROOME CV, FACKLAM RR, TIESJEMA RH AND PARKE JC. 1983. Considerations for formulating the 2nd-generation pneumococcal capsular polysaccharide vaccine with emphasis of the cross-reactive types within groups. J Infect Dis 148: 1136-1159.
- ROBBINS JB, CHU C, WATSON DC, SZU SC, DANIELS EM, LOWE CU AND SCHNEERSON R. 1991. O-specific side-chain toxin-protein conjugates as parenteral vaccines for the prevention of shigellosis and related diseases. Rev Infect Dis 13: S362-365.
- ROBBINS JB, CHU C AND SCHNEERSON R. 1992. Hypothesis for vaccine development: protective immunity to enteric diseases caused by nontyphoidal salmonellae and shigellae may be conferred by serum IgG antibodies to the O-specific polysaccharide of their lipopolysaccharides. Clin Infect Dis 15: 346-361.
- ROBIN G, KEISARI Y, SLEPON R, ASHKENAZI S AND COHEN D. 1999. Quantitative analysis of IgG class and subclass and IgA serum response to Shigella sonnei and Shigella flexneri 2a polysaccharides following vaccination with Shigella conjugate vaccines. Vaccine 17: 3109-3115.
- ROITT I. 1997. Essential Immunology, 9th ed. Oxford: BLACKwell Scientific.
- ROMERO-STEINER S, MUSHER DM, CENTRON MS, PAIS LB, GROOVER JE, FIORE AE, PLIKAYTIS BD AND CARLONE GM. 1999. Reduction in functional antibody activity against Streptococcus pneumoniae in vaccinated elderly individuals highly correlates with decreased IgG antibody avidity. Clin Infect Dis 29: 281-288.
- ROSENSTEIN NE, PERKINS BA, STEPHENS DS, POPOVIC T AND HUGHES JM. 2001. Meningococcal disease. N Eng J Med 344: 1378-1388.
- RUTHERFORD TJ, JONES C, DAVIES DB AND ELLIOTT AC. 1991. Location and quantitation of the sites of O-acetylation on the capsular polysaccharide from Streptococcus pneumoniae type 9V by 1H -n.m.r. spectroscopy: comparison with type 9A. Carbohydr Res 218: 175-184.
- SANTOSHAM M, WOLFF M, REID R, HOHENBOKEN M, BATEMAN M, GOEPP J, CORTESE M, SACK D, HILL J AND NEWCOMER W. 1991. The efficacy in Navajo infants of a conjugate vaccine consisting of Haemophilus influenzae type b polysaccharide and Neisseria meningitidis outer-membrane proteincomplex. N Engl J Med 324: 1767-1772.
- SANTOSHAM M, RIVIN B, WOLFF M, REID R, NEWCOMER W, LETSON GW, ALMEIDO-HILL J, THOMPSON C AND SIBER GR. 1992. Prevention of Haemophilus influenzae type b infections in Apache and Navajo children. J Infect Dis 165 (Suppl 1): S144-151.
- SAXENA M AND DI FABIO JL. 1994. Salmonella typhi O-polysaccharide-tetanus toxoid conjugated vaccine. Vaccine 12: 879-884.
- SCHLESINGER Y AND GRANOFF DM. 1992. Avidity and bactericidal activity of antibody elicited by different Haemophilus influenzae type b conjugate vaccines. The Vaccine Study Group. JAMA 267: 1489-1494.
- SCHNEERSON R, BARRERA O, SUTTON A AND ROBBINS JB. 1980. Preparation, characterization, and immunogenicity of Haemophilus influenzae type b polysaccharide-protein conjugates. J Exp Med 152: 361-376.
- SHAFER DE, TOLL B, SCHUMAN RF, NELSON BL, MOND JJ AND LEES A. 2000. Activation of soluble polysaccharides with 1-cyano-4-dimethylaminopyridinium tetrafluoroborate (CDAP) for use in protein-polysaccharide conjugate vaccines and immunological reagents. II. Selective crosslinking of proteins to CDAP-activated polysaccharides. Vaccine 18: 1273-1281.
- SHEN X, LAGERGARD T, YANG Y, LINDBLAD M, FREDRIKSSON M AND HOLMGREN J. 2000a. Effect of pre-existing immunity for systemic and mucosal immune responses to intranasal immunization with group B Streptococcus type III capsular polysaccharide-cholera toxin B subunit conjugate. Vaccine 19: 850-861.
- SHEN X, LAGERGARD T, YANG Y, LINDBLAD M, FREDRIKSSON M AND HOLMGREN J. 2000b. Group B Streptococcus capsular polysaccharide-cholera toxin B subunit conjugate vaccines prepared by different methods for intranasal immunization. Infect Immun 68: 5749-5755.
