Abstracts
Both the regio- and stereo-chemistries of the photoreactions of 1-naphthyl (R)-2-phenylpropanoate have been investigated in poly(vinyl acetate) films in their glassy (at 5ºC) and melted (at 50ºC) states and in ethyl acetate. These results are compared with those from irradiations in polyethylene films and in n-hexane. The regioselectivity of the intermediate 1-naphthoxy/(R)-2-phenylpropanoyl radical pair combinations is much higher in both the melt and glassy states of poly(vinyl acetate) films than that in the melt state of completely amorphous polyethylene films, but the stereoselectivity of intermediate prochiral 1-naphthoxy/1-phenylethyl radical pair combinations is much lower in poly(vinyl acetate). The results emphasize the need to control the ratio between the rates of radical tumbling and translation, as well as the ratio between the rates of in-cage motions and cage-escape, if high stereo- and regio-selectivities of combination products are to be achieved. A mechanistic picture of how the radicals of the intermediate pairs are affected by and interact with the various media is advanced.
radical pair; stereoselectivity; polymer relaxation; photo-Fries
A régio- e a estereoquímica das fotoreações de (R)-2-fenilpropanoato de 1-naftila foram investigadas em filmes de poli (acetato de vinila) nos estados vítreo (5ºC) efundido (50ºC) e em acetato de etila. Estes resultados foram comparados aos obtidos em filmes de polietileno e em n-hexano. A regioseletividade das combinações de pares radicalares intermediários 1-naftoxi/(R)-2-fenilpropanoila é muito maior nos estados fundido e vítreo do poli (acetato de vinila) do que no estado fundido de filmes de polietileno, mas a estereoseletividade das combinações de pares de radicais intermediários proquirais 1-naftoxi/(R)-2-fenilpropanoila é muito menor em poli (acetato de vinila). Os resultados enfatizam a necessidade de se controlar a relação entre as taxas de tombamento e de translação de radicais, assim como a relação entre as taxas dos movimentos no interior da gaiola ou de escape da gaiola, se for desejado atingir estéreo- e regioseletividades elevadas. Propõe-se um quadro mecanístico a respeito de como os radicais dos pares intermediários são afetados pelos vários meios e interagem com eles.
par de radicais; estereoseletividade; relaxação de polímeros; foto-Fries
CHEMICAL SCIENCES
Photo-Fries rearrangements of 1-naphthyl (R)-2-phenylpropanoate in poly(vinyl acetate) and ethyl acetate. Influence of medium polarity and polymer relaxation on motions of singlet radical pairs
Jinqi Xu; Mathew George; Richard G. Weiss
Department of Chemistry, Georgetown University, 37th and O Streets, NW, Washington, DC 20057-1227 USA
ABSTRACT
Both the regio- and stereo-chemistries of the photoreactions of 1-naphthyl (R)-2-phenylpropanoate have been investigated in poly(vinyl acetate) films in their glassy (at 5ºC) and melted (at 50ºC) states and in ethyl acetate. These results are compared with those from irradiations in polyethylene films and in n-hexane. The regioselectivity of the intermediate 1-naphthoxy/(R)-2-phenylpropanoyl radical pair combinations is much higher in both the melt and glassy states of poly(vinyl acetate) films than that in the melt state of completely amorphous polyethylene films, but the stereoselectivity of intermediate prochiral 1-naphthoxy/1-phenylethyl radical pair combinations is much lower in poly(vinyl acetate). The results emphasize the need to control the ratio between the rates of radical tumbling and translation, as well as the ratio between the rates of in-cage motions and cage-escape, if high stereo- and regio-selectivities of combination products are to be achieved. A mechanistic picture of how the radicals of the intermediate pairs are affected by and interact with the various media is advanced.
Key words: radical pair, stereoselectivity, polymer relaxation, photo-Fries.
RESUMO
A régio- e a estereoquímica das fotoreações de (R)-2-fenilpropanoato de 1-naftila foram investigadas em filmes de poli (acetato de vinila) nos estados vítreo (5ºC) efundido (50ºC) e em acetato de etila. Estes resultados foram comparados aos obtidos em filmes de polietileno e em n-hexano. A regioseletividade das combinações de pares radicalares intermediários 1-naftoxi/(R)-2-fenilpropanoila é muito maior nos estados fundido e vítreo do poli (acetato de vinila) do que no estado fundido de filmes de polietileno, mas a estereoseletividade das combinações de pares de radicais intermediários proquirais 1-naftoxi/(R)-2-fenilpropanoila é muito menor em poli (acetato de vinila). Os resultados enfatizam a necessidade de se controlar a relação entre as taxas de tombamento e de translação de radicais, assim como a relação entre as taxas dos movimentos no interior da gaiola ou de escape da gaiola, se for desejado atingir estéreo- e regioseletividades elevadas. Propõe-se um quadro mecanístico a respeito de como os radicais dos pares intermediários são afetados pelos vários meios e interagem com eles.
Palavras-chave: par de radicais, estereoseletividade, relaxação de polímeros, foto-Fries.
