Abstracts
Establishment of infection by Trypanosoma cruzi, the agent of Chagas' disease, depends on a series of events involving interactions of diverse parasite molecules with host components. Here we focus on the mechanisms of target cell invasion by metacyclic trypomastigotes (MT) and mammalian tissue culture trypomastigotes (TCT). During MT or TCT internalization, signal transduction pathways are activated both in the parasite and the target cell, leading to Ca2+ mobilization. For cell adhesion, MT engage surface glycoproteins, such as gp82 and gp35/50, which are Ca2+ signal-inducing molecules. In T. cruzi isolates that enter host cells in gp82-mediated manner, parasite protein tyrosine kinase as well as phospholipase C are activated, and Ca2+ is released from I P3-sensitive stores, whereas in T. cruzi isolates that attach to target cells mainly through gp35/50, the signaling pathway involving adenylate cyclase appears to be stimulated, with Ca2+ release from acidocalciosomes. In addition, T. cruzi isolate-dependent inhibitory signals, mediated by MT-specific gp90, may be triggered both in the host cell and the parasite. The repertoire of TCT molecules implicated in cell invasion includes surface glycoproteins of gp85 family, with members containing binding sites for laminin and cytokeratin 18, enzymes such as cruzipain, trans-sialidase, and an oligopeptidase B that generates a Ca2+-agonist from a precursor molecule.
Trypanosoma cruzi; trypomastigotes; cell invasion; signal transduction; Ca2+ mobilization
O estabelecimento da infecção por Trypanosoma cruzi, o agente da doença de Chagas, depende de uma série de eventos envolvendo interações de diversas moléculas do parasita com componentes do hospedeiro. Focalizamos aqui os mecanismos de invasão celular por tripomastigotas metacíclicos (TM) e por tripomastigotas de cultura de tecido (TCT). Durante a internalização de TM ou TCT, vias de transdução de sinal são ativadas tanto no parasita como na célula alvo, acarretando a mobilização de Ca2+. Para adesão, TM utiliza as glicoproteínas de superfície como a gp82 e gp35/50, que são moléculas indutoras de sinal de Ca2+. Em isolados de T. cruzi que entram na célula hospedeira de maneira dependente de gp82, a proteína tirosina quinase assim como a fosfolipase C do parasita são ativadas, e Ca2+ é liberado de reservatórios sensíveis a IP3, enquanto em isolados de T. cruzi que se ligam às células alvo através de gp35/50, a via de sinalização envolvendo adenilil ciclase parece ser estimulada, com liberação de Ca2+ de acidocalciossomos. Além disso, dependendo do isolado de T. cruzi, sinais inibitórios mediados por gp90 específica de TM podem ser desencadeados tanto na célula hospedeira como no parasita. O repertório de moléculas de TCT implicadas na invasão celular inclui glicoproteínas de superfície da família gp85, com membros contendo sitos de ligação à laminina e citoqueratina 18, enzimas como a cruzipaína, trans-sialidase, e uma oligopeptidase B que gera um agonista de Ca2+ a partir de uma molécula precursora.
Trypanosoma cruzi; tripomastigotas; invasão celular; transdução de sinal; mobilização de Ca2+
BIOMEDICAL AND MEDICAL SCIENCES
Molecular basis of mammalian cell invasion by Trypanosoma cruzi
Nobuko Yoshida
Departamento de Microbiologia, Imunologia e Parasitologia, Escola Paulista de Medicina Universidade Federal de São Paulo, R. Botucatu, 862 - 04023-062 São Paulo, SP, Brasil
ABSTRACT
Establishment of infection by Trypanosoma cruzi, the agent of Chagas' disease, depends on a series of events involving interactions of diverse parasite molecules with host components. Here we focus on the mechanisms of target cell invasion by metacyclic trypomastigotes (MT) and mammalian tissue culture trypomastigotes (TCT). During MT or TCT internalization, signal transduction pathways are activated both in the parasite and the target cell, leading to Ca2+ mobilization. For cell adhesion, MT engage surface glycoproteins, such as gp82 and gp35/50, which are Ca2+ signal-inducing molecules. In T. cruzi isolates that enter host cells in gp82-mediated manner, parasite protein tyrosine kinase as well as phospholipase C are activated, and Ca2+ is released from IP3-sensitive stores, whereas in T. cruzi isolates that attach to target cells mainly through gp35/50, the signaling pathway involving adenylate cyclase appears to be stimulated, with Ca2+ release from acidocalciosomes. In addition, T. cruzi isolate-dependent inhibitory signals, mediated by MT-specific gp90, may be triggered both in the host cell and the parasite. The repertoire of TCT molecules implicated in cell invasion includes surface glycoproteins of gp85 family, with members containing binding sites for laminin and cytokeratin 18, enzymes such as cruzipain, trans-sialidase, and an oligopeptidase B that generates a Ca2+-agonist from a precursor molecule.
Key words:Trypanosoma cruzi, trypomastigotes, cell invasion, signal transduction, Ca2+ mobilization.
RESUMO
O estabelecimento da infecção por Trypanosoma cruzi, o agente da doença de Chagas, depende de uma série de eventos envolvendo interações de diversas moléculas do parasita com componentes do hospedeiro. Focalizamos aqui os mecanismos de invasão celular por tripomastigotas metacíclicos (TM) e por tripomastigotas de cultura de tecido (TCT). Durante a internalização de TM ou TCT, vias de transdução de sinal são ativadas tanto no parasita como na célula alvo, acarretando a mobilização de Ca2+. Para adesão, TM utiliza as glicoproteínas de superfície como a gp82 e gp35/50, que são moléculas indutoras de sinal de Ca2+. Em isolados de T. cruzi que entram na célula hospedeira de maneira dependente de gp82, a proteína tirosina quinase assim como a fosfolipase C do parasita são ativadas, e Ca2+ é liberado de reservatórios sensíveis a IP3, enquanto em isolados de T. cruzi que se ligam às células alvo através de gp35/50, a via de sinalização envolvendo adenilil ciclase parece ser estimulada, com liberação de Ca2+ de acidocalciossomos. Além disso, dependendo do isolado de T. cruzi, sinais inibitórios mediados por gp90 específica de TM podem ser desencadeados tanto na célula hospedeira como no parasita. O repertório de moléculas de TCT implicadas na invasão celular inclui glicoproteínas de superfície da família gp85, com membros contendo sitos de ligação à laminina e citoqueratina 18, enzimas como a cruzipaína, trans-sialidase, e uma oligopeptidase B que gera um agonista de Ca2+ a partir de uma molécula precursora.
Palavras-chave:Trypanosoma cruzi, tripomastigotas, invasão celular, transdução de sinal, mobilização de Ca2+.
OVERVIEW
Trypanosoma cruzi is transmitted by insect vectors when these blood-sucking triatomines deposit on the skin their feces containing MT, the infective forms of the parasite. Through the ocular mucosa or lesions in the skin, the parasites find their way to invade host cells. Metacyclic forms may enter mammalian hosts also by oral route. According to Coura et al. (2002), more than half of the acute cases of Chagas disease recorded between 1968 and 2000 in Brazilian Amazon can be attributed to microepidemics of orally transmitted infection from contaminated food. The potential sources of contamination are whole triatomine insects or their feces containing infective parasites. Hoft (1996) has shown that MT are efficient in establishing infection in mice when given orally. They enter the gastric mucosal epithelium, transform into amastigotes and replicate intracellularly (Hoft et al. 1996). Trypomastigotes emerge from infected cells, circulate in the bloodstream and disseminate to diverse organs and tissues. These circulating parasites can be transmitted by blood transfusion or congenitally. Following the acute phase, in which the parasites go through multiple rounds of cell invasion and intracellular replication, their proliferation is controlled by the host immune response.
Mammalian cell invasion by T. cruzi has been extensively studied in vitro by using MT cultured in liquid media and TCT, as counterparts of insect-borne and bloodstream parasites respectively. Different T. cruzi isolates and a variety of cell types, mostly cells that are not professional phagocytes, have been used (Table I). The picture emerging from these studies is that T. cruzi penetration into host cells is a multi-step process involving various parasite and host cell molecules that, in a concerted series of events, leads to intracellular Ca2+ mobilization in both cells (Docampo et al. 1996, Burleigh and Andrews 1998, Yoshida 2003). Attachment of trypomastigotes is receptor-mediated and is restricted to cell surface domains (Schenkman et al. 1991a). For instance, parasites bind to and enter HeLa cells at their edges and invade polarized MDCK cells through their basolateral domains (Mortara 1991, Schenkman et al. 1988a). To invade mammalian cells, MT and TCT engage distinct sets of surface molecules that differentially interact with host components. The MT-specific glycoprotein gp82 binds to gastric mucin (Neira et al. 2003) whereas members of the TCT gp85 family have been shown to bind components of extracelullar matrix, such as fibronectin and laminin (Ouaissi et al. 1986a, Giordano et al. 1994). Although MT gp82 and TCT gp85 display differential adhesive properties and interact with different receptors on target cells, they are related molecules. They have considerable sequence identity (40-60%) and are included in the T. cruzi gp85/sialidase superfamily (Araya et al. 1994, Colli and Alves 1999). Mucin-like glycoproteins constitute another group of molecules implicated in host cell invasion that are differentially expressed in MT and TCT. Mucins from MT are protease-resistant gp35/50 molecules (Mortara et al. 1992, Schenkman et al. 1993a), whereas TCT mucins are larger molecules, migrating in SDS-PAGE as diffuse bands between 70 and 200 kDa (Almeida et al. 1994, Schenkman et al. 1991b).
MT SURFACE MOLECULES THAT MEDIATE BIDIRECTIONAL SIGNAL TRANSDUCTION AND HOST CELL INVASION
Studies with MT of 10 T. cruzi isolates, derived from different sources in distinct geographical regions, have revealed two groups of parasites that differentially express surface glycoproteins and display differential ability to invade mammalian cells in vitro (Fig. 1). The two groups are highly divergent. By molecular phylogenetic analysis, Briones et al. (1999) have found that the distance between the two T. cruzi lineages is larger than the distances among Leishmania spp.
MT of highly invasive isolates are deficient in gp90 and gp35/50, identified respectively by monoclonal antibodies (MAbs) 1G7 and 10D8, but express the variant forms of gp90 and gp35/50, identified by MAbs 5E7 and 2B10. MT of poorly invasive isolates express a gp90 molecule that is detectable by MAbs 1G7 and 5E7, and gp35/50 molecules recognized by both MAbs 10D8 and 2B10. Expression of gp82, which reacts with MAb 3F6, is ubiquitous among these isolates (Fig. 1).
Gp35/50, gp82 and gp90 bind to as yet undefined host cell receptors. The interaction of any of these molecules with its receptor triggers bidirectional signaling cascades. Whether the MT-target cell interaction results in productive infection depends on which surface molecule is predominantly engaged. To attach to and enter host cells, MT of highly invasive CL isolate, for instance, engage gp82 (Ramirez et al. 1993), which efficiently triggers Ca2+ signaling in MT and host cells (Ruiz et al. 1998), whereas MT of poorly invasive G isolate appear to rely mainly on gp35/50 for their internalization (Yoshida et al. 1989). Gp35/50 molecules are not as effective as gp82 in promoting invasion, due probably to their poor Ca2+ signal-inducing activity (Dorta et al. 1995). If the interaction is mediated by gp90, which is devoid of Ca2+ signaling activity, productive infection is precluded.
