Abstracts
Heart failure (HF) is a common endpoint for many forms of cardiovascular disease and a significant cause of morbidity and mortality. The development of end-stage HF often involves an initial insult to the myocardium that reduces cardiac output and leads to a compensatory increase in sympathetic nervous system activity. Acutely, the sympathetic hyperactivity through the activation of beta-adrenergic receptors increases heart rate and cardiac contractility, which compensate for decreased cardiac output. However, chronic exposure of the heart to elevated levels of catecholamines released from sympathetic nerve terminals and the adrenal gland may lead to further pathologic changes in the heart, resulting in continued elevation of sympathetic tone and a progressive deterioration in cardiac function. On a molecular level, altered beta-adrenergic receptor signaling plays a pivotal role in the genesis and progression of HF. beta-adrenergic receptor number and function are decreased, and downstream mechanisms are altered. In this review we will present an overview of the normal beta-adrenergic receptor pathway in the heart and the consequences of sustained adrenergic activation in HF. The myopathic potential of individual components of the adrenergic signaling will be discussed through the results of research performed in genetic modified animals. Finally, we will discuss the potential clinical impact of beta-adrenergic receptor gene polymorphisms for better understanding the progression of HF.
heart failure; sympathetic nervous system; adrenergic receptors
A insuficiência cardíaca (IC) é a via final comum da maioria das doenças cardiovasculares e uma das maiores causas de morbi-mortalidade. O desenvolvimento do estágio final da IC freqüentemente envolve um insulto inicial do miocárdio, reduzindo o débito cardíaco e levando ao aumento compensatório da atividade do sistema nervoso simpático (SNS). Existem evidências de que apesar da exposição aguda ser benéfica, exposições crônicas a elevadas concentrações de catecolaminas, liberadas pelo terminal nervoso simpático e pela glândula adrenal, são tóxicas ao tecido cardíaco e levam a deterioração da função cardíaca. Em nível molecular observa-se que a hiperatividade do SNS está associada a alterações na sinalização intracelular mediada pelos receptores beta-adrenérgicos. Sabe-se que tanto a densidade como a função dos receptores beta-adrenérgicos estão diminuídas na IC, assim como outros mecanismos intracelulares subjacentes à estimulação da via receptores beta-adrenérgicos. Nesta revisão, apresentaremos uma breve descrição da via de sinalização dos receptores beta-adrenérgicos no coração normal e as conseqüências da hiperatividade do SNS na IC. Daremos ênfase ao potencial miopático de diversos componentes da cascata de sinalização dos receptores beta-adrenérgicos discutindo estudos realizados com animais geneticamente modificados. Finalmente, discorreremos sobre o impacto clínico do conhecimento dos polimorfismos para o gene do receptor beta-adrenérgico para um melhor entendimento da progressão da IC.
insuficiência cardíaca; sistema nervoso simpático; receptores adrenérgicos
BIOMEDICAL AND MEDICAL SCIENCES
Neurohumoral activation in heart failure: the role of adrenergic receptors
Patricia C. Brum; Natale P.L. Rolim; Aline V.N. Bacurau; Alessandra Medeiros
Escola de Educação Física e Esporte da Universidade de São Paulo, Departamento de Biodinâmica do Movimento Humano, Laboratório de Fisiologia Cel. e Mol. do Exercício, Av. Professor Mello Moraes, 65, Butantã, 05508-900 São Paulo, SP, Brasil
ABSTRACT
Heart failure (HF) is a common endpoint for many forms of cardiovascular disease and a significant cause of morbidity and mortality. The development of end-stage HF often involves an initial insult to the myocardium that reduces cardiac output and leads to a compensatory increase in sympathetic nervous system activity. Acutely, the sympathetic hyperactivity through the activation of beta-adrenergic receptors increases heart rate and cardiac contractility, which compensate for decreased cardiac output. However, chronic exposure of the heart to elevated levels of catecholamines released from sympathetic nerve terminals and the adrenal gland may lead to further pathologic changes in the heart, resulting in continued elevation of sympathetic tone and a progressive deterioration in cardiac function. On a molecular level, altered beta-adrenergic receptor signaling plays a pivotal role in the genesis and progression of HF. beta-adrenergic receptor number and function are decreased, and downstream mechanisms are altered. In this review we will present an overview of the normal beta-adrenergic receptor pathway in the heart and the consequences of sustained adrenergic activation in HF. The myopathic potential of individual components of the adrenergic signaling will be discussed through the results of research performed in genetic modified animals. Finally, we will discuss the potential clinical impact of beta-adrenergic receptor gene polymorphisms for better understanding the progression of HF.