INTRODUCTION
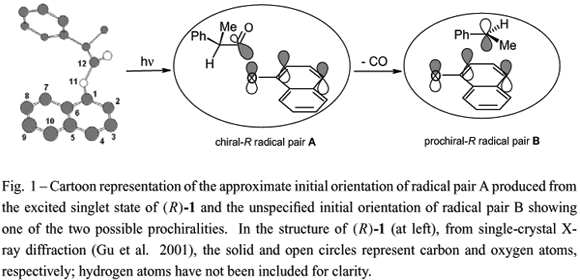
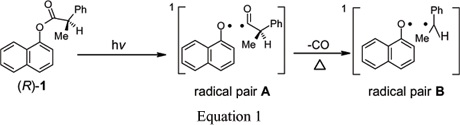
The rates and regio- and stereo-chemistries of radical pair combinations are known to be sensitive to local environmental effects (Porter and Krebs 1988, John 1993, Curran et al. 1995, Turro 2002, Ramamurthy and Schanze 2003). Some of these are the shapes, sizes, and flexibilities of the 'walls' of the reaction cages in which the combinations occur (Ramamurthy et al. 1993, Weiss et al. 1993). Others include the polarity of the cages and their ability to catalyze intersystem crossing of the radical pairs (Warrier et al. 2003). We have exploited the regiochemistry of photo-Fries reactions of aryl esters in which some decarbonylation products are formed to measure the in-cage rates of radical pair combinations (Gu and Weiss 2000). Those results provide important information about the influence of local environment on translational motions of a radical pair. Recently, we have extended that approach to develop a diagnostic system for following simultaneously the influences of time and space on radical pair combinations within reaction cages (Xuand Weiss 2003, 2005a, b, c). It is based on the fates of two related singlet radical pairs: 1-naphthoxy/(R)-2-phenylpropanoyl (chiral radical pair A) from lysis of the excited singlet state of 1-naphthyl(R)-2-phenylpropanoate ((R)-1) and 1-naphthoxy/1-phenylethyl (prochiral radical pair B) from decarbonylation of radical pair A (i.e., indirectly from (R)-1) (Equation 1). Thus far, the chemistries of the combinations of chiral radical pair A and pro-chiral radical pair B have been investigated in relatively low polarity media such as n-alkanes with different viscosities (Xu and Weiss 2005a), polyethylene (PE) films with a wide range of crystallinities above their glass transition temperatures (Tgs) (Xu and Weiss 2005b), and the solid phases of n-nonadecane (Xu and Weiss 2005c). As expected, both the regio- and stereo-selectivities decrease as temperature is raised and medium viscosity is lowered (Xu and Weiss 2003, 2005a, b, c).
The high microviscosity and "templating"effects (related to low 'wall' flexibility from long polymer chain relaxation times) of the PE reaction cavities restrict radical pair movements (Gu et al. 1999, 2000a, b, Gu and Weiss 2000, 2001a, b), enabling even prochiral radical pair B to retain partial orientational memory of its chiral radical pair A precursor (Xu and Weiss 2003, 2005a, b, c). These results suggest that the rates of tumbling of a 1-phenylethyl radical and of radical pair combination are similar in these reaction cages, and both are at least comparable to the rates of relaxation of the polymeric chains constituting the PE cavity walls (Gu et al. 1999, 2000a, b, Gu and Weiss 2000,2001a, b, Xu and Weiss 2003, 2005a, b, c).
Atactic poly(vinyl acetate) (PVAc) is a completely amorphous polymer whose polarity is higher than that of PE and similar to that of ethyl acetate, even though its polymer backbone is of low polarity (Charles 1996, Stevens 1998, Ravve 2000). Common commercial PVAc is highly branched due to chain transfer reactions that occur during radical polymerizations of vinyl acetate. The glass transition temperature of PVAc, near room temperature, is much higher than the Tg of PE films.
Previously, McCourt and coworkers (Wang et al. 1990) have used the photo-Fries reaction to demonstrate that the distribution of a guest molecule, 2-naphthyl acetate, is non-random in the glassy state of poly(methyl methacrylate). The concentrations of 2-naphthyl acetate employed there, ca. 0.1 M (1-2 wt%), are 10-100 times larger than those of (R)-1 in the current investigation. In addition, we have investigated the regiochemistry of photo-Fries rearrangements of some simple 1-naphthyl esters, including racemic 1, in the glassy (at 5ºC) and melted (at 50ºC) states of PVAc films (Gu et al. 2002). The regioselectivity of the singlet radical pair A combinations is much greater in PVAc films in both states than in ethyl acetate or in hexane at comparable temperatures. The enhanced regioselectivity has been attributed to strong interactions between radicals analogous to those in radical pair A (or radical pair B) and the acetate pendent groups of PVAc.
Here, we report both the regio- and stereo-chemistries of the photoreactions of (R)-1 in PVAc films in their glassy (at 5ºC) and melted (at 50ºC) states. Comparisons of these results with those from irradiations in PE films and in n-hexane and ethyl acetate (solvents of lower and comparablepolarity to that of PVAc) provide insights into the influences of the dynamics of polymeric chainmotions and solvent polarity on the radical paircombinations. Because the regiochemistry of thereactions of racemic 1 in PVAc have been reported previously (Gu et al. 2002), stereochemical aspects of the radical pair combinations are emphasized here. This information is of interest fundamentally-to understand the interplay between different molecular motions in confining spaces-and practically-to design media whose reaction cavities can be used for regio- and enantio-selective syntheses.
MATERIALS AND METHODS
INSTRUMENTATION
UV/Vis absorption spectra were measured on a Cary 300 Bio UV-Vis Spectrophotometer. Gas chromatography (GC) was conducted on a Hewlett-Packard 5890 gas chromatograph equipped with a flame ionization detector. An Alltech Scientific DB-5 (0.25µm film; 0.25 mm × 30 m) column was used to determine products purities and for periodic monitoring of reactions. HPLC analyses werecarried out on a HP series 1100 liquid chromatograph equipped with an autosampler and a UV-Vis diode array detector. Columns were silica gel (5µm, 4.6 × 250 mm from Allsphere and Phenomenex) for determinations of product distributions and cyano derivatized silica (5µm, 10 × 250 mm from IBMInstruments) for semi-preparative product separations (both using mixtures of hexanes and ethyl acetate as eluting solvents). A Chiralcel OJ-H (5µm, 4.6 × 250 mm from Chiral Technologies) with hexanes and 2-propanol as eluting solvent were used for determinations of enantiomeric excesses.
MATERIALS
Hexanes, cyclohexane, ethyl acetate, dichloromethane, n-pentane, and 2-propanol were HPLC grade from Fisher and were used as received. Anhydrous diethyl ether (reagent grade from Fisher Scientific) was passed through a column filled with basic aluminum oxide to remove the antioxidant, 2,6-di-tert-butyl-p-cresol.
A 15-20 g portion of poly(vinyl acetate) (áMwñ ca. 83,000, d = 1.180 g cm-3 from Aldrich; Tg » 32.5°C (Gu et al. 2002)) was dissolved in 100 mL of CH2Cl2 by refluxing the mixture with stirring for 3-5 h. The solution was cooled to room temperature and slowly poured into 300 mL of stirred hexanes. The resulting white precipitate was dried in air and then under vacuum ( < 1 Torr) for 2-3 days. Methylene chloride solutions of PVAc were cast into films on clean Teflon plates. After several days of air-drying, the films were immersed in at least 3 cyclohexane aliquots during more than one week to remove any remaining additives.