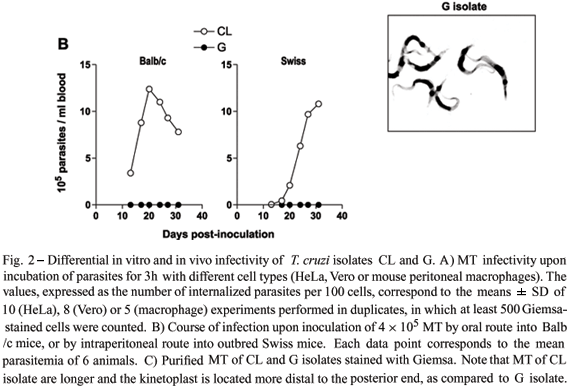
Extensive studies with MT of T. cruzi isolates CL and G, which represent the prototypes of highly and poorly invasive parasites, have shown that their infectivity is not related to the target cell type. Regardless of the mammalian cell type, the number of internalized parasites of CL isolate is always several fold higher than that of G isolate (Fig. 2A). These in vitro findings closely correlate to the observations in vivo. Regardless of the mouse strain or the route of parasite administration, MT of CL isolate produce high parasitemias, in contrast to MT of G isolate that invariably produce subpatent infection (Fig. 2B). Presumably the mechanisms of host cell entry acting in vitro prevail in vivo. Invasion of epithelial HeLa cells by MT may be equivalent to the invasion of gastric mucosal epithelium, upon oral infection. Likewise, MT entry into mouse peritoneal macrophages, whether the parasites are seeded onto cultured cells or are inoculated intraperitoneally, may be equivalent.

One interesting observation is that MT ofCL and G isolates are morphologically different(Fig. 2C). As compared to CL isolate, parasites of G isolate are shorter and the kinetoplast is more proximal to the posterior end. Isolate-dependent morphological variations are also observed in blood trypomastigotes and the different morphologies apparently denote physiological differences (Brener 1973). Slender forms appear to penetrate host cells better than the stout forms (Brener 1969). In some T. cruzi isolates, slender trypomastigotes are prevalent during the first days of infection, whereas in other isolates stout forms predominate during the entire infection in mice (Brener 1973).
GP82, THE CA2+ SIGNAL-INDUCING MOLECULE THAT PROMOTES CELL INVASION OF HIGHLY INVASIVE T. cruzi ISOLATES
The role of the metacyclic stage-specific surface molecule gp82 in mammalian cell invasion was first determined by inhibition of MT internalization using MAb 3F6 or the purified native glycoprotein (Ramirez et al. 1993). Subsequent studies indicated that gp82 promotes MT entry into host cells by inducing the activation of signaling cascades and Ca2+ mobilization in both cells (Ruiz et al. 1998, Yoshida et al. 2000). Recent studies with T. cruzi clone CL-14, which is poorly infective, have further reinforced the role played by gp82. When compared to the parental CL isolate, the sole difference in the surface profile of clone CL-14 MT was the deficient expression of gp82 (Atayde et al. 2004).
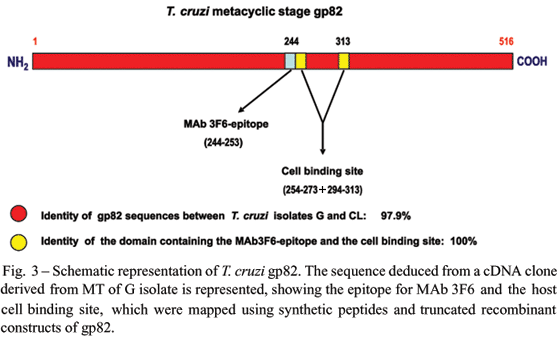
Gp82 is a glycoprotein containing N-linkedoligosaccharides (Ramirez et al. 1993) that is anchored to MT plasma membrane by glycosylphosphatidylinositol (GPI) moiety (Cardoso de Almeida and Heise 1993). The identity of amino acid sequences of gp82 deduced from cDNA clones of G and CL metacyclic forms is 97.9%, and 100% as regards the central domain containing the mammalian cell binding site (Ruiz 1998). Using synthetic peptides and truncated recombinant gp82 constructs, Manque et al. (2000) found that the cell binding site is contiguous to and partially overlaps the epitope for MAb 3F6, and appears to be conformational, being possibly formed by juxtaposition of two sequences separated in the linear molecule by a hydrophobic stretch (Fig. 3).
The central domain of MT gp82 shares 60-65% identity with the C-terminal region of TCT glycoproteins of gp85 family. However, within the sequences containing the cell binding site and the epitope for MAb 3F6, there are significant differences between MT and TCT molecules, and these include substitutions of acidic amino acids for uncharged or positively charged residues, substitutions of uncharged residues for lysine or arginine, and substitutions of residues with polar side chains for those with nonpolar side chains and vice-versa.
Binding of gp82 triggers in host cells a transient increase in intracellular Ca2+ concentration, in the same manner as the soluble extracts of MT (Fig. 4). Non infective epimastigotes do not induce Ca2+ signaling (Tardieux et al. 1994, Dorta et al. 1995) unless they are transfected with T. cruzi expression vector carrying gp82 cDNA (Manque et al. 2003). Gp82 triggers Ca2+ response in mammalian cells susceptible to T. cruzi infection, such as HeLa and Vero cells, but not in T. cruzi-resistant human leukemic K562 cells.
The kinetics of Ca2+ mobilization in MT is distinct from that observed in host cells and can be triggered by soluble extracts of HeLa cells (Fig. 4), but not of K562 cells, in a manner inhibitable by native or recombinant gp82. Upon binding to host cells, gp82 relays a signal to the parasite that results in protein tyrosine kinase (PTK) activation and phosphorylation of p175, a protein that is undetectable in noninfective epimastigotes (Favoreto et al. 1998). Activation of PTK and Ca2+ response are associated events, being both inhibited by gp82, as well as by genistein, a specific inhibitor of PTK (Yoshida et al. 2000). The signaling cascade induced by gp82 includes the participation of phospholipase C (PLC), that generates inositol 1,4,5-triphosphate (IP3), as inferred from experiments in which the parasite infectivity was impaired by PLC inhibitor U73122, as well as by drugs, such as heparin, a competitive IP-receptor blocker, and caffein, which affects Ca release from IP3-sensitive stores (Yoshida et al. 2000). Our hypothesis based on these findings is that in MT of T. cruzi isolates that engage gp82 to enter target cells, such as the CL isolate, a signaling cascade is initiated at the parasite cell surface by gp82 and proceeds downstream through a sequence involving PTK, PLC and IP3, leading to Ca2+ mobilization, possibly from endoplasmic reticulum (Fig. 5).
Many questions concerning the gp82-mediated signaling cascade remain to be elucidated. One of these is how gp82, which is GPI-anchored and therefore associates only with the outer leaflet of the MT lipid bilayer, activates PTK. In mammalian cells, association of GPI-anchored proteins and PTKs has been reported. A neuron survival factor neurturin, for instance, signals through multicomplex receptors that consist of receptor tyrosine kinase and a member of a GPI-linked family of receptors that determine ligand specificity (Buj-Bello et al. 1997). In T cells, PTKs can be found in complexes immunoprecipitated with antibodies directed to GPI-anchored proteins (Stefanova et al. 1991, Shenoy-Scaria et al. 1992, Thomas and Samelson 1992). An alternative possibility is the interaction of GPI-anchored proteins and kinases mediated by transmembrane linker proteins, as suggested by Brown (1993). Also awaiting elucidation is the question concerning the connection between PTK or p175 and PLC.
GP82, A MEDIATOR OF INVASION OF GASTRIC MUCOSAL EPITHELIUM
Recently, the involvement of gp82 in oral T. cruzi infection was inferred from experiments in which the infectivity of CL isolate MT was greatly reduced upon treatment with anti-gp82 MAb 3F6 (Neira et al. 2003). In addition to playing a critical role in promoting invasion of gastric mucosal epithelium, the ability of gp82 to bind to gastric mucin may be important for the establishment of infection by oral route. Shigella dysenteriae, for instance, whose pathogenic potential is correlated with its ability to invade and multiply within the cells of colonic epithelium, preferentially adheres to colonic mucin (Sudha et al. 2001).
The role of gp82 in establishing efficient T. cruzi infection by oral route has been reinforced by studies using gp82-deficient isolates, such as 569 and 588 (Cortez et al. 2003). When administered orally into mice, MT of these gp82-deficient T. cruzi isolates produce patent parasitemia, but to greatly reduced levels when compared to isolate CL. Although the in vivo infectivity of isolates 569 and 588 is lower than that of the CL isolate, the ability to enter host cells in vitro is similar. MT of isolates 569 and 588 can efficiently invade cells in vitro because they express gp30, a surface glycoprotein detectable by MAb 3F6 and displaying Ca2+ signal-inducing activity. Gp30-mediated HeLa cell entry of isolates 569 and 588, which is inhibited by MAb 3F6 as well as by native gp30, is dependent on the parasite signal transduction involving PTK activation and Ca2+ mobilization from thapsigargin-sensitive stores, similarly to the gp82-mediated signaling. Notwithstanding the functional similarity between gp82 and gp30, MT of gp82-deficient isolates are less infective by oral route than the CL isolate, and this is probably due to the low efficiency of gp30 in binding to gastric mucin. Accordingly, in the presence of high concentration of gastric mucin, to mimic the in vivo condition, the rate of HeLa cell invasion of gp82-deficient MT, but not of CL isolate, was significantly reduced (Cortez et al. 2003). These findings indicate that the host cell invasion by MT can be mediated either by gp82 or gp30, but efficient mucosal infection depends on the expression of gp82, which promotes the adhesion to gastric mucin, the first step towards the penetration into the underlying epithelial cells.
GP35/50, THE CA2+ SIGNAL-INDUCING MOLECULE THAT PROMOTES CELL INVASION OF POORLY INVASIVE T. cruzi ISOLATES
The involvement of mucin-like gp35/50 molecules in mammalian cell invasion was determined by inhibition of MT internalization using MAb 10D8 or the purified native glycoprotein (Yoshida et al. 1989, Ruiz et al. 1993). Like gp82, gp35/50 bind to target cells in a receptor-mediated manner and triggers bidirectional Ca2+ response, but to a lower degree than gp82 (Ruiz et al. 1998).