Key words: heart failure, sympathetic nervous system, adrenergic receptors.
RESUMO
A insuficiência cardíaca (IC) é a via final comum da maioria das doenças cardiovasculares e uma das maiores causas de morbi-mortalidade. O desenvolvimento do estágio final da IC freqüentemente envolve um insulto inicial do miocárdio, reduzindo o débito cardíaco e levando ao aumento compensatório da atividade do sistema nervoso simpático (SNS). Existem evidências de que apesar da exposição aguda ser benéfica, exposições crônicas a elevadas concentrações de catecolaminas, liberadas pelo terminal nervoso simpático e pela glândula adrenal, são tóxicas ao tecido cardíaco e levam a deterioração da função cardíaca. Em nível molecular observa-se que a hiperatividade do SNS está associada a alterações na sinalização intracelular mediada pelos receptores beta-adrenérgicos. Sabe-se que tanto a densidade como a função dos receptores beta-adrenérgicos estão diminuídas na IC, assim como outros mecanismos intracelulares subjacentes à estimulação da via receptores beta-adrenérgicos. Nesta revisão, apresentaremos uma breve descrição da via de sinalização dos receptores beta-adrenérgicos no coração normal e as conseqüências da hiperatividade do SNS na IC. Daremos ênfase ao potencial miopático de diversos componentes da cascata de sinalização dos receptores beta-adrenérgicos discutindo estudos realizados com animais geneticamente modificados. Finalmente, discorreremos sobre o impacto clínico do conhecimento dos polimorfismos para o gene do receptor beta-adrenérgico para um melhor entendimento da progressão da IC.
Palavras-chave: insuficiência cardíaca, sistema nervoso simpático, receptores adrenérgicos.
INTRODUCTION
Heart failure, a syndrome of poor prognosis, is developed as a consequence of cardiac disease, and recognized clinically by a constellation of signs and symptoms produced by complex circulatory and neurohormonal responses (Packer 1992, Katz 2002, 2003). Concisely, heart failure can be described as an inability of the heart to maintain adequate blood circulation in peripheral tissues and the lungs. It is a common endpoint for many forms of cardiovascular diseases, and a major clinical and public health problem. More than 20 million people worldwide are estimated to have heart failure (Cleland et al. 2001, Tendera 2004), and its increasing prevalence is associated with the rise in the median life span of the population (Bonneux et al. 1994). In Brazil, heart failure leads to approximately 25,000 deaths per year (Albanesi Filho 2003), and is associated with considerable morbidity, since patients with heart failure undergo frequent hospital readmissions, which are higher in those of lower socioeconomic strata (Albanesi Filho 2003).
Throughout most of the 20th century, heart failure has been regarded as a hemodynamic disorder. According to this view, the impaired pump performance led to increased pulmonary and venous pressures, decreased cardiac output associated with progression of underlying disease, and ultimately to the death of the patient (Katz 2001). By attempting to improve the hemodynamic derangements, the goals of heart failure therapy were to lower the increased venous pressure with diuretics, to unload the failing heart by using peripheral vasodilators and to increase cardiac contractility by administering inotropic agents.
Unfortunately, clinical trials performed with diuretics, peripheral vasodilators and inotropic agents obtained disappointing results, since in spite of causing a short-term hemodynamic improvement, long-term use failed to prolong survival in heart failure patients (Packer 1992, Katz 2002,2003).
The importance of non-hemodynamic mechanisms in inducing cardiac failure emerged in the early 1980s, when the neurohumoral response to low cardiac output was found to have a major adverse effect on long-term survival (Francis et al. 1984). This finding highlighted the importance of prognosis in heart failure, which had been overlooked until then. More interestingly, the understanding of heart failure passed through a paradigm shift, and currently the syndrome is viewed primarily as a neurohumoral disorder.