The syntheses of racemic and the (R)-enantiomer of 1 and of their photoproducts have been reported previously (Xu and Weiss 2003). (R)-1 was > 99% chemically and enantiomerically pure by HPLC analyses.
GENERAL PROCEDURES FOR FILM DOPING AND IRRADIATIONS
Solutions of 2 mM (R)-1 in ethyl acetate (containing a small amount of n-dodecane as an internal standard) were irradiated with the Pyrex and water filtered output of a 450W Hanovia medium pressure mercury lamp for a few seconds at 5, 23, and 50ºC.
PVAc films were immersed overnight in a solution of ca. 20 mM (R)-1 in 85/15 (v/v) n-pentane/diethyl ether. After being wiped gently with a hexanes-saturated tissue paper to remove any surface-occluded (R)-1, the films were dried under vacuum ( < 0.5 Torr) for > 3 h before irradiation. The concentrations of (R)-1 doped in the films (ca. 2-6 mmol/kg) were determined by averaging the optical densities from at least 3 UV-Vis absorption spectra measured at different positions of each film. The molecular extinction coefficient of 1 in PVAc at 280 nm is assumed to be 6950 M-1cm-1, the value in ethyl acetate (Gu et al. 2002).
Each doped PVAc film was placed in a septum-sealed Pyrex tube and purged with flowing N2 gas for > 45 min. It was irradiated as above for periods from a few seconds to several minutes depending on the temperature and degree of conversion sought. Control of temperature at and above 23(±1) °C was achieved by immersing the sample tubes in a stirred water-bath in a Pyrex beaker that was heated on a hot plate. Samples in sealed tubes were immersed in a stirred ice-water bath to achieve 5(±1)°C irradiation temperatures.
DETERMINATION OF PRODUCT DISTRIBUTIONS AND ENANTIOMERIC EXCESSES
Irradiated PVAc films were extracted repeatedlyfor 2-3 h with 50-100 mL aliquots of 20/80 (v/v) n-pentane/diethyl ether until the last one contained no detectable UV-Vis absorbances ascribable to aromatic species. The combined extracts were divided into 2 parts in a 1/3 (v/v) ratio. One-fourth of the extracts was reduced to residue by simple distillation at ca. 50°C (hot water bath). The residue was dissolved in hexanes and analyzed by HPLC (silica column). Analyses from at least three injections were averaged to obtain the conversion and relative product distribution (Xu and Weiss 2003); the mass balance was always > 80%.
The remaining extract was concentrated to residue by bubbling N2 through it. The residue was dissolved in hexanes and its components were separated on a semi-preparative HPLC column. The ee of the hexane solution of each separated chiral product was then determined by HPLC on a Chiralcel OJ-H column using the averaged peak area ratios of its (R) and (S) enantiomers at four different detection wavelengths with 1 or 2 injections. All of the HPLC peaks were compositionally homogeneous in that the shapes of their UV-Vis absorption spectra did not change as they were recorded at various points during the peak elution (Xu and Weiss 2003).
A small aliquot of irradiated ethyl acetate solution was analyzed by HPLC with a silica column to obtain the conversion and relative product distribution. The remaining portion was concentrated (N2 bubbling) and the residue was analyzed as described above to determine ee values of photoproducts and remaining 1.
RESULTS AND DISCUSSION
COMBINATIONS OF RADICAL PAIRS A AND B FROM (R)-1. GENERAL CONSIDERATIONS
After photo-induced lysis of (R)-1, in-cage combination of radical pair A can reform the startingester or form the keto intermediates of the eventually isolated photo-Fries rearrangement products, 1-[2-(1-hydroxynaphthyl)]-2-phenyl-1-propanone (2AN) and 1-[4-(1-hydroxynaphthyl)]-2-phenyl-1-propanone (4AN) (Scheme 2) (Bellus 1971, Martin 1992, Miranda 1995, Miranda and Galindo 2003a, b); tautomerization of the keto intermediates (Mori et al. 2004b) occurs rapidly on the time scale of our laboratory manipulations, even in saturated paraffins. The apparent and actual 1,3- and 1,5-sigmatropic H-shifts occur in the range of microsecond to seconds, depending strongly on the nature of the solvent, and many are intermolecular (Chiang et al. 1984a, b, Arai et al. 1994, 1995).
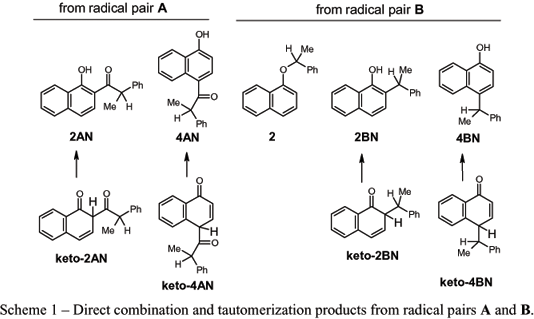
Alternatively, one of the radicals of the pair may escape from the cage, resulting in the eventual formation of out-of-cage products (vide infra). Finally, radical pair A may become singlet prochiral radical pair B if the 2-phenylpropanoyl radical cleaves into carbon monoxide and a 1-phenylethyl radical more rapidly than it adds to its 1-naphthoxy partner or escapes from the cage. Analogous in-cage combinations of radical pair B produce ether 2 as well as keto intermediates that tautomerize into 2-(1-phenylethyl)-1-naphthol (2BN) and 4-(1-phenylethyl)-1-naphthol (4BN). The structures of the keto intermediates and their enol tautomers from radical pairs A and B, as well as the directly formed ether 2 from radical pair B, are shown in Scheme 1.