Gp35/50 mucins are expressed in metacyclic forms and epimastigotes of all T. cruzi isolates examined to date. They are encoded by large multigene family (Di Noia et al. 1998) and are detected in SDS-PAGE gels as two-three bands displaying isolate-dependent size polymorphism (Mortara et al. 1992). Gp35/50 molecules are GPI-anchored glycoproteins rich in threonine. Glycans are O-linked to threonine residues in the mucin protein core through N-acetylglucosamine, rather than N-acetylgalactosamines as usually found in vertebrate mucins and, depending on the T. cruzi isolate, may contain galactofuranose residues in addition to galactopyranose (Previato et al. 1994, Acosta-Serrano et al. 1995, Salto et al. 2000). MAb 10D8, that recognize gp35/50 mucins in MT of poorly invasive isolates, such as G and Tulahuen, and partially neutralize their infectivity (Yoshida et al. 1989), reacts with epitopes containing galactofuranose, whereas MAb 2B10 appears to react with galactopyranose-containing epitopes present in all isolates.
T. cruzi mucins are the main acceptors ofsialic acid in trans-sialidase (TS)-mediated reaction (Schenkman and Eichinger 1993). TS is an enzyme that specifically transfers (a 2-3)-linked sialic acid from extrinsic host-derived macromolecules to O-linked oligosaccharides of T. cruzi mucin-like glycoproteins (Schenkman et al. 1991b, Schenkman and Eichinger 1993). The enzyme, that is up to 30 times more active in TCT than in MT (Acosta-Serrano et al. 2001), was first identified in TCT and bloodstream trypomastigotes as a developmentally regulated sialidase that releases sialic acid from human erythrocytes and plasma glycoprotein (Pereira 1983). Using T. cruzi epimastigotes, Previato et al. (1985) found that sialic acid from exogenous sialylated glycoconjugates is incorporated into parasite macromolecules. TS preferentially tranfers sialyl residues to available galactose acceptors and acts as a sialidase in the absence of appropriate amountsof suitable acceptors (Schenkman and Eichinger 1993). In MT, sialyl residues are transferred exclusively into gp35/50 mucins (Schenkman et al. 1993a).
Sialyl residues of gp35/50 mucins are not required for MT invasion, they may rather impair the interaction with target cells. Treatment of MT of G isolate with bacterial neuraminidase, for instance, removes sialic acid from gp35/50 and increases the parasite infectivity (Yoshida et al. 1997). Resialylation of gp35/50, by incubation of parasites with T. cruzi TS and sialyl lactose, restores the reactivity with lectin or monoclonal antibody specific for sialic acid and, accordingly, reduces the rate of MT entry into target cells to levels similar to those before desialylation. Compatible with this finding, the capacity to bind to host cells and to trigger Ca2+ response was found to be higher in desialylated gp35/50 as compared to its sialylated counterpart (Yoshida et al. 1997). Infectivity of CL isolate MT is not affected by neuraminidase treatment because these parasites rely on gp82, rather than on gp35/50, for their internalization.
In MT that preferentially engage gp35/50 to invade host cells, as is the case of G isolate, the signaling cascade triggered in the parasite is distinct from that induced by gp82 (Fig. 5). PTK and PLC are not implicated, instead cyclic AMP (cAMP) may play a role, as deduced from the increased parasite infectivity upon treatment with adenylyl cyclase activator forskolin (Neira et al. 2002). The Ca2+ required for cell invasion appears to be released from acidocalcisomes, the vacuoles containing a Ca2+/H2+ exchange system (Docampo et al. 1995), provided that treatment of MT with a combination of ionomycin plus NH4Cl or nigericin, that releases Ca2+ from these acidic compartments, significantly diminishes target cell entry (Neira et al. 2002). How gp35/50, an GPI-anchored molecule, relays the external signal to the parasite interior, and what are the components required for that process are questions that remain unanswered.
GP90, THE MT-SPECIFIC DOWN REGULATOR OF HOST CELL INVASION
The property of gp90 as a negative regulator of target cell invasion was demonstrated by experiments using antisense oligonucleotides targeted to gp90 gene sequences. Treatment of MT of G isolate with antisense oligonucleotides reduced the expression of gp90 and increased the parasite ability to enter host cells, whereas their sense counterparts or the mismatched sequences had no effect (Málaga and Yoshida 2001).
GP90 is a metacyclic stage-specific glycoprotein containing N-linked oligosaccharides (Yoshida et al. 1990). It binds to mammalian cells in a receptor-mediated manner but, unlike gp82 or gp35/50, does not trigger Ca2+ signal, and intracellular Ca2+ is not mobilized in the parasite upon binding of anti-gp90 MAb 1G7, in contrast to what happens upon interaction with monoclonal antibodies directed to gp82 or gp35/50 (Ruiz et al. 1998). In MT, the interaction of gp90 with its receptor may trigger an inhibitory pathway, similarly to cells of the immune system where, in addition to activation signals, signaling cascades acting as negative regulators can be induced (Veillette et al. 2002, Vivier et al. 2004). Like NK cell surface inhibitory receptors that antagonize activation pathways using protein tyrosine phosphatases (Vivier et al. 2004), gp90 mediates the activation of MT tyrosine phosphatase that counteracts the action of PTK by dephosphorlylating p175 (Manque et al. 2003).
Like gp82 and gp35/50, gp90 is also anchored to the plasma membrane via GPI (Schenkman et al. 1988b). The type of association of these GPI-anchored molecules with other components of the plasma membrane is unknown. One possibility is the interaction through their extracellular domains. Another possibility is the association through GPI lipid moiety, and in this case the nature of the lipid may influence which plasma membrane molecule is recruited. Of note in this regard is that the lipid portion of gp35/50 GPI from noninfective epimastigotes is composed essentially of 1-O-hexadecyl-2-O-hexadecanoyl-PI and of 1-O-hexadecyl-2-O-octadecanoyl-PI, whereas that of metacyclic stage gp35/50 is mainly ceramide-PI (Acosta-Serrano et al. 1995).
MECHANISMS OF MT INVASION OF GASTRIC MUCOSAL EPITHELIUM UPON ORAL INFECTION
MT, but not blood trypomastigotes, have uniquely specialized functions for mucosal invasion and efficiently enter gastric mucosal epithelium (Hoft 1996, Hoft et al. 1996). This is in accord with the notion that metacyclic and bloodstream trypomastigotes are morphologically similar but are physiologically distinct (Tyler and Engman 2001). Hoft (1996) suggested that MT express stage-specific surface molecules required for adhesion to mucosal epithelial surface receptors and/or for penetration of mucin coat. This hypothesis has been supported by results from experiments of oral infection in mice and in vitro cell invasion assays mimicking the in vivo conditions (Neira et al. 2003, Cortez et al. 2003). Based on these data, the following sequence of events can be visualized (Fig. 6). When MT reach the stomach, they resist destruction because they are protected by protease-resistant gp35/50 mucins, which are abundant on the parasite surface. Pepsin digestion leaves intact the gp82 domain containing both the target cell and the gastric mucin-binding sites. The parasites bind to gastric mucin through gp82 (Fig. 6A) and traverse the mucus to reach the underlying epithelial cells. It has as yet to be demonstrated, but it is possible that T. cruzi has mucinase activity, like Entamoeba histolytica, which expresses cyteine proteinases that disrupt the polymeric structure of colonic mucin (Moncada et al. 2003), or Trichomonas vaginalis, which invades vaginal mucous layer by secreting mucinase (Lehker and Sweeney 1999). Preliminary experiments indicate that T. cruzi secrets an enzyme that acts on gastric mucin.

Once the mucous barrier is overcome, MT of T. cruzi isolates such as CL efficiently invade gastric epithelial cells by engaging gp82 (Fig. 6B) and triggering bidirectional Ca2+ response. MT of G isolate may reach the epithelial cells as effectively as the CL isolate MT, but their entry may be impaired by gp90. Some parasites manage to be internalized by engaging gp35/50 (Fig. 6B).
On the other hand, MT of T. cruzi isolatesexpressing gp30, but deficient in gp82, such as 569 and 588, would have difficulty in penetrating the thick mucin coat, because gp30 binds poorly to gastric mucin. Binding to mucin may be critical for the effective action of mucinase inasmuch as, without binding, the enzyme would be secreted into the medium, not onto the mucin layer. The gp82-deficient parasites that manage to translocate through the mucin coat, and reach the epithelial cells, are then internalized in gp30-mediated manner, probably as efficiently as the gp82-expressing MT.
TCT MOLECULES IMPLICATED IN HOST CELL INVASION
Diverse TCT molecules have been implicated in host cell invasion. These include surface and/or secreted components such as Tc-85 containing binding sites for laminin and cytokeatin 18 (Giordano et al. 1994, Magdesian et al. 2001), mucins and TS (Schenkman et al. 1991b), cystein proteinases (Meirelles et al. 1992) and members of the prolyl oligopeptidase family of serine proteases (Burleigh et al. 1997, Grellier et al. 2001).
TC-85 FAMILY AND OTHER TCT COMPONENTS WITH AFFINITY FOR EXTRA CELLULAR MATRIX
Tc-85, the surface glycoprotein expressed in TCT was first implicated in mammalian cell invasionusing MAbs that partially block parasite internalization (Alves et al. 1986). Among the membersof Tc-85 family, the best characterized is the molecule that contains binding sites for laminin (Giordano et al. 1994, 1999) and host cell cytokeratin 18 (Magdesian et al. 2001). The Tc-85 cell binding site colocalizes to the most conserved motif (VTVXNVFLYNR) of the gp85/TS superfamily, at the C-terminal domain, and does not contain the laminin-binding site (Magdesian et al. 2001). In the sequence deduced from laminin-binding Tc-85 cDNA, there is a stretch of 122 amino acid residues downstream to this conserved motif (Giordano et al. 1999), whereas in MT gp82 the same motif localizes closer to the C- terminus, being followed by 38 residues, of which 14 correspond to the hydrophobic GPI-anchor sequence (Araya et al. 1994). It is possible that this subterminal localization of conserved motif in MT gp82 precludes its interaction with cytokeratin C18.
The involvement of fibronectin in target cell invasion by TCT was deduced from experiments in which the peptide RGDS, corresponding to fibronectin cell attachment site, was found to bind to the parasite surface and to inhibit its internalization (Ouaissi et al. 1986b). By using affinity chromatography, the TCT ligand for fibronectin was purified and identified as an 85 kDa protein that interacts with cells bearing fibronectin molecules, such as human monocytes and neutrophils as well as 3T3 fibroblasts (Ouaissi et al. 1986a).
Another TCT surface molecule with affinity for extracellular matrix components is penetrin, a 60 kDa protein that selectively binds to heparin, heparin sulfate and collagen, and promotes fibroblast adhesion and penetration (Ortega-Barria and Pereira 1991). An intriguing observation is that the recombinant penetrin, expressed in Escherichia coli and localized on its surface, induced the bacterial attachment to and penetration into Vero cells in a proteoglycan- and collagen-inhibitable manner (Ortega-Barria and Pereira 1991). Assays to probe the role of host cell heparin and heparan sulfate glycosaminoglycans in T. cruzi invasion showed that proteoglycan-deficient mutants of Chinese hamster ovary (CHO) cells are poor targets for TCT penetration (Herrera et al. 1994). Penetrin has not been further characterized and its structure remains undefined.