- SHINEFIELD HR, BLACK S, RAY P, CHANG I, LEWIS N, FIREMAN B, HACKELL J, PARADISO PR, SIBER G, KOHBERGER R, MADORE DV, MALINOWSKI FJ, KIMURA A, LE C, LANDAW I, AGUILAR J AND HANSEN J. 1999. Safety and immunogenicity of heptavalent pneumococcal CRM197 conjugate vaccine in infants and toddlers. Ped Infect Dis J 18: 757-763.
- SIBER GR. 1994. Pneumococcal disease prospects for a new generation of vaccines. Science 265: 1385-1387.
- SINGH M, GANGULY NK, KUMAR L AND VOHRA H. 1999. Protective efficacy and immunogenicity of Vi-porin conjugate against Salmonella typhi Microbiol Immunol 43: 535-542.
- SKOV-SØRENSEN UB, BLOM J, BIRCH-ANDERSEN A AND HENRICHSEN J. 1988. Ultrastructural localization of capsules, cell wall polysaccharide, cell wall proteins and F antigen in pneumococci. Infect Immun 56: 1890-1896.
- SNAPPER CM AND MOND JJ. 1996. A model for induction of T cell-independent humoral immunity in response to polysaccharide antigens. J Immunol 157: 2229-2233.
- SNAPPER CM, ROSAS FR, KEHRY MR, MOND JJ AND WETZLER LM. 1997. Neisserial porins may provide critical second signals to polysaccharide-activated murine B cells for induction of immunoglobulin secretion. Infect Immun 65: 3203-3208.
- STROOP CJM, XU Q, RETZLAFF M, ABEYGUNAWARDANA C AND BUSH CA. 2002. Structural analysis and chemical depolymerisation of the capsular polysaccharide of Streptococcus pneumoniae type 1. Carbohydr Res 337: 335-344.
- STURGESS AW, RUSH K, CHARBONNEAU RJ, LEE JI, WEST DJ, SITRIN RD AND HENNESSY JP Jr. 1999. Haemophilus influenzae type b conjugate vaccine stability: catalytic depolymerization of PRP in the presence of aluminum hydroxide. Vaccine 17: 1169-1178.
- SUN J, CHEN J, CHEN Z, ROBBINS JB, BATTEY JF AND GU XX. 2000. Biological activities of antibodies elicited by lipooligosaccharide based-conjugate vaccines of nontypeable Haemophilus influenzae in an otitis media model. Vaccine 18: 1264-1272.
- SUN YY AND HU XJ. 1999. Preparation and immunogenicity of serogroup B meningococcal OS-OMPC conjugates. Biomed Environ Sci 12: 296-303.
- SZU SC, STONE AL, ROBBINS JD, SCHNEERSON R AND ROBBINS JB. 1987. Vi capsular polysaccharide protein conjugates for prevention of typhoid fever preparation, characterisation and immunogenicity in laboratory animals. J Exp Med 166: 1510-1524.
- SZU SC, LI X AND SCHNEERSON R. 1989. Comparative immunogenicities of Vi polysaccharide-protein conjugates composed of cholera-toxin or its B subunit as a carrier bound to high molecular weight of lower molecular weight Vi. Infect Immun 57: 3823-3827.
- SZU SC, TAYLOR DN, TROFA AC, CLEMENTS JD, SHILOACH J, SADOFF JC, BRYLA DA AND ROBBINS JB. 1994. Laboratory and preliminary clinical characterization of Vi capsular polysaccharide-protein conjugate vaccines. Infect Immun 62: 4440-4444.
- TAI JY, VELLA PP, MCLEAN AA, WOODHOUR AF, MCALEER WJ, SHA A, DENNIS-SYKES C AND HILLEMAN MR. 1987. Haemophilus influenzae type b polysaccharide-protein conjugate vaccine. Proc Soc Exp Biol Med 184: 154-161.
- TAKEDA S, PIER GB, KOJIMA Y, MULLER E, TOSTESON T AND GOLDMANN DA. 1991. Protection against endocarditis due to Staphylococcus epidermidis by immunization with capsular polysaccharide/adhesion. Circulation 84: 2539-2546.
- THEILACKER C, COLEMAN FT, MUESCHENBORN S, LLOSA N, GROUT M AND PIER GB. 2003. Construction and characterization of a Pseudomonas aeruginosa mucoid exopolysaccharide-alginate conjugate vaccine. Infect Immun 71: 3875-3884.
- TOLLERSRUD T, ZERNICHOW L, ANDERSEN SR, KENNY K AND LUND A. 2001. Staphylococcus aureus capsular polysaccharide type 5 conjugate and whole cell vaccines stimulate antibody responses in cattle. Vaccine 19: 3896-3903.