The decarbonylation process is very rapid, requiring only ca. 24 ns at 23°C in isooctane (Braun et al. 1962, Turro et al. 1983, Tsentalovich and Fischer 1994, Zhang and Nau 2000, W.M. Nau et al., unpublished data). Its rate constant (k-CO) is independent of viscosity in paraffinic media (Tsentalovich and Fischer 1994, W.M. Nau et al., unpublished data) and depends only slightly on the polarity of its environment (Lunazzi et al. 1983, Tsentalovich and Fischer 1994). For instance, k-CO for decarbonylation of the phenylacetyl radical at room temperature has been measured to be 5.3 ×106 s-1 in hexane (e = 1.88), 2.6 ×106 s-1 in methanol (e = 32.7) and 1.7 ×106 s-1 in acetonitrile (e = 37.5) (Riddick et al. 1986, Tsentalovich and Fischer 1994). The value of k-CO has not been measured in PVAc or in ethyl acetate. Because the relative permittivities (Riddick et al. 1986, Lindemann 1999) of isooctane (e = 1.94), ethyl acetate (e = 6.02), and PVAc films (e = 3.5 at 50°C) are similar, we use k-CO in isooctane for the values in ethyl acetate and PVAc.
As mentioned above, radicals in both of theradical pairs can diffuse from their initial solvent cages and undergo out-of-cage reactions that result in fragmentation products (3-5), radical pair B-like combination products (2, 2BN, and 4BN but from different original partners), and a crossed combination product, 2,3-diphenylbutane (6) (Scheme 2). The latter is derived from dimerization of 1-phenylethyl radicals that escape from their initial cages, and it is a clear indicator that some of the other photoproducts (N.B., 2, 2BN, and 4BN) also are formed by radicals that re-encounter each other after escaping from the cages in which they were 'born'. Since 4 and 5 can be produced through other pathways besides encounter of two 1-phenylethyl radicals, we have used only 6 as a monitor of the importance of re-encounter processes by radical pairs A or B (Gu et al. 1999, Gu and Weiss 2000).
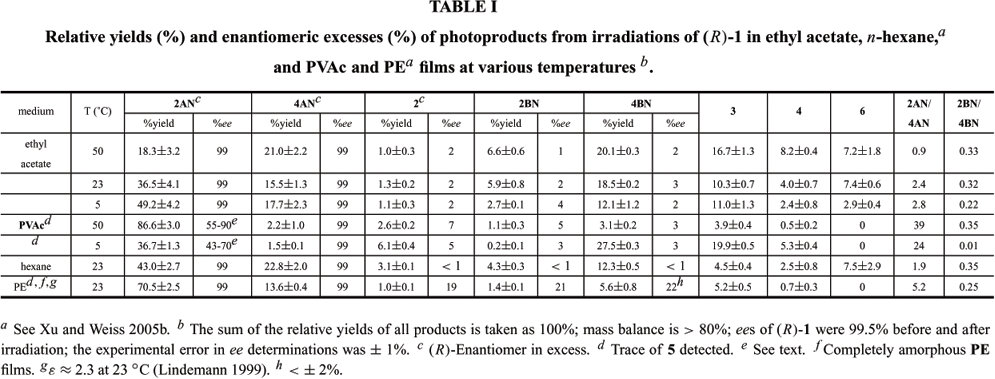
Small yields of a decarboxylation product, 1-(1-naphthyl)-1-phenylethane were detected also. It is produced by a concerted extrusion of CO2 from the excited singlet state of (R)-1 (Finnegan andKnutson 1965, 1967, Gu et al. 2001, Mori et al. 2003). In addition, abstraction of an H-atom by the constituents of radical pairs A and B, especially from the backbone of the PVAc polymer or from ethyl acetate, can lead to products in which a radical is attached to the polymer or solvent. Based upon our mass balances and UV-Vis absorption spectra of the films after irradiation and extraction, the yields of such products must be small and their specific structures were not explored. In addition, the relative product yields do not change when irradiations of (R)-1 are allowed to proceed up to 30% conversion (Gu et al. 1999, 2000a). The relative yields and ees of photoproducts from irradiations of (R)-1 in PVAc films and ethyl acetate, as well as in completely amorphous PE films and n-hexane, are shown in Table I. The relative yields of photoproducts in Table I differ slightly from the distributions reported previously for irradiations of racemic 1 in PVAc and ethyl acetate (Gu et al. 2002). In large part, the disparity can be traced to a less precise method of analysis, gas chromatography, being used in the previous study.
In the absence of secondary photolytic reactions (vide infra), the regio- and stereo-chemistries of the photoreactions of (R)-1 are dependent on steps in Scheme 2 up to and including the radical pair combinations yielding 2 and the keto intermediates of 2AN, 4AN, 2BN, and 4BN. Enolization should affect neither the relative yields nor the chiral centers of the photoproducts. To simplify subsequent discussions, only the final enolized products will be indicated rather than the first-formed keto intermediates.
RADICAL PAIR COMBINATIONS IN ETHYL ACETATE; COMPARISONS WITH RESULTS IN n-HEXANE
Based on the relative yields of 6 produced upon irradiation of (R)-1 in ethyl acetate (Table I), 2, 2BN, and 4BN are primarily from in-cage combinations of radical pair B at and below room temperature, but some reaction from out-of-cage encounters, especially at 50ºC, occurs as well. This assertion is supported by the regio- and stereo-selectivities of combinations of radical pairs A and B.
The regioselectivity of radical pair combinations depends in part on the relative spin densities at the positions of the 1-naphthoxy radical where its partner adds (C(4) > C(2) > O(11); see Figure 1 for the atom numbering of 1) (Dixon et al. 1973, Gu and Weiss 2001b), the initial orientation of the radical pair, and the degree to which the initial orientation of the radical pair is retained throughoutits lifetime. The latter factor is very dependent upon the nature of the environment surrounding the radical pair.