A member of the prolyl oligopeptidase (POP) family of serine proteases, with specificity for human collagen types I and IV, has been identified in cell-free extracts of trypomastigotes, amastigotes and epimastigotes (Santana et al. 1997, Grellier et al. 2001). The 80 kDa enzyme, denominated POP Tc80, also hydrolyses fibronectin and appears to be implicated in host cell invasion. Selective and irreversible inhibitors of POP Tc80 were found to block TCT entry into nonphagocytic mammalian cells (Grellier et al. 2001, Bastos et al. 2005).
In T. cruzi infection in vivo, the ability ofdiverse trypomastigote molecules in binding to laminin, fibronectin, collagen, heparin, heparan sulfate, heparin, in addition to the hydrolytic activity of some of them, may be essential for the parasite transit through the extracellular matrix towards target cells.
TS AND SIALIC ACID ACCEPTOR MOLECULES
The role of TS in mammalian cell invasion has as yet to be fully clarified. On the basis that polyclonal antibodies that block TS activity enhance invasion of host cells by TCT in vitro, Cavallesco and Pereira (1988) proposed that the enzyme negatively modulates T. cruzi infection. They also found that, compared to the minor subset of trypomastigotes recognized by anti-TS antibodies (TS+), the TS- population showed enhanced ability to enter host cells. By using anti-TS monoclonal antibodies and various cell types and parasite isolates, Prioli et al. (1990) confirmed the previous results, reinforcing the hypothesis that TS down regulates T. cruzi infection. Subsequently, however, contradictory data were reported by the same group. Pereira et al. (1996) prepared pure TS+ and TS- populations and tested them for host cell invasion. They found that TS+ trypomastigotes were highly invasive whereas TS- parasites were extremely inefficient in invading epithelial cells and fibroblasts. Furthermore, introduction of small amounts of TS- into suspensions of nonpenetrating TStrypomastigotes converted them to a highly invasive phenotype (Pereira et al. 1996).
Immunocytochemical studies localized TS on the surface of TCT and in association with the flagellar pockets, suggesting that the enzyme is also secreted (Souto-Padrón et al. 1990). The shed acute phase antigen (SAPA), present in T. cruzi-infected patients and identified as TS, is the secreted form of enzyme that generates strong immune response (Pollevick et al. 1991). Whether the secreted form of TS and the enzyme expressed on TCT surface act simultaneously during interaction with target cells, or the action of one of them predominates depending on the circumstances, is not clear. According to Ming et al. (1993), TS may function as a counter-receptor for parasite binding to a 2,3-sialyl receptors on host cells as a prelude to TCT invasion. Alternatively, after binding of TCT to target cells through another molecule, secreted TS may transfer sialic acid from the mammalian cell membrane to the parasite mucins. These transference reactions could disrupt the binding of sialoadhesins, allowing the parasites to detach and find a new binding site in order to proceed towards their internalization (Schenkman and Eichinger 1993).
In TCT, the sialic acid is incorporated by TS mainly into mucins migrating in SDS-PAGE as a broad band of 70-200 kDa (Schenkman et al. 1991a, b). Mucins and sialyl residues apparently are not primary requirements for TCT invasion (Schenkman et al. 1993b), although monoclonal antibodies to sialic acid-containing epitopes inhibit parasite adhesion to host cells (Schenkman et al. 1991b). On the other hand, the involvement of target cell sialic acid in T. cruzi internalization was reported by different groups, using sialic acid-deficient CHO (Lec2) cells. Trypomastigotes entered Lec2 cells to a much lower extent than parental CHO cells, but sialylation by TS restored parasite adhesion and invasion (Ciavaglia et al. 1993, Ming et al. 1993, Schenkman et al. 1993b).
CRUZIPAIN, THE MAJOR T. cruzi CYSTEINE PROTEINASE
Murta et al. (1990) identified T. cruzi antigen gp57/ 51 as a cysteine proteinase which is active across pH range 5-7.5. This enzyme, named cruzipain, is expressed in all developmental forms of different T. cruzi isolates (Murta et al. 1990, Paiva et al. 1998). By using peptidyl diazomethane derivatives, a class of irreversible inhibitors of cysteine proteinase, the involvement of cruzipain enzyme in host cell invasion and intracellular development was inferred (Meirelles et al. 1992). The role of cruzipain in vivo was also determined. Engel et al. (1998) cured experimental T. cruzi infection by treating mice with peptide-fluoromethyl ketones, inhibitors that inactivate cruzipain and arrest intracellular replication as well as intercellular transmission in vitro (Harth et al. 1993).
The participation of cruzipain in host cell invasion by TCT is associated with its ability to generate bradykinin, according to Scharfstein et al. (2000), who investigated the involvement of B type ofbradykinin receptor (B2R) using human umbilical vein endothelial cells (HUVECs) or CHO cells overexpressing B2R (CHO-B2R). They found that captopril, an inhibitor of bradykinin degradation by kininase II, potentiated TCT entry into HUVECs and CHO-B2R, but not into mock-transfected CHO cells, whereas the B2R antagonist HOE 140 or monoclonal antibody to bradykinin blocked these effects. Purified cruzipain enhanced parasite invasion and triggered Ca2+ mobilization in CHO-B2R in a manner inhibitable by HOE 140 or cruzipain inhibitor E-64, indicating that the enzyme plays a role in generating the kinin agonist from cell-bound kininogen. This kinin-mediated signal transduction route is not ubiquitous, its activation depending on the cell type and the parasite isolate used (Scharfstein et al. 2000).
It has also been shown that MGTA, an inhibitor of kininase I, selectively decreases TCT infectivity for B1R-expressing cells and that addition of B1R or B2R antagonists to host cells coexpressing these receptors inhibit parasite infectivity to a similar extent (Todorov et al. 2002). Because the combined application of both antagonists had no additive effect on both cardiomyocytes and HUVECs, the authors deduced that B1R and B2R operate interdependently to meet the intracellular Ca2+ concentration threshold required for efficient TCT internalization.
Processing of kininogens presumably takesplace within the secluded spaces formed by juxtaposition of parasite and the target cell, inasmuch as membrane-permeable but not soluble cruzipain inhibitors block parasite invasion of cells that naturally overexpress kinin receptors (Scharfstein et al. 2000, Todorov et al. 2002). Cruzipain appears to be modulated by both the host and T. cruzi components. On the premise that kininogen molecules may be displayed on cell surfaces by binding to glycosaminoglycans, Lima et al. (2002) examined whether the ability of cruzipain to release kinins from high molecular weight kininogen is modulated by heparin sulfate. In the presence of heparin sulfate, they found an enhancement of 6-fold in cruzipain activity towards synthetic substrates and of up to 35-fold by direct measurement of bradykinin. On the other hand, a tight-binding cysteine proteinase inhibitor, chagasin, was identified in T. cruzi (Monteiro et al. 2001). It is localized in the flagellar pocket and cytoplasmic vesicles of TCT, and its expression is inversely correlated with that of cruzipain.
T. cruzi OLIGOPEPTIDASE B
A soluble factor of unknown structure, secreted by TCT, is so far the sole component of this T. cruzi developmental form reported to directly trigger Ca2+ response in host cells. This soluble factor is generated by the action of a 120 kDa alkaline peptidase on precursors present only in infective trypomastigotes (Burleigh and Andrews 1995). The purified peptidase is devoid of Ca2+ signaling activity on its own and is also present in noninfective epimastigotes (Burleigh and Andrews 1995).
By cloning and sequencing of the corresponding cDNA, the TCT peptidase was found to be a cytosolic enzyme closely related to members of the prolyl oligopeptidase family of serine endopeptidases, and was denominated T. cruzi oligopeptidase B (Burleigh et al. 1997). The oligopeptidase B null mutant trypomastigotes are defective in mobilizing Ca2+ from thapsigargin-sensitive stores in mammalian cells, and in establishing infection in vitro and in vivo (Caler et al. 1998). Based on experimental evidences, it has been proposed that the Ca2+ agonist generated by oligopeptidase B is exported from the parasite, binds to a receptor on the surface of target cells, activating phospholipase C and generating IP, which binds to its receptor on the membrane of the endoplasmic reticulum and promotes Ca2+ release.
OTHER T. cruzi MOLECULES
Several other T. cruzi molecules have been implicated in host cell invasion. Surface antigens with metalloprotease activity, which are homologous to Leishmania gp63, were identified in MT and TCT and affinity-purified antibodies to these antigens inhibited host cell invasion by ~ 50% (Cuevas et al. 2003).
Moro et al. (1995) characterized a secreted T. cruzi protein with peptidyl-prolyl cis-trans isomerase activity, susceptible to inhibition by the immunosuppressant FK506 and related drugs, and showed that the addition of the recombinant protein to simian epithelial or HeLa cells enhances parasite invasion. The monomeric protein has a peptidyl-prolyl cis-trans isomerase core, encompassing the characteristic rotamase hydrophobic active site, and its mechanism of action may be the triggering of host cell signal, with or without the contribution of rotamase activity (Pereira et al. 2002).
A 67 kDa lectin-like glycoprotein, which binds to desialylated human erythrocyte membranes in a galactose-dependent way and recognizes receptors in mouse cardiac tissue and human cardiac aortic endothelium, has been described and the cell invasion inhibitory effect of anti-gp67 antibodies reported (Silber et al. 2002).
SIGNAL TRANSDUCTION IN HOST CELL DURING T. cruzi INVASION
CA2+ SIGNALING AND LYSOSOME RECRUITMENT IN TARGET CELLS
In 1994, Tardieux et al. (1994) reported that TCT, or their isolated membranes, but not the noninfective epimastigotes, induce repetitive cytosolic-free Ca2+ transients in normal rat kidney (NRK) fibroblasts, and that parasite entry is inhibited by depletion of host cell cytosolic-free Ca2+ or pretreatment with Cachannel blockers. In addition to NRK cells, soluble fraction of TCT induced Ca2+ response in a variety of cell types, such as hamster CHO and Dede, dog MDCK, monkey CV-1, human A7 (Burleigh and Andrews 1995), L6E9 myoblasts (Moreno et al. 1994) and isolated primary canine cardiac myocytes (Barr et al. 1996). MT were also found to trigger Ca2+ signaling in diverse cell types which included human epithelial HeLa cells and macrophages (Dorta et al. 1995, Wilkowsky et al. 1996). IP3, generated upon PLC activation, mediated intracellular Ca2+ mobilization triggered by TCT soluble factor (Rodriguez et al. 1995).
According to Andrews (1995), host cell Ca2+ response induces the recruitment of lysosomes to the site of T. cruzi penetration. At that site, lysosomal markers are immediately incorporated into parasitophorous vacuole without accumulation of polymerized actin around the recently internalized parasites, and invasion is facilitated by disruption of microfilaments (Tardieux et al. 1992). Lysosome redistribution and TCT invasion of NRK or L6E9 cells is inhibited upon treatment with microtubule-binding drugs nocodazole, colchicine, vinblastine and taxol, or after microinjection with antibodies to kinesin, indicating that lysosome transport is mediated by microtubule/kinesin (Rodriguez et al. 1996). Recently, Jaiswal et al. (2002) reported that lysosomes that fused were predominantly predocked at the plasma membrane, Ca2+ being primarily responsible for fusion and not recruitment of lysosomes to the cell surface. By fusing with the plasma membrane, lysosomes would contribute to formation of the parasitophorous vacuole (Andrews 1995).