The sympathetic nervous system is a critical component of neurohumoral response observed in heart failure. In the early stages of the syndrome, an intrinsic decrease in myocardial function leads to an increase in sympathetic activity. Acutely, through the activation of cardiac beta-adrenergicreceptors (b-adrenergic receptors), heart rate and cardiac contractility are increased and compensate for decreased cardiac output, which is returned to a more idealized level. However, as heart failure worsens, sympathetic activity is further increased in an attempt to compensate for a progressive loss of cardiac function. Unfortunately, chronic exposure of the heart to elevated levels of catecholamines released from sympathetic nerve terminals and the adrenal gland may lead to further pathologic changes in the heart, resulting in continued elevation of sympathetic tone and a progressive deterioration in cardiac structure and function (Post et al. 1999, Port and Bristow 2001, Brum et al. 2002, Lohse et al. 2003a).
It has long been suspected that increased activity of the sympathetic nerves is present in heart failure (Starling 1897, Chidsey and Braunwald 1966). In fact, sympathetic nerve activity bears a direct relationship to both severity and prognosis of the heart failure (Cohn et al. 1984, Negrão et al. 2001) (Fig. 1). Likewise, the cardiotoxic effects of catecholamines have been recognized since the beginning of the 20th century (Josue 1907). However, the molecular mechanisms underlying these cardiotoxic effects are just beginning to be understood. Cardiac deleterious effect of sympathetic overactivity seems to be related mainly to the activation of b1AR pathway (Communal et al. 1999, Lohse et al. 2003a, Xiao et al. 2004). Accordingly, there has been considerable interest in the possibility that therapy directed at bAR and adrenergic signaling pathway has the potential to treat the pathophisiologic mechanisms involved in the progression of the heart failure.
In the present review, the long-term consequences of sustained adrenergic activation in the context of heart failure will be reviewed. We will present an overview of the normal bAR pathway in the heart and then discuss the alterations in b-adrenergic receptor signaling in heart failure. Data from genetic modified animals that demonstrate the myopathic potential of individual components of the adrenergic signaling will also be emphasized. Finally, we will discuss the potential clinical impact of b-adrenergic receptor polymorphisms for better understanding the progression of cardiovascular diseases, such as heart failure.
THE CARDIAC b-ADRENERGIC PATHWAY
Adrenergic receptors form the interface between the sympathetic nervous system and cardiovascular system. As mentioned above, cardiovascular function is tightly regulated by the sympathetic nervous system. In response to a variety of stimuli, including exercise and blood loss, an increase in cardiac output is met by a commensurate increment in sympathetic nervous system that through b-adrenergic receptors increase the heart rate (chronotropism), the force cardiac contraction (inotropism), the rate of cardiac relaxation (lusitropism), and automaticity.
Ahlquist first classified the adrenergic receptors in 1948 as a (for excitatory) and b (for inhibitory) based on their control of blood vessel contractility (Ahlquist 1948). His initial observations suggested that a-adrenergic activation, for example, generally lead to smooth-muscle contraction, as evidenced by vasoconstriction or uterine contraction, whereas b-adrenergic stimulation produced the opposite effect of relaxing smooth muscle. Ahlquist's classification was expanded by Lands and collaborators (1967), who recognized that both a- and b-adrenergic receptors could be further categorized into 2 distinct subtypes based on their relative potencies for the ligands available at that time. More recently, molecular cloning has led to the identification of 9 adrenergic receptor subtypes, namely a1A, a1B, a1D, a2A, a2B, a2C, b1, b2, and b3 (Bylund et al. 1994).
In the heart, b1- and b2-adrenergic receptor subtypes are expressed at a ratio of 70:30, and both increase cardiac frequency and contractility (Wallukat 2002) (Fig. 2). In addition, b3-adrenergic receptors have been described to mediate negative inotropic effects in cardiac myocytes (Devic et al. 2001).