In part because the viscosities of n-hexane (0.3036 cP) and ethyl acetate (0.426 cP) (Timermans 1965) are similar at 23ºC, the regioselectivities of the radical pair combinations in the two solvents are somewhat similar: 2AN/4AN >> 1 and 2BN/4BN << 1 and the 2AN/4AN ratios suggest that radical pair A combines predominantly in-cage in both solvents (Bellus 1971, Martin 1992, Miranda 1995, Miranda and Galindo 2003a, b). The initial orientation of radical pair A places the radical center of 2-phenylpropanoyl closer to C(2) than to C(4) of its 1-naphthoxy radical partner (Figure 1) (Xu and Weiss 2005a). Furthermore, the much higher 2AN/4AN product ratios in ethyl acetate than in n-hexane indicate that in-cage motions of the radicals are also mediated by their strong interactions with the polar ethyl acetate solvent (Terazima 2000, Gu et al. 2002). An additional consequence of the strong solvent-radical interactions is that cage-escape and the tumbling motions of radical pair B leading to loss of enantiomeric excess of 2, 2BN, and 4BN (vide infra) are attenuated. The short lifetimes of the radical pairs and the high activation energy (ca. 54.4 kJ/mol (Conradi et al. 1979)) for another possible route to loss of chiral identity, internal rotation about the phenyl-benzylic C-C bond of 1-phenylethyl, precludes it from competing with combinations within a cage. Although the ee values of the decarbonylated products in ethyl acetate are small, they are clearly larger than those in n-hexane (in which ee values of 2, 2BN, and 4BN products are virtually 0%) and indicate that, within a cage, the combination rates of radical pair B are comparable to the rate of tumbling of a 1-phenylethyl radical.
REGIOCHEMISTRY OF RADICAL PAIR COMBINATIONS IN PVAC FILMS AT 5 AND 50?C. COMPARISON WITH RESULTS IN AMORPHOUS PE FILMS
The absence of photoproduct 6 upon irradiationsof (R)-1 at 5ºC (i.e., in the glassy state) and at 50ºC (i.e., in the melt state) are an indicator that the combinations of both radical pairs A and B are almost exclusively in-cage in both the glassy and melt phases of PVAc. At both temperatures, the 2AN/4AN ratios are much higher than in ethyl acetate or PE films. The van der Waals volume of (R)-1 (ca. 254 Å3) (Bondi 1964) is much largerthan the mean free hole volume in PVAc bothbelow and above the glass transition temperature (ca. 85 Å3 at 5ºC and ca. 105 Å3 at 50ºC) (Kobayashi et al. 1989). Since PVAc is swelled to dope(R)-1 into the films, the guest molecules are surrounded tightly by many polymeric chains after removal of the solvent, so that the size and shape of the reaction cavities are defined in large part by the size and shape of (R)-1; because the relaxation processes of polymeric chains in PVAc films are slower, especially in the glassy state, than the rate of radical pair combinations (Angell 1995, Yoshii et al. 2000, Seymour and Carraher 2003), the initial conformation of (R)-1 at the moment of its electronic excitation can have a large influence on the chemical course of product formation (i.e., there is a "templating" effect) (Weiss et al. 1993, Gu et al. 1999, Gu and Weiss 2000). In both PVAc and PE films, the greater proximity of the radical center of 2-phenylpropanoyl to C(2) than to C(4) of 1-naphthoxy immediately after the lysis of the excited singlet state of (R)-1 (Figure 1) and the attenuation of motions of the radicals caused by the templating effect and high local viscosity of the reaction cages promote preferential formation of 2AN.
The rate constants for formation of the ketointermediates of the AN products from radical pair A can be calculated approximately using the decarbonylation rate of 2-phenylpropanoyl (k-CO) in isooctane (see above) and the relative yields ofthe AN and BN products (Equation 2) (Gu andWeiss 2000). Using this method, k2AN = (0.8-1.3) ×109 s-1 for 2AN and k4AN = (2.1-3.3)× 107 s-1 for 4AN in PVAc films at 50ºC. Although it is possible to perform similar calculations using the data from experiments in PVAc at 5ºC, the numbers have questionable physical meaning because the events leading to photoproducts in the glassy state are stochastic in nature (Wang et al. 1990). By comparison, from data obtained in unstretched, completely amorphous PE films (Xu and Weiss2005b), k2AN = (0.5-1.2) ×109 s-1 at 60ºC and (2.2-3.7) ×108 s-1 at 23ºC and k4AN = (1.3-3.0) ×108 s-1 at 60ºC and (4.2-7.1)× 107 s-1 at 23ºC. The ranges of values are due to our uncertainty about the origin of 1-naphthol (3). If all of 3 is from 1-naphthoxy radicals within radical pair A (see out-of-cage route in Scheme 2), [3'] = 0 in eq 2 and the larger value is applicable; if 3 emanates from 1-naphoxy radicals of radical pair B (see out-of-cage' route in Scheme 2), [3'] is the same as the total yield of 3 and the lower value is more correct. Since 3 is probably produced via both routes, the rate constants should take intermediate values.
Regardless, the values clearly demonstrate (1) that k2AN is significantly larger than k4AN in each of the films, (2) that k2AN is almost the same in the two films at comparable temperatures, and (3) that the rate of formation of 4AN is slowed much more in the melt of PVAc than in amorphous PE. The latter observation may be attributed to an additional factor that retards radical motions within PVAc cages-strong dipole-dipole interactions of the radicals with surrounding acetate pendent groups (Terazima 2000, Gu et al. 2002). Apparently, the much smaller motions required to bring the radical center of a 2-phenylpropanoyl radical to a position where it can form a bond with C2 of its 1-naphthoxy within radical pair A is not affected by the polarity of its environment to the same extent as when the bond is made at the more distant C4. However, formation of 2AN requires the radicals of radical pair A to move at least 3.1 Å with respect to each other (Xu and Weiss 2005b).

Contrary to the results noted for combinations of radical pair A, combinations of radical pair B favor 2 and 4BN in both the glassy and melt states of PVAc (Table I). The decreased yield of 2BN is most striking in the glassy state at 5ºC where exceedingly long relaxation times of the polymer chains must amplify inhomogeneities of reaction cavities-reaction cavities may be described better in the glassy state stochastically rather than as an ensemble average (Wang et al. 1990). In the glassy state of the polymer, the motions of long chain segments become very slow and have time constants, on the order of 100 s (Angell 1995, Yoshii et al. 2000, Seymour and Carraher 2003), that are much longer than submicrosecond time scales for combination of the radical pairs. Therefore, only motions involving very short chain segments and pendent groups can facilitate the movement of radicals leading to their combination. As a consequence, radicals are still capable of reorientation, but they can move only short distances over relatively long periods. Those 2-phenylpropanoyl radicals that live long enough to lose CO (leading to radical pair B) must be 'born' from excited singlet states of (R)-1 whose conformations are locked by the glassy reaction cavities into positions which do not promote AN-type product formation. We suspect that the long lifetimes of the ensuing radical pair B here compensates for the lack of radical mobility. The net result is that memory of the initial positions of radical pair A is lost in radical pair B and electronic considerations prevail in the eventual combination events.