Elevation in intracellular free Ca2+ concentration triggered lysosome fusion and exocytosis, as deduced from the appearance on the plasma membrane of the lysosomal glycoprotein lgp120, and the release of the lysosomal enzyme beta-hexosaminidase or the lysosomally processed form of cathepsin D (Rodriguez et al. 1997). Ca2+-dependent exocytosis of lysosomes is cAMP-regulated and is enhanced by isopreternol, a b-adrenergic agonist that activates adenylyl cyclase through heterotrimeric G protein Gs (Rodriguez et al. 1999). Sinaptotagmin VII, a ubiquitously expressed sinaptotagmin isoform that is localized on the membrane of lysosomes in different cell types and regulates exocytosis of these organelles, appears to mediate T. cruzi invasion. TCT entry was impaired in CHO cells loaded with antibodies that recognize the Ca2+-binding domain of sinaptotagmin VII and inhibit the Ca2+-triggered exocytosis of lysosomes (Caler et al. 2001).
Targeted lysosome exocytosis may not be the predominant mechanism by which TCT gain access to non-professional phagocytic cells. It has been found that only a minimal fraction of invading TCT associate with host cell lysosomes whereas the majority of parasites induce plasma membrane invagination and the TCT-containing vacuoles gradually acquire lysosomal markers (Woolsey et al. 2003). The newly forming T. cruzi compartments first interact with an early endosome compartment and subsequently with other late endosomes, before interaction with lysosomes (Wilkowsky et al. 2002).
HOST CELL ACTIN CYTOSKELETON
As a rapid and transient reorganization of host cell microfilaments is induced by TCT soluble factor and live trypomastigotes, probably as a direct consequence of increased intracellular Ca2+ concentration, it has been proposed that this disassembly of the cortical actin cytoskeleton plays a role in T. cruzi invasion (Rodriguez et al. 1995). In accord with this hypothesis is the observation that treatment of NRK cells with cytochalasin D, a drug that disrupts microfilaments, enhances TCT internalization (Tardieux et al. 1992, Caler et al. 2000). However, such an effect has not been detected by other groups. Cytochalasin D had little or no effect on TCT entry into MDCK or HeLa cells (Schenkman et al. 1991c) whereas marked inhibiton was detected in diverse cell types, including Vero, LLCMK 2, HFSF fibroblast, L-6 skeletal muscle myoblast and resident peritoneal macrophages (Rosestolato et al. 2002), in addition to heart muscle cells (Barbosa and Meirelles 1995). As regards MT invasion, it was significantly inhibited by treatment of HeLa cells with cytochalasin B or latrunculin B (Osuna et al. 1993), but unaffected by cytochalasin D (Schenkman and Mortara 1992). It is not clear why the results from different groups differ so widely, provided that apparently the experimental conditions and the drug concentration used are similar.
Recent data implicate the actin cytoskeleton in the intracellular retention of parasites. Woolsey and Burleigh (2004) demonstrated that cytochalasin D treatment of host cells inhibits early lysosome association with invading TCT by uncoupling the cell penetration step from lysosome recruitment and/or fusion, and prolonged disruption of actin microfilaments results in significant loss of internalized parasites from infected cells. That a significant fraction of the internalized parasite is not retained inside host cells for a productive infection was confirmed by Andrade and Andrews (2004), by blocking lysosome-mediated TCT invasion through phosphoinositide 3-kinase inhibition.
An interesting observation, that reinforces the role of the cortical actin cytoskeleton disassembly in T. cruzi invasion, has been made in human placenta syncytiotrophoblasts. Using immunohistochemical techniques, Sartori et al. (2003a) observed the presence of actin in the syncytiotrophoblasts throughout the brush border in placentae from non-chagasic women but, after culture with trypomastigotes, this labeling disappeared, indicating that the parasite induced disassembly of the cortical actin cytoskeleton.
PHOSPHOINOSITIDE (PI)-3 KINASES, PROTEIN KINASES AND PHOSPHATASES
Among the mechanisms of T. cruzi invasion are those dependent on lipid as well as protein kinases. Infection of macrophages with trypomastigotes stimulates the formation of the lipid products of PI 3-kinases and treatment with wortmannin, an inhibitor of PI 3-kinases, impaires parasite internalization (Todorov et al. 2000). Immunofluorescence microscopy, using antibodies against p85, the regulatory subunit of PI 3-kinase, localized the enzyme at the site of parasite interaction with macrophages, which was rich in F-actin (Vieira et al. 2002). In addition to phagocytic human macrophages and J774 murine cells, nonphagocytic Vero, L6E9 and 3T3 cells become less susceptible to T. cruzi infection upon treatment with wortmannin (Wilkowsky et al. 2001). According to Woolsey et al. (2003), host cell PI 3-kinases activated by TCT early in the cell invasion process regulate lysosome-dependent parasite entry. Treatment of T. cruzi with wortmannin also inhibited parasite internalization (Wilkowsky et al. 2001), indicating that both parasite and target cell PI 3-kinase activities are implicated in cell invasion.
The involvement of protein tyrosine kinase(PTK) in T. cruzi invasion of macrophages has been reported by Vieira et al. (1994). By treating either TCT or macrophages with genistein, a specific PTK inhibitor, these authors found a significant decrease in parasite endocytosis. Monoclonal anti-phosphotyrosine antibodies revealed an accumulation of tyrosine-phosphorylated residues at the site of parasite association with the macrophage surface, colocalizing with host cell F-actin-rich domains (Vieira et al. 2002). Activation of parasite PTK is required for MT and TCT entry into nonphagocytic cells (Favoreto et al. 1998) but, in contrast to what is seen in macrophages, host cell PTK activity is not involved, as inferred from the lack of inhibition of TCT or MT internalization upon treatment of RNK or HeLa cells with genistein (Rodriguez et al. 1995, Favoreto et al. 1998).
Other protein kinases also participate in T. cruzi entry into host cells. Wilkowsky et al. (2001) found that infection of transiently transfected 3T3 cells containing an inactive mutant protein kinase B are less susceptible to invasion, as compared to the active mutant-transfected cells. Activation of protein kinase C and enhancement of parasite uptake by macrophages have been detected upon incubation with recombinant gp83, a TCT surface ligand (Villalta et al. 1999).
Activation of the host cell PTK is not required for T. cruzi invasion of nonphagocytic cells. On the other hand, protein tyrosine phosphatases appear to be involved. Invasion of TCT induced tyrosine dephosphorylation of several proteins in L6E9 myoblasts, and the parasite internalization was greatly reduced in the presence of protein tyrosine phosphatase inhibitors and in the presence of excess phosphotyrosine, but not of phosphoserine or phosphothreonine (Zhong et al. 1998). In human HEp2 tumor cells, infection with T. cruzi resulted in the alterations of their placental alkaline phosphatase activity as well as in a different pattern of actin organization, compared to control cells, and the interference in the enzyme activity before infection decreased the invasion rate (Sartori et al. 2003b).
TARGET CELL SURFACE MOLECULES IMPLICATED IN SIGNAL TRANSDUCTION AND TCT INVASION
In addition to Ca2+ signaling through bradykinin receptor, the signaling route mediated by transforming growth factor-b (TGFb) receptor may be activated during TCT invasion of target cells. In a series of experiments, Ming et al. (1995) showed the requirement of TGFb pathway for T. cruzi invasion of epithelial cells. They found that TCT attached to TGFb receptor-deficient epithelial cell lines, but were unable to penetrate. Susceptibility to T. cruzi infection was restored by transfection with TGFb receptor genes, and treatment with TGFb greatly enhanced parasite internalization. As a TGFb-responsive reporter gene is induced in TGFb-sensitive cell lines by TCT, but not by noninvasive epimastigotes, Ming et al. (1995) postulated that T. cruzi may directly trigger activation of the TGFb-signaling pathway required for invasion. The putative TGFb-like factor from TCT has never been characterized.
In macrophages, the heterodimeric b1 integrins, which belong to a ubiquitous family of integral membrane proteins that link the extracellular matrix to the cortical cytoskeleton, may be involved in signal transduction and T. cruzi internalization. Fernandez et al. (1993) observed that, when added to human macrophages, monoclonal antibodies to b1 subunit of VLA integrin family specifically blocked T. cruzi uptake, without interfering with the uptake of Leishmania pinfanoi or Escherichia coli. As that inhibition correlated with the ability to block fibronectin binding to macrophages, it is uncertain whether the parasite interacts with VLA directly or through the binding to fibronectin.
Galectin 3, which increases K-Ras activation and triggers a Ras signal (Elad-Sfadia et al. 2004), is another host cell component that may participate in T. cruzi invasion. In experiments with human coronary artery smooth muscle cells, that express galectin-3 on the surface and also secret it, Kleshchenko et al. (2004) found that exogenously added galectin-3 increases trypomastigote binding. T. cruzi adhered poorly to cells with reduced expression of galectin-3, but the adhesion property was restored by exogenous galectin-3.
MECHANISMS OF TARGET CELL INVASION BY BLOOD TRYPOMASTIGOTES
From the data of in vitro studies with TCT, the possible mechanisms that the bloodstream trypomastigotes may use to invade host cells can be envisaged (Fig. 7). Before reaching the target cells, in many instances the parasites have to overcome the barrier of extracellular matrix, in the same manner as MT encounter the mucin coat in the stomach. Through the surface molecules of gp85/TS superfamily, the parasites bind to fibronectin/laminin (Fig. 7A) and pave the way for the action of enzymes, such as the serine protease POP Tc80 that hydrolyses collagen/fibronectin (Fig. 7B).
Upon encountering the target cells, trypomastigotes attach to them in a manner mediated by Tc-85, TS or mucin (Fig. 7C). This interaction induces the activation of T. cruzi oligopeptidase B that generates a Ca2+ signaling factor from a precursor molecule. Triggering of host cell Ca2+ mobilization by this secreted parasite factor promotes invasion. Alternatively, or simultaneously, cruzipain is secreted by attached trypomastigotes within the confines of parasite-target cell juxtaposition, and its action on the kininogen generates bradykinin, which also induces Ca2+ response by interacting with its receptor (Fig. 7D). Trypomastigotes of T. cruzi isolates that trigger both signaling pathways may be more invasive. By testing different T. cruzi isolates, Aparicio et al. (2004) have found a correlation between the level of secreted cruzipain and the capacity of trypomastigotes to invade host cells. The low invasive capacity of G isolate, with low levels of cruzipain, may result from the lack of activation of cruzipain-mediated signaling pathway. In addition to the referred signaling routes, parasite-target cell interactions mediated by TGFb receptors and b1 integrins may take place, further contributing to the process of parasite internalization.