All three b-adrenergic receptors subtypes are members of the large family of seven membrane-spanning, GTP-binding protein (G-protein)-coupled receptors. Their activation by an agonist catalyze the exchange of GTP for GDP on the Ga-subunit of G proteins, resulting in the dissociation of the heterotrimer into active Ga- and Gbg-subunits, which are competent to signal independently (Lefkowitz et al. 2002, Wallukat 2002). The heterogeneity of G-protein a subunits of which there are ~ 20 subtypes (Gs, Gi, Gq, Go, etc) is a central basis of G-protein coupled receptor signaling (Morris and Malbon 1999). Additional levels of signaling specificity are conferred by combinatorial permutations of various bg-heterodimeric subunits (5b and 11g subunits) with the a subunits. Even though all three subtypes of b-adrenergic receptors are expressed in cardiac myocyte, they possess distinct intracellular signaling pathways and functional properties (Lohse et al. 2003b, Xiang and Kobilka 2003b).The positive chronotropic effects of b1 receptor activation are clearly mediated via the stimulatory G protein (Gs) in myocytes. Even though it has been recently proposed that b1 adrenergic receptor can switch from Gs to Gi-coupling in a PKA-dependent manner (Martin et al. 2004), the intracellular effect of this switching is not known. In contrast, dual coupling of b2 receptors to Gs and inhibitory G protein (Gi) is evident in cardiac myocytes from newborn mice (Kuschel et al. 1999, Xiao et al. 2003). The b2 receptor coupling to Gi is reported to be involved in its anti-apoptotic properties in cardiac myocytes (Communal et al. 1999, Zhu et al. 2001). In neonatal cardiac myocytes from b1 and b2 receptor double knockout mice, dual coupling of the b3 subtype to both Gs and Gi, with the Gi component dominating, has been described (Devic et al. 2001). These specific signaling properties of b receptor subtypes have been linked to subtype-selective association with intracellular scaffolding and signaling proteins, and distinct subcellular localization (Steinberg 1999, Hall and Lefkowitz 2002, Xiang et al. 2002, Xiang and Kobilka 2003a).
Figure 3A is a schematic representation of b-adrenergic receptor signaling in the heart. b-adrenergic receptors stimulate the effect or enzyme, adenylyl cyclase (AC) of which there are at least 9 isoforms, being AC's V and VI the main isoforms expressed in the heart. Stimulation of AC results in catalysis of ATP into the second messenger adenosine 3', 5'-cyclic monophosphate (cAMP), which in turn binds to the regulatory subunits of cAMP-dependent protein kinase (PKA). In doing so, the catalytic subunits of PKA are rendered competent to phosphorylate several intracellular protein targets at serine and threonine residues. In several tissues, including the heart, PKA and its targets are in close proximity because of A-kinase anchoring proteins (AKAPs). An association of b2-adrenergic receptors and AKAPs have been previously reported (for review see Wong and Scott 2004), however there is a lack of information about the interaction of b1-adrenergic receptors and AKAPs in cardiac myocytes. Clearly, unique interaction between b1 and b2 adrenergic receptors and specific AKAPs would give an additional support to specific signaling properties of these subtypes.

Besides playing a very important role phosphorylating b-adrenergic receptors, which results in partial uncoupling and desensitization of the receptor to further agonist stimulation (heterologous desensitization) PKA has other roles in adrenergic receptor signaling. Some prominent targets of PKA phosphorylation in the adrenergic receptor signaling pathway are: a) L-type calcium channels and ryanodine receptors, both leading to an increase in Ca2+ entry into the cells (Zhao et al. 1994, Gerhardstein et al. 1999); b) phospholamban, a modulator of the sarcoplasmic reticulum associated ATP-dependent calcium pump (SERCA), which accelerates Ca2+ reuptake by the sarcoplasmic reticulum resulting in an accelerated cardiac relaxation (Simmerman and Jones 1998); and c) troponin I and myosin binding protein-C (MyBP-C), which reduce myofilament sensitivity to Ca2+ (Sulakhe and Vo 1995, Kunst et al. 2000, Xiao 2000). Lastly, PKA phosphorylates b-adrenergic receptors, resulting in partial uncoupling and desensitization of the receptor to further agonist stimulation (heterologous desensitization).