- TWUMASI Jr PA, KUMAH S, LEACH A, O'DEMPSEY TJ, CEESAY SJ, TODD J, BROOME CV, CARLONE GM, PAIS LB, HOLDER PK, PLIKAYTIS BD AND GREENWOOD BM. 1995. A trial of a group A plus group C meningococcal polysaccharide-protein conjugate vaccine in African infants. J Infect Dis 171: 632-638.
- VEREZ-BENCOMO V, FERNANDEZ-SANTANA V, HARDY E, TOLEDO ME, RODRIGUEZ MC, HEYNNGNEZZ L, RODRIGUEZ A, BALY A, HERRERA L, IZQUIERDO M, VILLAR A, VALDES Y, COSME K, DELER ML, MONTANE M, GARCIA E, RAMOS A, AGUILAR A, MEDINA E, TORANO G, SOSA I, HERNANDEZ I, MARTINEZ R, MUZACHIO A, CARMENATES A, COSTA L, CARDOSO F, CAMPA C, DIAZ M AND ROY R. 2004. A synthetic conjugate polysaccharide vaccine against Haemophilus influenzae Type b. Science 305: 522-525.
- WALDOR MK, COLWELL R AND MEKELANOS JJ. 1994. The Vibrio cholerae O139 serogroup antigen includes an O-antigen capsule and lipopolysaccharide virulence determinants. Proc Natl Acad Sci USA 91: 11388-11392.
- WANG Y, HUEBNER J, TZIANABOS AO, MARTIROSIAN G, KASPER DL AND PIER GB. 1999. Structure of an antigenic teichoic acid shared by clinical isolates of Enterococcus faecalis and vancomycin-resistant Enterococcus faecium Carbohydr Res 316: 155-160.
- WARD J, LIEBERMAN JM AND COCHI SL. 1994. Haemophilus influenzae vaccines. In: PLOTKIN SA AND MORTIMER EA (Ed.) Vaccines, Philadelphia: W.B. Saunders and Co., p. 337-386.
- WATSON DC, ROBBINS JB AND SZU SC. 1992. Protection of mice against Salmonella typhimurium with an O-specific polysaccharide-protein conjugate vaccine. Infect Immun 60: 4679-4686.
- WELCH PG, FATTOM A, MOORE JR J, SCHNEERSON R, SHILOACH J, BRYLA DA, LI X AND ROBBINS JB. 1996. Safety and immunogenicity of Staphylococcus aureus type 5 capsular polysaccharide-Pseudomonas aeruginosa recombinant exoprotein A conjugate vaccine in patients on hemodialysis. J Am Soc Nephrol 7: 247-253.
- WESSELS MR, PAOLETTI LC, KASPER DL, DIFABIO JL, MICHON F, HOLME K AND JENNINGS HJ. 1990. Immunogenicity in animals of a polysaccharide-protein conjugate vaccine against type III group B Streptococcus. J Clin Invest 86: 1428-1433.
- WESSELS MR, PAOLETTI LC, GUTTORMSEN HK, MICHON F, D'AMBRA AJ AND KASPER DL. 1998. Structural properties of group B streptococcal type III polysaccharide conjugate vaccines that influence immunogenicity and efficacy. Infect Immun 66: 2186-2192.
- WU TH AND GU XX. 1999. Outer membrane proteins as a carrier for detoxified lipooligosaccharide conjugate vaccines for nontypeable Haemophilus influenzae Infect Immun 67: 5508-5513.
- WUORIMAA T, DAGAN R, VAKEVAINEN M, BAILLEUX F, HAIKALA R, YAICH M, ESKOLA J AND KAYTHY H. 2001. Avidity and subclasses of IgG after immunization of infants with an 11-valent pneumococcal conjugate vaccine with or without aluminum adjuvant. J Infect Dis 184: 1211-1215.
- XU Q, KLEES J, TEYRAL J, CAPEN R, HUANG M, STURGESS AW, HENNESSEY JP, WASHABAUGH M, SITRIN R AND ABEYGUNAWARDANA C. 2005. Quantitative nuclear magnetic resonance analysis and characterization of the derivatised Haemophilus influenzae type b polysaccharide intermediate for PedvaxHIB. Analyt Biochem 337: 235-245.
- YANG HH, KILGORE PE, YANG LH, PARK JK, PAN YF, KIM Y, LEE YJ, XU ZY AND CLEMENS JD. 2001. An outbreak of typhoid fever, Xing-An County, People's Republic of China, 1999: estimation of the field effectiveness of Vi polysaccharide typhoid vaccine. J Infect Dis 183: 1775-1780.
Publication Dates
-
Publication in this collection
09 May 2005 -
Date of issue
June 2005
History
-
Received
14 Feb 2005 -
Accepted
16 Feb 2005