Polarity changes cannot be responsible for these observations because the yield of 2AN decreases dramatically between 50 and 5ºC in PVAc but increases significantly over the same temperature change in ethyl acetate. The large increase in the yield of 1-naphthol (3) at 5ºC in PVAc is also consistent with a large fraction of the radical pairs being unable to move easily in their glassy reaction cages (Wang et al. 1990, Gu et al. 2002). The very slow radical movement in PVAc films at 5ºC provides the 1-naphthoxy radical more opportunities for hydrogen abstraction (principally from the polymer) to form photoproduct 3 and make the 2-phenylpropanoyl radical less likely to combine at the C(2) position of 1-naphthoxy at 5ºC than at 50ºC, even though decarbonylation of the 2-phenylpropanoyl radical is also slowed significantly at 5ºC (Turro et al. 1983). At this point, we view the very small 2BN/4BN ratio in PVAc at 5ºC as another indication that the glassy states increases templating effects and the distribution of cage types experienced by the radical pairs.
STEREOCHEMISTRY OF RADICAL PAIR COMBINATIONS IN GLASSY AND MELT PHASE PVAC FILMS. COMPARISON WITH RESULTS IN AMORPHOUS PE FILMS
The formation of a large amount of (S)-2AN can be attributed to the enhanced restrictions to motions within the reaction cages in the glassy and melt phases of PVAc films. Analogous loss of enantiomeric purity of 2AN has been observed during irradiations of (R)-1 in PE films with relatively high crystallinities (unlike the PE film employed here) (Xu and Weiss 2005b). It can be traced to protraction of the lifetime of keto-(R)-2AN, increasing the probability of its excitation (Jiménez et al. 1997, Mori et al. 2004a) and reversible intramolecular g-H-atom abstraction at the chiral center (i.e., a secondary reaction analogous to the first step in the Norrish-Yang reaction (Yang and Elliott 1969, Stephenson et al. 1971)) before tautomerization to (R)-2AN can occur. An analogous intramolecular H-abstraction upon excitation of keto-4AN is not possible for steric reasons, and 4AN remains enantiomerically pure under all of the irradiation conditions we have explored. The range of ees for 2AN from PVAc films reported in Table I is a consequence of somewhat different irradiation times, fluxes, and conversions of (R)-1 in the several experiments conducted.
Based on the ee values for 2, 2BN, and 4BN (Table I), the stereoselectivity of radical pair B combinations in both PVAc films and ethyl acetate is much lower than in PE films. Thus, although the reaction cages of PVAc, especially in its glassy state, have stiffer walls than the cages of the PE films,they are unable to afford higher stereoselectivity in products from prochiral radical pair B combinations. As noted above, the regiochemical results suggest that the absolute rates of both translational and tumbling motions (leading to interconversion of the prochiral-R and prochiral-S radical pairs B; Scheme 2) are slowed in PVAc films. However, the relative retardation of tumbling by the 1-phenylethyl radical must be less within the reaction cages of PVAc than those of PE.
CONCLUSIONS AND COMMENTS ON THE INFLUENCE OF ENVIRONMENT ON THE FATES OF RADICAL PAIRS
Radicals such as those in radical pairs A and B are more polarizable than their parent species (i.e., molecules produced by adding an H-atom to the radical site). As a result, they diffuse more slowly, especially in polar media where dipole-dipole and H-bonding interactions may be important (Terazima 2000). The magnitude of the decreased mobility depends on several factors, including the properties of the solvent and the radicals, and the characteristics of the radical motions. In addition, the slower relaxation processes of polymeric chains in PVAc films than in PE films affect the chemistry of radical pair combinations. For these reasons, the regioselectivity of radical pair A combinations is much higher in both the melt and glassy states of PVAc films than that in completely amorphous PE films at 23ºC (i.e., in their melt state, far above Tg), but the stereoselectivity of prochiral radical pair B combinations is much lower in PVAc. In addition, even though reaction cavities of glassy PVAc, with their stiffer walls, control better the regiochemistry of radical pair combinations than those of melt PVAc, they do not control as well the stereochemistry. This behavior is reminiscent of the stiffer reaction cages within the interfacial regions of partially crystalline PE films, where radical pair combinations are less stereoselective than in the amorphous cages of the same film (Xu and Weiss 2003, 2005a, b, c).
From a synthetic standpoint, the results obtained here emphasize the need to control the relationship between the rates of radical tumbling and translation, as well as the relationship between the rates of in-cage motions and cage-escape, if high stereo- and regio-selectivities are to be achieved. It should be possible eventually to tune the important factors that modulate the motions of the radicals (N.B., the polarity and physical state of the medium, the shape, size and electronic propertiesof radicals themselves, and temperature) to maximize the yields of regio- and stereo-isomers of desired products. The research performed here contributes to our understanding how to reach that goal.
ACKNOWLEDGMENTS
The authors are grateful to the U. S. National Science Foundation for financial support of this research and to Prof. George S. Hammond for helpful suggestions.
Manuscript received on August 2, 2005; accepted for publication on September 27, 2005; contributed by RICHARD G. WEISS* * Member, Academia Brasileira de Ciências Correspondence to: Prof. Richard G. Weiss E-mail: weissr@georgetown.edu
Dedicated to the memory of Professor Christopher S. Foote (1935-2005), an excellent scientist and teacher and an admirer of Brasil.
- ANGELL CA. 1995. Formation of glasses from liquids and biopolymers. Science 267: 1924-1935.
- ARAI T, TOBITA S AND SHIZUKA H. 1994. Direct measurements of the rates of 1,3-sigmatropic and 1,5-sigmatropic hydrogen shifts in the photo-fries rearrangements of phenyl acetate. Chem Phys Lett 223: 521-526.