As described above the stimulation b-adrenergic receptors leads to dissociation G-proteins in Ga and G bg subunits. One important function of the dissociated G bg subunit is to facilitate the juxtaposition of b-adrenergic receptors and G-protein receptor kinases (GRK2 also known as b-adrenergic receptor kinase 1, bARK1), which ultimately mediates phosphorylation of b-adrenergic receptors and further desensitization in an agonist occupancy-dependent manner (homologous desensitization).
Finally, b-adrenergic receptors in cardiac myocytes can regulate other effectors independently of AC activation, including voltage-sensitive calcium channels and sodium channels (Reiter 1988, Kaumann 1991, Matsuda et al. 1992).
THE CARDIAC b-ADRENERGIC PATHWAY IN HEART FAILURE
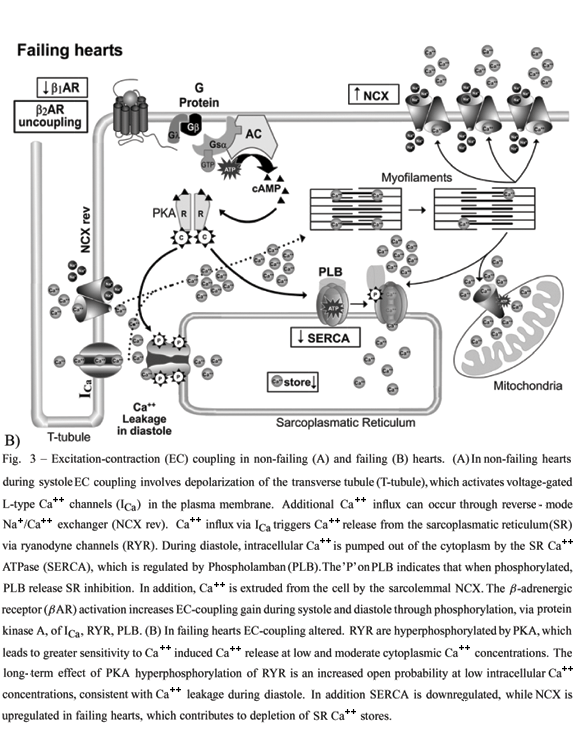
Heart failure caused by diverse etiologies is characterized by a sympathetic hyperactivity, paralleled by a reduction in b-adrenergic receptor density, and desensitization of remaining b-adrenergic receptor, leading to a markedly blunted cardiac contractile response to b-adrenergic receptor activation (Bristow et al. 1982) (Fig. 3B). A reduction in b-adrenergic receptor density was first demonstrated in 1982 by Bristow et al. (Bristow et al. 1982) in failing hearts explanted at the time of transplantation. In addition, they also showed b-adrenergic receptor desensitization in the setting of heart failure. Alterations in b-adrenergic receptor signaling have been also observed at Gi, AC and GRK2 (Post et al. 1999) levels.
As previously described, b1-adrenergic receptor subtype comprises approximately 70 to 80% of total cardiac b-adrenergic receptors in non-failing hearts. In heart failure, b1-adrenergic receptor is selectively down-regulated resulting in an approximate 50:50 ratio of b1 to b2 subtypes (Wallukat et al. 2003). In addition, b2-adrenergic receptor seems to be uncoupled from activation of AC (Post et al. 1999, Port and Bristow 2001, Lohse et al. 2003b). This latter effect seems to be due to b-adrenergic receptor phosphorylation by specific kinases, namely: a) GRK2 (bARK1) that phosphorylates both b-adrenergic receptor subtypes in an agonist-dependent manner, and b) PKA and PKC that phosphorylate b-adrenergic receptors in an agonist-independent manner (Sibley et al. 1986,Hausdorff et al. 1990). Interestingly, elevated levels of GRK2 in failing human hearts have also been reported (Ungerer et al. 1993).