CONCLUDING REMARKS
Activation of signal transduction pathways triggered in the parasite and the host cell, leading to intracellular Ca2+ mobilization, Ca2+-induced reorganization of the host cell actin cytoskeleton and lysosome recruitment, constitutes the general mechanism by which T. cruzi trypomastigotes invade mammalian cells. With a plethora of T. cruzi molecules that have been identified and characterized structural and functionally, plus the identification of target cell components involved, the whole process is beginning to be understood at the molecular level. The picture is complex. Not only MT and TCT engage different molecules to interact with host cells, but different T. cruzi isolates may use distinct sets of molecules, activating distinct signaling pathways. In addition, some molecular interactions may trigger inhibitory signals that down regulate trypomastigote invasion. Furthermore, as the in vivo infection is concerned, the interaction of parasites with host components before reaching the target cells has also to be considered. Great progress has been made towards understanding the mammalian cell invasion by T. cruzi, but a lot more work has to be done before we can draw a more complete and detailed picture of that process.
ACKNOWLEDGMENTS
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Desenvolvimento Científicoe Tecnológico (CNPq). I thank Dr. Sergio Schenkman, Dr. José Daniel Lopes and Dr. Mauro Cortez for reading the manuscript.
Manuscript received on February 25, 2005; accepted for publication on February 28, 2005; presented by LUCIA MENDONÇA PREVIATO
E-mail: nyoshida@ecb.epm.br
- ACOSTA-SERRANO A, SCHENKMAN S, YOSHIDA N, MEHLER A, RICHARDSON JM AND FERGUSON MAJ. 1995. The lipid structure of the GPI-anchored mucin-like sialic acid acceptors of Trypanosoma cruzi changes during parasite differentiation from epimastigotes to infective metacyclic trypomastigote forms. J Biol Chem 270: 27244-27253.
- ACOSTA-SERRANO A, ALMEIDA IC, FREITAS JUNIOR LH, YOSHIDA N AND SCHENKMAN S. 2001. The mucin-like glycoprotein super-family of Trypanosoma cruzi: structure and biological roles. Mol Biochem Parasitol 114: 143-150.
- ALMEIDA IC, FERGUSON MA, SCHENKMAN S AND TRAVASSOS LR. 1994. Lytic anti-a-galactosyl antibodies from patients with chronic Chagas' disease recognize novel O-linked oligosaccharides on mucin-like glycosyl-phosphatidylinositol glycoproteins of Trypanosoma cruzi Biochem J 304: 793-802.
- ALVES MJ, ABUIN G, KUWAJIMA VY AND COLLI W. 1986. Partial inhibition of trypomastigote entry into cultured mammalian cells by monoclonal antibodies against a surface glycoprotein of Trypanosoma cruzi Mol Biochem Parasitol 21: 75-82.
- ANDRADE LO AND ANDREWS NW. 2004. Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J Exp Med 200: 1135-1143.
- ANDREWS NW. 1995. Lysosome recruitment during host cell invasion by Trypanosoma cruzi Trends Cell Biol 5: 133-137.
- APARICIO IM, SCHARFSTEIN J AND LIMA AP. 2004. A new cruzipain pathway of human cell invasion by Trypanosoma cruzi requires trypomastigote membranes. Infect Immun 72: 5892-5902.
- ARAYA JE, CANO MI, YOSHIDA N AND FRANCO DA SILVEIRA J. 1994. Cloning and characterization of a gene for the stage-specific 82-kDa surface antigen of metacyclic trypomastigotes of Trypanosoma cruzi Mol Biochem Parasitol 65: 161-169.
- ATAYDE VD, NEIRA I, CORTEZ M, FERREIRA D, REYMULLER E AND YOSHIDA N. 2004. Molecular basis of non virulence of Trypanosoma cruzi clone CL-14. Int J Parasitol 34: 851-860.
- BARBOSA HS AND MEIRELLES MN. 1995. Evidence of participation of cytoskeleton of heart muscle cells during the invasion of Trypanosoma cruzi Cell Struct Funct 20: 275-284.
- BARR SC, HAN W, ANDREWS NW, LOPEZ JW, BALL BA, PANNABECKER TL AND GILMOUR-JR RF. 1996. A factor from Trypanosoma cruzi induces repetitive cytosolic free Ca2+ transients in isolated primary canine cardiac myocytes. Infect Immun 64: 1770-1777.
- BASTOS IM ET AL. 2005. Molecular, functional and structural properties of the prolyl oligopeptidase of Trypanosoma cruzi (POP Tc80) that is required for parasite entry into mammalian cells. Biochem J 388: 29-38.
- BRENER Z. 1969. The behavior of slender and stout forms of Trypanosoma cruzi in the blood-stream of normal and immune mice. Ann Trop Med Parasitol 63: 215-220.
- BRENER Z. 1973. Biology of Trypanosoma cruzi Ann Rev Microbiol 27: 347-382.
- BRIONES MRS, SOUTO RP, STOLF BS AND ZINGALEZ B. 1999. The evolution of two Trypanosoma cruzi subgroups inferred from rRNA genes can be correlated with the interchange of American mammalian faunas in the Cenozoic and has implications to pathogenicity and host specificity. Mol Biochem Parasitol 104: 219-232.
- BROWN D. 1993. The tyrosine kinase connection: how GPI-anchored proteins activate T cells. Curr Opinion Immunol 5: 349-354.
- BUJ-BELLO A, ADU J, PIÑON LGP, HORTON A, THOMPSON J, ROSENTHAL A, CHINCHETRU M, BUCHMAN VL AND DAVIES AM. 1997. Neurturin responsiveness requires a GPI-linked receptor and the Ret receptor tyrosine kinase. Nature 387: 721-724.
- BURLEIGH B AND ANDREWS NW. 1995. A 120-kDa alkaline peptidase from Trypanosoma cruzi is involved in the generation of a novel Ca2+-signaling factor for mammalian cells. J Biol Chem 270: 5172-5180.
- BURLEIGH B AND ANDREWS NW. 1998. Signaling and host cell invasion by Trypanosoma cruzi Curr Opinion Microbiol 1: 461-465.
- BURLEIGH B, CALER EV, WEBSTER P AND ANDREWS NW. 1997. A cytosolic serine endopeptidase from Trypanosoma cruzi is required for the generation of Ca2+ signaling in mammalian cells. J Cell Biol 136: 609-620.
- CALER EV, VAENA DE AVALOS S, HAYNES PA, ANDREWS NW AND BURLEIGH B. 1998. Oligopeptidase B-dependent signaling mediates host cell invasion by Trypanosoma cruzi EMBO J 17: 4975-4986.
- CALER EV, MORTY RE, BURLEIGH B AND ANDREWS NW. 2000.NW. 2000. Dual role of signaling pathways leading to Ca2+ and cyclic AMP elevation in host cell invasion by Trypanosoma cruzi Infect Immun 68: 6602-6610.
- CALER EV, CHAKRABARTI S, FOWLER KT, RAO S AND ANDREWS NW. 2001. The exocytosis-regulatory protein sinaptotagmin VII mediates cell invasion by Trypanosoma cruzi J Exp Med 193: 1097-1104.
- CARDOSO DE ALMEIDA ML AND HEISE N. 1993. Proteins anchored via glycosylphosphatidylinositol and solubilizing phospholipases in Trypanosoma cruzi Biol Res 26: 285-312.
- CAVALLESCO R AND PEREIRA MEA. 1988. Antibody to Trypanosoma cruzi neuraminidase enhances infection in vitro and identifies a subpopulation of trypomastigotes. J Immunol 140: 617-625.
- CIAVAGLIA MC, CARVALHO TU AND SOUZA W. 1993. Interaction of Trypanosoma cruzi with cells with altered glycosylation patterns. Biochem Biophys Res Commun 193: 718-721.
- COLLI W AND ALVES MJM. 1999. Relevant glycoconjugates on the surface of Trypanosoma cruzi Mem Inst Oswaldo Cruz 94 (Suppl. 1): 37-49.
- CORTEZ M, NEIRA I, FERREIRA D, LUQUETTI AO, RASSI A, ATAYDE VD AND YOSHIDA N. 2003. Infection by Trypanosoma cruzi metacyclic forms deficient in gp82 but expressing a related surface molecule gp30. Infect Immun 71: 6184-6191.
- COURA JR, JUNQUEIRA ACV, FERNANDES O, VALENTE SAS AND MILES MA. 2002. Emerging Chagas disease in Amazonian Brazil. Trends Parasitol 18: 171-176.
- CUEVAS IC, CAZZULO JJ AND SANCHEZ DO. 2003. gp63 homologues in Trypanosoma cruzi: surface antigens with metalloprotease activity and a possible role in host cell infection. Infect Immun 71: 5739-5749.
- DI NOIA JM, D'ORSO I, ASLUND L, SANCHEZ DO AND FRASCH AC. 1998. The Trypanosoma cruzi mucin family is transcribed from hundreds of genes having hypervariable regions. J Biol Chem 273: 10843-10850.
- DOCAMPO R, SCOTT DA, VERCESI AE AND MORENO SNJ. 1995. Intracellular Ca2+ storage in acidocalcisomes of Trypanosoma cruzi Biochem J 310: 1005-1012.
- DOCAMPO R, SCOTT DA, VERCESI AE AND MORENO SNJ. 1996. The role of Ca2+ in the process of cell invasion by intracellular parasites. Parasitol Today 12: 61-65.
- DORTA ML, FERREIRA AT, OSHIRO MEM AND YOSHIDA N. 1995. Ca2+ signal induced by Trypanosoma cruzi metacyclic trypomastigote surface molecules implicated in mammalian cell invasion. Mol Biochem Parasitol 73: 285-289.
- ELAD-SFADIA G, HAKLAI R, BALAN E AND KLOOG Y. 2004. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J Biol Chem 279: 34922-34930.
- ENGEL JC, DOYLE OS, HSICH I AND MCKERROW JH. 1998. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J Exp Med 188: 725-734.
- FAVORETO-JR S, DORTA ML AND YOSHIDA N. 1998. Trypanosoma cruzi 175 kDa protein tyrosine phosphorylation is associated with host cell invasion. Exp Parasitol 89: 188-194.
- FERNANDEZ MA, MUNOZ-FERNANDEZ MA AND FRESNO M. 1993. Involvement of beta 1 integrins in the binding and entry of Trypanosoma cruzi into human macrophages. Eur J Immunol 23: 552-557.
- GIORDANO R, CHAMMAS R, VEIGA SS, COLLI W AND ALVES MJM. 1994. An acidic component of the heterogeneous Tc-85 protein family from the surface of Trypanosoma cruzi is a laminin binding glycoprotein. Mol Biochem Parasitol 65: 85-94.
- GIORDANO R, FOUTS DL, TEWARI D, COLLI W, MANNING JE AND ALVES MJM. 1999. Cloning of a surface membrane glycoprotein specific for the infective form of Trypanosoma cruzi having adhesive properties to laminin. J Biol Chem 274: 3461-3468.
- GRELLIER P ET AL. 2001. Trypanosoma cruzi prolyl oligopeptidase Tc80 is involved in nonphagocytic mammalian cell invasion by trypomastigotes. J Biol Chem 276: 47078-47080.