- ARAI T, TOBITA S AND SHIZUKA H. 1995. Tunneling effects on the 1,3- and 1,5-sigmatropic hydrogen shifts in the ground state of photo-Fries rearranged intermediates of phenyl acetate studied by laser flash photolysis. J Am Chem Soc 117: 3968-3975.
- BELLUS D. 1971. Photo-Fries rearrangement and related photochemical [1,j]-shifts (j = 3, 5, 7) carbonyl and sulfonyl groups. Adv Photochem 8: 109-159.
- BONDI A. 1964. Van der Waals volumes and radii. J Phys Chem 68: 441-451.
- BRAUN W, RAJBENBACH L AND EIRICH FR. 1962. Peroxide decomposition and cage effect. J Phys Chem 66: 1591-1595.
- CHARLES CE. 1996. Polymer Chemistry, 4th ed., New York: Marcel Dekker, 688 p.
- CHIANG Y, KRESGE AJ, TANG YS AND WIRZ J. 1984a. The pKa and keto-enol equilibrium constant of acetone in aqueous solution. J Am Chem Soc 106: 460-462.
- CHIANG Y, KRESGE AJ AND WIRZ J. 1984b. Flash-photolytic generation of acetophenone enol. The keto-enol equilibrium constant and pKa of acetophenone in aqueous solution. J Am Chem Soc 106: 6392-6395.
- CONRADI MS, ZELDES H AND LIVINGSTON R. 1979. Electron spin resonance determination of hindered rotation in benzyl radicals. Resonance stabilization energy. J Phys Chem 83: 2160-2161.
- CURRAN DP, PORTER NA AND GIESE B. 1995. Stereochemistry of radical reactions: concepts, guidelines, and synthetic applications, Weinheim: VCH (andreferences cited therein), p. 242-250.
- DIXON WT, FOSTER WEJ AND MURPHY D. 1973. Electron spin resonance spectra of naphthoxy radicals. J Chem Soc Perkin Trans II 15: 2124-2127.
- FINNEGAN RA AND KNUTSON D. 1965. Photodecarboxylation process: preparation of some hindered aryl ketones. Chem Ind 44: 1837-1838.
- FINNEGAN RA AND KNUTSON D. 1967. Stereochemistry of photodecarboxylation and photodecarbonylation reactions of aryl esters. The photolysis of (S)-(+)-3,5-di-t-butylphenyl 2-methylbutanoate. J Am Chem Soc 89: 1970-1972.
- GU W AND WEISS RG. 2000. Mediation of photochemical reactions of 1-naphthyl phenylacylates by polyethylene films. A 'radical clock' to measure rates of radical-pair cage recombinations in 'viscous space'. Tetrahedron 56: 6913-6925.
- GU W AND WEISS RG. 2001a. Photo-Fries rearrangements of phenyl phenylacylates in polyethylenefilms. Comparison of reactivity and selectivity with 1-naphthyl phenylacylates. J Org Chem 66: 1775-1780.
- GU W AND WEISS RG. 2001b. Extracting fundamental photochemical and photophysical information from photorearrangements of aryl phenylacylates and aryl benzyl ethers in media comprised of polyolefinic films. J Photochem Photobiol C: Photochem Rev 2: 117-137.
- GU W, WARRIER M, RAMAMURTHY V AND WEISS RG. 1999. Photo-Fries reactions of 1-naphthyl esters in cation-exchanged zeolites Y and polyethylene media. J Am Chem Soc 121: 9467-9468.
- GU W, HILL AJ, WANG XC, CUI CX AND WEISS RG. 2000a. Photorearrangements of five 1- and 2-naphthyl acylates in three unstretched and stretched polyethylene films. Does reaction selectivity correlate with free volumes measured by positron annihilation lifetime spectroscopy? Macromolecules 33: 7801-7811.
- GU W, WARRIER M, SCHOON B, RAMAMURTHY V AND WEISS RG. 2000b. Understanding the influence of active (zeolite) and passive (polyethylene) reaction cages on photo-Claisen rearrangements of aryl benzyl ethers. Langmuir 16: 6977-6981.
- GU W, ABDALLAH DJ AND WEISS RG. 2001. Conformational control of photoinduced deacrboxylation of simple aryl esters. Enhancement by templating effects in polyethylene films. J Photochem Photobiol A: Chem 139: 79-87.
- GU W, BI S AND WEISS RG. 2002. Photo-Fries rearrangements of 1-naphthyl esters in the glassy and melted states of poly(vinyl acetate). Comparisons with reactions in less polar polymers and low-viscosity solvents. Photochem Photobiol Sci 1: 52-59.
- JIMÉNEZ MC, MIRANDA MA, SCAIANO JC AND TORMOS R. 1997. Two-photon processes in the photo-Claisen and photo-Fries rearrangements. Direct observation of dienic ketenes generated by photolysis of transient cyclohexa-2,4-dienones. Chem Commun, p. 1487-1488.
- JOHN LE. 1993. An introduction to free radicals. J Wiley & Sons, New York, USA (and references cited therein), p. 56-76,
- KOBAYASHI Y, ZHENG W, MEYER EF, MCGERVEY JD, JAMIESON AM AND SIMHA R. 1989. Free volume and physical aging of poly(vinyl acetate) studied by positron annihilation. Macromolecules 22: 2302-2306.
- LINDEMANN MK. 1999. Physical constants of poly(vinyl acetate). In: BRANDRUP J, IMMERGUT EH AND GRULKE EA (Eds), Polymer Handbook, 4th ed., J Wiley & Sons, New York, USA, p. V77-V85.
- LUNAZZI L, INGOLD KU AND SCAIANO JC. 1983. Absolute rate constants for the decarbonylation of the phenylacetyl radical. J Phys Chem 87: 529-530.
- MARTIN R. 1992. Uses of the Fries rearrangement for the preparation of hydroxyaryl ketones. A review. Org Prep Proc Int 24: 369-435.
- MIRANDA MA. 1995. Photo-Fries reactions and related processes. In: HORSPOOL WM ET AL. (Eds), Handbook of organic photochemistry and photobiology, Boca Raton: CRC Press, p. 570-578.