Sustained b-adrenergic receptor activation can also influence G protein and AC expression. It has been demonstrated that Gi expression levels are increased in human failing hearts of different etiologies (Feldman et al. 1988, Neumann et al. 1988, Eschenhagen et al. 1992, Ping and Hammond 1994) leading to a decreased Gs:Gi ratio. Likewise, AC's V and VI isoforms were reported to be down regulated both in mRNA and protein levels of failing hearts (Ishikawa et al. 1994). Collectively, b-adrenergic receptor down regulation and desensitization, as well as decreased Gs:Gi ratio and AC's V and VI isoform expression will culminate with less production of cAMP. Decreased formation of cAMP, in turn, leads to diminished activation of PKA. However, decreased PKA activity is not always correlated with less phosphorylation of its intracellular effectors in failing hearts. For example, the cardiac ryanodine receptor (RyR2), a calcium release channel localized in sarcoplasmic reticulum and target of calcium-induced calcium release triggered by L-type calcium channels, has been shown to be hyperphosphorylated in failing hearts (Marxet al. 2000, Reiken et al. 2003, Wehrens et al. 2005). The hyperphosphorylated RyR2 is associated with a dissociation of FKBP12.6 (a protein which stabilizes the closed state of RyR2 channels) from RyR2 channels, and results into calcium leakage during diastole, and further depletion of sarcoplasmic reticulum calcium stores (Fig. 3B). The hyperphosphorylated state of RyR2 receptors is likely due to the down-regulation of phosphatases PP1 and PP2A association with RyR2 (Reiken et al. 2003, Wehrens and Marks 2003). In a similar manner, PKA phosphorylation of phospholamban appears to be unchanged in heart failure (Bohm et al. 1994, Kirchhefer et al. 1999), whereas its overall phosphorylation seems to be decreased (Fig. 3B) (Schwinger et al. 1999). The decreased phosphorylation of phospholamban results in a greater inhibition of the sarcoplasmic reticulum calcium ATPase (SERCA2), and thereby in a decreased cardiac relaxation. Concomitantly to decreased phospholamban phosphorylation, SERCA2 expression both at mRNA and protein levels seems to be decreased in heart failure leading to an additional impairment of Ca2+ reuptake to sarcoplasmic reticulum and consequently diastolic dysfunction (Fig. 3B) (Hajjar et al. 1998, Schwinger et al. 1999). Together all changes in b-adrenergic receptor signaling pathways described above result in decreased cardiac inotropic and lusitropic responses to adrenergic stimulation.
Abnormalities in b-adrenergic receptor signal transduction are not involved only in cardiac functional impairment, they also play a role in cardiac structural changes observed in heart failure being involved in the transition from compensated cardiac hypertrophy to decompensated heart failure (Morisco et al. 2001, Lowes et al. 2002). The exposure to high levels of circulating cathecholamines has been reported to be toxic to cardiac myocytes (Rona 1985, Mann et al. 1992), leading to myofibrillar degradation and increased cardiac collagen volume fraction mediated by b-adrenergic receptor stimulation. However, substantial evidence from the literature points to significant differences between b1 and b2-adrenergic receptor subtypes and their ability to stimulate apoptosis, or programmed cell death, in isolated cardiac myocytes (Xiao et al. 2004) and in vivo experiments performed in knockout mice lacking b1, b2 or both subtypes (Patterson et al. 2004). b1-adrenergic receptor stimulation results in an increased cardiac myocyte apoptosis via cAMP-dependent mechanism (Communal et al. 1998, 1999), whereas stimulation of b2-subtype inhibits apoptosis via a Gi-coupled pathway involving PI3K and Akt-PKD (Chesley et al. 2000, Zaugg et al. 2000, Zhu et al. 2001). These findings have interesting clinical implications for heart failure therapy, since they provide cellular and molecular mechanisms that underline the beneficial therapeutic effects of some b-adrenergic receptor antagonists, and provide the rationale for combining b1-subtype specific blockade with b2- subtype activation.
MYOPATHIC POTENTIAL OF INDIVIDUAL COMPONENTS OF ADRENERGIC RECEPTOR PATHWAYS
The advances in mice engineering technologies and a better knowledge of the genome structure have provided a wealth of information with regard to understanding the role of adrenergic receptors signaling in heart failure. Moreover, mice have proven to be a valid model for studying heart failure because of the similarities between this disorder in mice and humans (Chien 1996). In this section we briefly describe some data obtained from both the "gain of function" (transgenic animals) and "loss of function" (knockout animals) of individual cardiac genes in an attempt to better understand the molecular mechanisms underlying the adrenergic receptor pathways in heart failure. Table I summarizes the genetically altered mouse models that we will discuss.