- HARTH G, ANDREWS N, MILLS AA, ENGEL JC, SMITH R AND MCKERROW JH. 1993. Peptide-fluoromethyl ketones arrest intracellular replication and intercellular transmission of Trypanosoma cruzi Mol Biochem Parasitol 58: 17-24.
- HERRERA EM, MING M, ORTEGA-BARRIA E AND PEREIRA MEA. 1994. Mediation of Trypanosoma cruzi invasion by heparan sulfate receptors on host cells and penetrin counter-receptors on the trypanosomes. Mol Biochem Parasitol 65: 73-83.
- HOFT DF. 1996. Differential mucosal infectivity of different life stages of Trypanosoma cruzi Am J Trop Med Hyg 55: 360-364.
- HOFT DF, FARRAR PL, KRATZ-OWENS K AND SHAFFER D. 1996. Gastric invasion by Trypanosoma cruzi and induction of protective mucosal immune responses. Inf Immun 64: 3800-3810.
- JAISWAL JK, ANDREWS NW AND SIMON SM. 2002. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol 159: 625-635.
- KLESHCHENKO YY, MOODY TN, FURTAK VA, OCHIENG J, LIMA MF AND VILLALTA F. 2004. Human galectin-3 promotes Trypanosoma cruzi adhesion to human artery smooth muscle cells. Infect Immun 72: 6717-6721.
- LEHKER MW AND SWEENEY D. 1999. Trichomonad invasion of the mucous layer requires adhesins, mucinases, and mortality. Sex Transm Infect 75: 231-238.
- LIMA APCA, ALMEIDA PC, TERSARIOL ILS, SCHMITZ V, SCHMAIER AH, JULIANO L, HIRATA IY, MÜLLER-ESTERL W, CHAGAS JR AND SCHARFSTEIN J. 2002. Heparan sulfate modulates kinin release by Trypanosoma cruzi through the activity of cruzipain. J Biol Chem 277: 5875-5881.
- MAGDESIAN MH, GIORDANO R, JULIANO MA, JULIANO L, SCHUMACHER RI, COLLI W AND ALVES MJM. 2001. Infection by Trypanosoma cruzi: identification of a parasite ligand and its host-cell receptor. J Biol Chem 276: 19382-19389.
- MÁLAGA S AND YOSHIDA N. 2001. Targeted reduction in expression of Trypanosoma cruzi surface glycoprotein gp90 increases parasite infectivity. Infect Immun 69: 353-359.
- MANQUE PM, EICHINGER D, JULIANO MA, JULIANO L, ARAYA J AND YOSHIDA N. 2000. Characterization of the cell adhesion site of Trypanosoma cruzi metacyclic stage surface glycoprotein gp82. Infect Immun 68: 478-484.
- MANQUE PM, NEIRA I, ATAYDE VD, CORDERO E, FERREIRA AT, FRANCO DA SILVEIRA J, RAMIREZ M AND YOSHIDA N. 2003. Cell adhesion and Ca2+ signaling activity in stably transfected Trypanosoma cruzi epimastigotes expressing the metacyclic stage-specific surface molecule gp82. Infect Immun 71: 1561-1565.
- MEIRELLES MN, JULIANO L, CARMONA E, SILVA SG, COSTA EM, MURTA AC AND SCHARFSTEIN J. 1992. Inhibitors of the major cysteinyl proteinase (gp57/51) impair host cell invasion and arrest the intracellular development of Trypanosoma cruzi in vivo. Mol Biochem Parasitol 52: 175-184.
- MING M, CHUENKOVA M, ORTEGA-BARRIA E AND PEREIRA MEA. 1993. Mediation of Trypanosoma cruzi invasion by sialic acid on the host cell and trans-sialidase on the trypanosome. Mol Biochem Parasitol 59: 243-252.
- MING M, EWEN ME AND PEREIRA MEA. 1995. Trypanosome invasion of mammalian cells requires activation of the TGF b signaling pathway. Cell 82: 287-296.
- MONCADA D, KEELLER K AND CHADEE K. 2003. Entamoeba histolytica cysteine proteinases disrupt the polymeric structure of colonic mucin an alter its protective function. Infect Immun 71: 838-844.
- MONTEIRO ACS, ABRAHAMSON M, LIMA APCA, VANNIER-SANTOS MA AND SCHARFSTEIN J. 2001. Identification, characterization and localization of chagasin, a tight-binding cysteine protease inhibitor in Trypanosoma cruzi J Cell Science 114: 3933-3942.
- MORENO SNJ, SILVA J, VERCESI AE AND DOCAMPO R. 1994. Cytosolic-free calcium elevation in Trypanosoma cruzi is required for cell invasion. J Exp Med 180: 1535-1540.
- MORO A, RUIZ-CABELLO F, FERNANDEZ-CANO A, STOCK RP AND GONZALEZ A. 1995. Secretion by Trypanosoma cruzi of a peptidyl-prolyl cis-trans isomerase involved in cell infection. EMBO J 14: 2483-2490.
- MORTARA RA. 1991. Trypanosoma cruzi: amastigotes and trypomastigotes interact with different structures on the surface of HeLa cells. Exp Parasitol 73: 1-14.
- MORTARA RA, SILVA S, ARAGUTH MF, BLANCO SA AND YOSHIDA N. 1992. Polymorphism of the 35- and 50-kilodalton surface glycoconjugates of Trypanosoma cruzi metacyclic trypomastigotes. Infect Immun 60: 4673-4678.
- MURTA AC, PERSECHINI PM, PADRON TS, SOUZA W, GUIMARÃES JÁ AND SCHARFSTEIN J. 1990. Structural and functional identification of gp57/51 antigen of Trypanosoma cruzi as a cysteine proteinase. Mol Biochem Parasitol 43: 27-38.
- NEIRA I, FERREIRA AT AND YOSHIDA N. 2002. Activation of distinct signal transduction pathways in Trypanosoma cruzi isolates with differential capacity to invade host cells. Int J Parasitol 32: 405-414.
- NEIRA I, SILVA FA, CORTEZ M AND YOSHIDA N. 2003. Involvement of Trypanosoma cruzi metacyclic trypomastigote surface molecule gp82 in adhesion to gastric mucin and invasion of epithelial cells. Infect Immun 71: 557-561.
- ORTEGA-BARRIA E AND PEREIRA MEA. 1991. A novel Trypanosoma cruzi heparin binding protein promotes fibroblast adhesion and penetration on engineered bacteria and trypanosomes into mammalian cells. Cell 67: 411-421.
- OSUNA A, RODRIGUEZ-CABEZAS N, BOY M, CASTANYS S AND GAMARRO F. 1993. The invasion mechanism of the metacyclic forms of Trypanosoma cruzi in nonphagocytic host cells. Biol Res 26: 19-26.
- OUAISSI MA, CORNETTE J AND CAPRON A. 1986a. Identification and isolation of Trypanosoma cruzi trypomastigote cell surface protein with properties expected of a fibronectin receptor. Mol Biochem Parasitol 19: 201-211.
- OUAISSI MA, CORNETTE J, AFCHAIN D, CAPRON A, GRAS-MASSE H AND TARTAR A. 1986b. Trypanosoma cruzi infection inhibited by peptides modeles from a fibronectin cell attachment domain. Science 234: 603-607.
- PAIVA CN, SOUTO-PADRÓN T, COSTA DA AND GATTASS CR. 1998. High expression of a functional cruzipain by a non-infective and non-pathogenic Trypanosoma cruzi clone. Parasitol 117: 483-490.
- PEREIRA ME. 1983. A developmentally regulated neuraminidase in Trypanosoma cruzi Science 219: 1444-1446.
- PEREIRA ME, ZHANG K, GONG Y, HERRERA EM AND MING M. 1996. Invasive phenotype of Trypanosoma cruzi restricted to a population expressing trans-sialidase. Infect Immun 64: 3884-3892.
- PEREIRA PJB, VEGA MC, GONZÁLEZ-REY E, FERNADEZ-CARAZO R, MACEDO-RIBEIRO S, GOMIS-RÜTH EX, GONZALEZ A AND COLL M. 2002. Trypanosoma cruzi macrophage infectivity potentiator has a rotamase core and a highly exposed a-helix. EMBO reports 3: 88-94.
- POLLEVICK GD, AFFRANCHINO JL, FRASCH AC AND SANCHEZ DO. 1991. The complete sequence of a shed acute-phase antigen of Trypanosoma cruzi Mol Biochem Parasitol 47: 247-250.
- PREVIATO JO, ANDRADE AF, PESSOLANI MC AND MENDONÇA-PREVIATO L. 1985. Incorporation of sialic acid into Trypanosoma cruzi macromolecules. A proposal for a new metabolic route. Mol Biochem Parasitol 16: 85-96.
- PREVIATO JO, JONES C, GONÇALVES LP, WAIT R, TRAVASSOS LR AND MENDONÇA-PREVIATO L. 1994. O-glycosidically linke N-acetylglucosamine-bound oligosaccharides from glycoproteins of Trypanosoma cruzi Biochem J 301: 151-159.
- PRIOLI RP, MEJIA JS AND PEREIRA MEA. 1990. Monoclonal antibodies against Trypanosoma cruzi neuraminidase reveal enzyme polymorphism, recognize a subset of trypomastigote, and enhance infection in vitro. J Immunol 144: 4384-4391.
- RAMIREZ MI, RUIZ RC, ARAYA JE, FRANCO DA SILVEIRA J AND YOSHIDA N. 1993. Involvement of the stage-specific 82-kilodalton adhesion molecule of Trypanosoma cruzi metacyclic trypomastigotes in host cell invasion. Infect Immun 61: 3636-3641.
- RODRIGUEZ A, RIOULT MG, ORA A AND ANDREWS N. 1995. A trypanosome-soluble factor induces IP3 formation, intracellular Ca2+ mobilization and microfilament rearrangement in host cells. J Cell Biol 129: 1263-1273.
- RODRIGUEZ A, SAMOFF E, RIOULT MG, CHUNG A AND ANDREWS N. 1996. Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesin-mediated transport. J Cell Biol 134: 349-362.
- RODRIGUEZ A, WEBSTER P, ORTEGO J AND ANDREWS NW. 1997. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol 137: 93-104.
- RODRIGUEZ A, MARTINEZ I, CHUNG A, BERLOT CH AND ANDREWS N. 1999. cAMP regulates Ca2+-dependent exocytosis of lysosomes and lysosome-mediated cell invasion by trypanosomes. J Biol Chem 274: 16754-16759.
- ROSESTOLATO CTF, DUTRA JMF, SOUZA W AND CARVALHO TMU. 2002. Participation of host cell actin filaments during interaction of trypomastigote forms of Trypanosoma cruzi with host cells. Cell Struct Funct 27: 91-98.
- RUIZ RC. 1998. Trypanosoma cruzi: associaçăo entre infectividade e expressăo de glicoproteínas com atividade sinalizadora de cálcio. Ph.D. thesis. Universidade Federal de Săo Paulo, SP, Brasil.