- MIRANDA MA AND GALINDO F. 2003a. Photo-Fries reactions and related processes. In: HORSPOOL WM ET AL. (Eds), Handbook of organic photochemistry and photobiology 2nd ed., Boca Raton: CRC Press, p. 42/1-42/11.
- MIRANDA MA AND GALINDO F. 2003b. The photo-Fries rearrangement. In: RAMAMURTHY V ET AL. (Eds), Photochemistry of organic molecules in isotropic and anisotropic media, New York: Marcel Dekker, p. 43-132.
- MORI T, INOUE Y AND WEISS RG. 2003. Enhanced photodecarboxylation of an aryl ester in polyethylene films. Org Lett 5: 4661-4664.
- MORI T, TAKAMOTO M, SAITO H, FURO T, WADA T AND INOUE Y. 2004a. Isolation of cyclohexadienone intermediates in the photo-Fries rearrangement of 2,4-dimethylnaphth-1-yl and 1,4-dimethylnaphth-2-yl 2,4,6-trimethylbenzoates. Chem Lett 33: 254-255.
- MORI T, TAKAMOTO M, SAITO H, FURO T, WADA T AND INOUE Y. 2004b. Remarkable differences in photo and thermal (acid-catalyzed) reactivities between ortho- and para-acylcyclohexadienonesas essential factors determining the overall efficiency of the photo-Fries rearrangement. Chem Lett 33: 256-257.
- PORTER NA AND KREBS PJ. 1988. Stereochemical aspects of radical pair reactions. In: ELIEL EL ET AL. (Eds), Topics in stereochemistry. J Wiley & Sons, New York, USA (and references cited therein) 18: 97-127.
- RAMAMURTHY V AND SCHANZE KS. 2003. Photochemistry of organic molecules in isotropic and anisotropic media, NewYork: Marcel Dekker, 408 p.
- RAMAMURTHY V, WEISS RG AND HAMMOND GS. 1993. A model for the influence of organized media on photochemical reactions. In: VOLMAN DH ET AL. (Eds), Advances in photochemistry, J Wiley & Sons, New York, USA 18: 67-234.
- RAVVE A. 2000. Principles of polymer chemistry, 2 ed., New York: Kluwer/Plenum, 708 p.
- RIDDICK JA, BUNGER WB AND SAKANO TK. 1986. Organic solvents (Techniques of organic chemistry series, Vol. II), 4th ed., J Wiley & Sons, New York, USA, Interscience, 1344 p.
- SEYMOUR RB AND CARRAHER CE. 2003. Polymer chemistry, 6th ed., New York: Marcel Dekker, 960 p.
- STEPHENSON LM, CAVIGLI PR AND PARLETT JL. 1971. Chemistry of 1,4-diradicals. I. Intermediates in the Norrish type II photocleavage. J Am Chem Soc 93: 1984-1988.
- STEVENS MP. 1998. Polymer Chemistry, 3rd ed., New York: Oxford University Press, 549 p.
- TERAZIMA M. 2000. Is the translational diffusion of organic radicals different from that of closed-shell molecules? Acc Chem Res 33: 687-694.
- TIMERMANS J. 1965. Physico-chemical constants of pure organic compounds, New York: Elsevier, 693 p.
- TSENTALOVICH YP AND FISCHER H. 1994. Solvent effect on the decarbonylation of acyl radicals studied by laser flash photolysis. J Chem Soc Perkin Trans 2 4: 729-733.
- TURRO NJ. 2002. Supramolecular organic photochemistry: control of covalent bond formation through noncovalent supramolecular interactions and magnetic effects. Proc Natl Acad Sci USA 99: 4805-4809.
- TURRO NJ, GOULD IR AND BARETZ BH. 1983. Absolute rate constants for decarbonylation of phenylacetyl and related radicals. J Phys Chem 87: 531-532.
- WANG Z, HOLDEN DA AND MCCOURT FRW. 1990. Generation of nonrandom chromophore distributions by the photo-Fries reaction of 2-naphthyl acetate in poly(methyl methacrylate). Macromolecules 23: 3773-3779.
- WARRIER M, KAANUMALLE LS AND RAMAMURTHY V. 2003. Alkali metal ion controlled product selectivity during photorearrangements of 1-naphthyl phenyl acylates and dibenzyl ketones. Can J Chem 81: 620-631.
- WEISS RG, RAMAMURTHY V AND HAMMOND GS. 1993. Photochemistry in organized and confined media: a model. Acc Chem Res 26: 530-536.
- XU J AND WEISS RG. 2003. Enantioselectivity of prochiral radical-pair recombinations. Reaction cavity differentiation in polyethylene films. Org Lett 5: 3077-3080.
- XU J AND WEISS RG. 2005a. Analyses of in-cage singlet radical-pair motions from irradiations of 1-naphthyl (R)-1-phenylethyl ether and 1-naphthyl(R)-2-phenylpropanoate in n-alkanes. J Org Chem 70: 1243-1252.
- XU J AND WEISS RG. 2005b. Combinations of chiral and prochiral singlet radical-pairs in reaction cavities of polyethylene films. Control and analysis of radical tumbling and translation. Photochem Photobiol Sci 4: 348-358.
- XU J AND WEISS RG. 2005c. Reaction cavities of liquid and solid phases of a long n-alkane, n-nonadecane, as probed by the regio- and stereo-chemical fates of two singlet radical pairs. Photochem Photobiol Sci 4: 210-215.
- YANG C AND ELLIOTT SP. 1969. Photochemistry of (S)-(+)-5-methyl-2-heptanone. J Am Chem Soc 91: 7550-7551.
- YOSHII K, MACHIDA S AND HORIE K. 2000. Local free volume and structural relaxation studied with photoisomerization of azobenzene and persistent spectral hole burning in poly(alkyl methacrylate)s at low temperatures. J Polym Sci Part B: Polym Phys 38: 3098-3105.
- ZHANG X AND NAU WM. 2000. Aryl substituent effects and solvent effects on the decarbonylation of phenacetyl radicals. J Phys Org Chem 13: 634-639.
Publication Dates
-
Publication in this collection
08 Mar 2006 -
Date of issue
Mar 2006
History
-
Accepted
27 Sept 2005 -
Received
02 Aug 2005