- RUIZ RC, RIGONI VL, GONZALEZ J AND YOSHIDA N. 1993. The 35/50 kDa surface antigen of Trypanosoma cruzi metacyclic trypomastigotes, an adhesion molecule involved in host cell invasion. Parasite Immunol 15: 121-123.
- RUIZ RC, FAVORETO S, DORTA ML, OSHIRO MEM, FERREIRA AT, MANQUE PM AND YOSHIDA N. 1998. Infectivity of Trypanosoma cruzi strains is associated with differential expression of surface glycoproteins with differential Ca2+ signaling activity. Biochem J 330: 505-511.
- SALTO ML, GALLO-RODRIGUEZ C, LIMA C AND LEDERKREMER RM. 2000. Separation of galfbeta1Ž XGlcNAc and galpbeta1Ž XGlcNAc (X = 3, 4, and 6) as the alditols by high-pH aninon-exchange chromatography and thin-layer chromatography: characterization of mucins from Trypanosoma cruzi Anal Biochem 279: 79-84.
- SANTANA JM, GRELLIER P, SCHREVEL J AND TEIXEIRA AR. 1997. A Trypanosoma cruzi-secreted 80 kDa proteinase with specificity for human collagen types I and IV. Biochem J 325: 129-137.
- SARTORI MJ, PONS P, MEZZANO L, LIN S AND FABRO SP. 2003a. Trypanosoma cruzi infection induces microfilament depletion in human placenta syncytiotrophoblasts. Placenta 24: 767-771.
- SARTORI MJ, MEZZANO L, LIN S, MUNOZ S AND FABRO SP. 2003b. Role of placental alkaline phosphatase in the internalization of trypomastigotes of Trypanosoma cruzi into HEp2 cells. Trop Med Int Health 8: 832-839.
- SCHARFSTEIN J, SCHMITZ V, MORANDI V, CAPELLA MMA, LIMA APCA, MORROT A, JULIANO L AND MÜLLER-ESTERL W. 2000. Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B2 receptors. J Exp Med 192: 1289-1299.
- SCHENKMAN RPF, VANDEKERCKHOVE F AND SCHENKMAN S. 1993b. Mammalian cell sialic acid enhances invasion by Trypanosoma cruzi Infect Immun 61: 898-902.
- SCHENKMAN S AND EICHINGER D. 1993. Trypanosoma cruzi trans-sialidase and cell invasion. Parasitol Today 9: 218-222.
- SCHENKMAN S AND MORTARA RA. 1992. HeLa cells extend and internalize pseudopodia during active invasion by Trypanosoma cruzi trypomastigotes. J Cell Sci 101: 895-905.
- SCHENKMAN S, ANDREWS NW, NUSSENZWEIG V AND ROBBINS ES. 1988a. Trypanosoma cruzi invade a mammalian epithelial cell in a polarized manner. Cell 55: 157-165.
- SCHENKMAN S, YOSHIDA N AND CARDOSO DE ALMEIDA ML. 1988b. Glycophosphatidylinositol - anchored proteins in metacyclic trypomastigotes of Trypanosoma cruzi Mol Biochem Parasitol 29: 141-152.
- SCHENKMAN S, DIAS C AND NUSSENZWEIG V. 1991a. Attachment of Trypanosoma cruzi trypomastigotes to receptors at restricted cell surface domains. Exp Parasitol 72: 76-86.
- SCHENKMAN S, JIANG M, HART GW AND NUSSENZWEIG V. 1991b. A novel cell surface trans-sialidase of Trypanosoma cruzi generates a stage-specific epitope required for invasion of mammalian cells. Cell 65: 1117-1125.
- SCHENKMAN S, ROBINS E AND NUSSENZWEIG V. 1991c. Attachment of Trypanosoma cruzi mammalian cells requires parasite energy, and invasion can be independent of the target cell cytoskeleton. Infect Immun 59: 645-654.
- SCHENKMAN S, FERGUSON MAJ, HEISE N, CARDOSO DE ALMEIDA ML, MORTARA RA AND YOSHIDA N. 1993a. Mucin-like glycoproteins linked to the membrane by glycosylphosphatidylinositol anchor are the major acceptors of sialic acid in a reaction catalysed by trans-sialidase in metacyclic forms of Trypanosoma cruzi Mol Biochem Parasitol 59: 293-304.
- SHENOY-SCARIA AM, KWONG J, FUJITA T, OLSZOWY MW, SHAW AS AND LUBLIN DM. 1992. Signal transduction through decay-acceleratting factor: interaction of glycosylphosphatidylinositol anchor and protein tyrosine kinases p56lck and p59fyn. J Immunol 149: 3535-3541.
- SILBER AM, MARCIPAR IS, ROODVELDT C, CABEZA MECKERT P, LAGUENS R AND MARCIPAR AJ. 2002. Trypanosoma cruzi: identification of a galactose-binding protein that binds to cell surface of human erythrocytes and is involved in cell invasion by the parasite. Exp Parasitol 100: 217-225.
- SOUTO-PADRÓN T, HARTH G AND SOUZA W. 1990. Immunocytochemical localization of neuraminidase in Trypanosoma cruzi Infect Immun 58: 586-592.
- STEFANOVA I, HOREJSI V, ANSOTEGUI IJ, KNAPP W AND STOCKINGER H. 1991. . GPI-anchored cell surface molecules complexed to protein tyrosine kinase. Science 254: 1016-1019.
- SUDHA PS, DEVARAJ H AND DEVARAJ N. 2001. Adherence of Shigella dysenteriae 1 to human colonic mucin. Curr Microbiol 42: 381-387.
- TARDIEUX I, WEBSTER P, RAVESLOOT J, BORON W, LUNN JA, HEUSER JE AND ANDREWS NW. 1992. Lysosome recruitment and fusion are early events required for Trypanosoma invasion of mammalian cells. Cell 71: 1117-1130.
- TARDIEUX I, NATHANSON MH AND ANDREWS NW. 1994. Role in host cell invasion of Trypanosoma cruzi- induced cytosolic free Ca2+ transients. J Exp Med 179: 1017-1022.
- THOMAS PM AND SAMELSON LE. 1992. The glycosylphosphatidylinositol anchored Thy-1 molecule interacts with the p60fyn protein tyrosine kinase in T cells. J Biol Chem 276: 12317-12322.
- TODOROV AG, EINICKER-LAMAS M, CASTRO SL, OLIVEIRA MM AND GUILHERME A. 2000. Activation of host cell phosphatidylinositol 3-kinases by Trypanosoma cruzi infection. J Biol Chem 275: 32182-32186.
- TODOROV AG ET AL. 2002. Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor subtypes (B1/B2). FASEB J Published online November 1.
- TYLER K AND ENGMAN DM. 2001. The life cycleof Trypanosoma cruzi revisited. Int J Parasitol 31: 472-481.
- VEILLETTE A, LATOUR S AND DAVIDSON D. 2002. Negative regulation of immunoreceptor signaling. Annu Rev Immunol 20: 669-707.
- VIEIRA M, DUTRA JM, CARVALHO TM, CUNHA-ESILVA NL, SOUTO-PADRÓN T AND SOUZA W. Cellular signaling during the macrophage invasion by Trypanosoma cruzi Histochem Cell Biol 118: 491-500.
- VIEIRA MCF, CARVALHO TU AND SOUZA W. 1994. Effect of protein kinase inhibition on the invasion process of macrophages by Trypanosoma cruzi Biochem Biophys Res Commun 203: 967-971.
- VILLALTA F, ZHANG Y, BIBB KE, PRATAP S, BURNS JR JM AND LIMA MF. 1999. Signal transduction in human macrophages by gp83 ligand of Trypanosoma cruzi: trypomastigote gp83 ligand up-regulated trypanosome entry through protein kinase C activation. Mol Cell Biol Res Commun 2: 64-70.
- VIVIER E, NUNÈS JA AND VÉLY F. 2004. Natural killer cell signaling pathways. Science 306: 1517-1519.
- WILKOWSKY SE, WAINSZELBAUM MJ AND ISOLA ELD. 1996. Trypanosoma cruzi: participation of intracellular Ca2+ during metacyclic trypomastigote-macrophage interaction. Biochem Biophys Res Commun 222: 386-389.
- WILKOWSKY SE, BARBIERI MA, STAHL P AND ISOLA ELD. 2001. Trypanosoma cruzi phosphatidylinositol 3-kinase and protein kinase B activation is associated with parasite invasion. Exp Cell Res 264: 211-218.
- WILKOWSKY SE, BARBIERI MA, STAHL P AND ISOLA ELD. 2002. Regulation of Trypanosoma cruzi invasion of nonphagocytic cells by the endocytically active GTPases dynamin, Rab5, and Rab7. Biochem Biophys Res Commun 291: 516-521.
- WOOLSEY AM AND BURLEIGH B. 2004. Host cell actin polymerization is required for cellular retention of Trypanosoma cruzi and early association with endosomal/lysosomal compartments. Cell Microbiol 6: 829-838.
- WOOLSEY AM, SUNWOO L, PETERSEN CA, BRACHMANN SM, CANTLEY LC AND BURLEIGH BA. 2003. Novel PI 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J Cell Sci 116: 3611-3622.
- YOSHIDA N. 2003. Trypanosoma cruzi cell invasion mechanisms. In: World Class Parasites: vol 7 American Trypanosomiasis. Tyler KM and Miles MA (Eds), Kluwer Academic Publishers, p. 69-79.
- YOSHIDA N, MORTARA RA, ARAGUTH MF, GONZALEZ JC AND RUSSO M. 1989. Metacyclic neutralizing effect of monoclonal antibody 10D8 directed to the 35- and 50-kilodalton surface glycoconjugates of Trypanosoma cruzi Infect Immun 57: 1663-1667.
- YOSHIDA N, BLANCO SA, ARAGUTH MF, RUSSO M AND GONZALEZ J. 1990. The stage-specific 90-kilodalton surface antigen of metacyclic trypomastigotes of Trypanosoma cruzi Mol Biochem Parasitol 39: 39-46.
- YOSHIDA N, DORTA ML, FERREIRA AT, OSHIRO MEM, MORTARA RA, ACOSTA-SERRANO A AND FAVORETO-JR S. 1997. Removal of sialic acid from mucin-like surface molecules of Trypanosoma cruzi metacyclic trypomastigotes enhances parasite-host cell interaction. Mol Biochem Parasitol 84: 57-67.
- YOSHIDA N, FAVORETO-JR S, FERREIRA AT AND MANQUE PM. 2000. Signal transduction induced in Trypanosoma cruzi metacyclic trypomastigotes during the invasion of mammalian cells. Braz J Med Biol Res 33: 269-278.
- ZHONG L, LU HG, MORENO SN AND DOCAMPO R. 1998. Tyrosine phosphate hydrolysis of host proteins by Trypanosoma cruzi is linked to cell invasion. FEMS Microbiol Lett 161: 15-20.
Publication Dates
-
Publication in this collection
08 Mar 2006 -
Date of issue
Mar 2006
History
-
Received
25 Feb 2005 -
Accepted
28 Feb 2005