ADRENERGIC RECEPTORS
The notion that b1- and b2-adrenergic receptor signaling and functional properties are distinctly different has been emphasized by studies performed in transgenic mouse models. Mice overexpressing the b2-subtype at relatively high abundance ( ~ 50 to 200 fold increases in receptor numbers) produced no or limited histopathology at time points up to four months of age while maintaining a hyperdynamic state characterized by increased basal AC activity, enhanced cardiac contractility and left ventricle function (Milano et al. 1994). The gene dose effect of the overexpression of the b2-subtype in a study by Liggett (Liggett 2000a) has provided data that showed a expression level dependence with regard to the development of cardiomyopathy. Animals expressing more than 60 times the b2-subtype maintained their hyperdynamic state for more than one year, without an apparent increase in mortality. In contrast, overexpression levels above 100 fold resulted in progressive cardiac enlargement, the development of heart failure, and premature death.
In marked contrast to results obtained from b2-subtype that needs a high level of expression to develop of heart failure, mice overexpressing b1-subtype, as low as five fold the endogenous expression level, present progressive hypertrophy and ventricular dysfunction, which culminate with heart failure by the age of 35 weeks (Engelhardt et al. 1999). The pathways mediating the cardiac deleterious effects of b1-subtype overexpression seems to involve an altered calcium handling (Engelhardt et al. 2001, 2004) and increased Na+-H+ exchanger (Engelhardt et al. 2002). In addition, overexpression of b1-subtype in mice leads to upregulation of pro-apoptotic proteins, such as Bax (Bisognano et al. 2000), and chronic stimulation of b1-subtype has been shown to increase rate of apoptosis (Communal et al. 1998, Xiao 2001, Zhu et al. 2001). Taken together these results recapitulate findings obtained from selective stimulation of b1-and b2-subtypes in cardiac myocytes culture, where b1-subtype seems to induce apoptosis whereas b2-subtype is antiapoptotic.
The role of myocardial adrenergic signaling in cardiac function has been additionally explored in transgenic hearts overexpressing b-adrenergic receptor kinase (b-ARK1 or GRK2) or a peptide inhibitor of b-ARK1 (b-ARKct). Mice overexpressing b-ARK1 demonstrated attenuation of isoproterenol-stimulated left ventricular contractility in vivo, dampening of myocardial adenylyl cyclase activity, and reduced functional coupling of b-adrenergic receptors. Conversely, mice overexpressing b-ARKct showed enhanced cardiac contractility both with or without propranolol (Koch et al. 1995). Moreover, inhibition of b-ARK1 with b-ARKct prevented cardiac dysfunction in several models of heart failure (Rockman et al. 1998, Cho et al. 1999, Harding et al. 2001). Recently, it was demonstrated that the levels of b-ARK1 inhibition determines degree of cardiac dysfunction after chronic pressure overload-induced heart failure (Tachibana et al. 2005).
In addition to overexpression of b-adrenergic receptor, targeted disruption of selective b1- and b2-subtypes have been described. As b1-subtype mediate the chronotropic and inotropic effects of catecholamines, chronotropic reserve in b1-deficient mice is markedly limited and heart rate responses to exercise is depressed (Rohrer et al. 1996). In contrast, deletion of b2-receptor gene did not alter cardiac responsiveness to catecholamines, but altered metabolic response to exercise (Chruscinski et al. 1999).
The pro-apoptotic response to b1-adrenergic receptor chronic stimulation was recently reinforced by data obtained in selective b1 and b2 adrenergic receptor knockout mice (Patterson et al. 2004). b2-knockout mice (b1-receptor is the main subtype remained in cardiac myocytes) treated with isoproterenol for 14 days presented an increased mortality rate and cardiac dysfunction and apoptosis, whereas b1-knockout mice had cardiac function and ultrastructure preserved.
Another very interesting genetic model that recapitulate several aspects of heart failure in humans is based on disruption of a2-adrenergic receptor in mice. Both a2A and a2C adrenergic receptor subtypes modulate sympathetic tone (MacMillan et al. 1996, Altman et al. 1999), and disruption of both a2A and a2C adrenergic receptors in mice leads to chronically elevated sympathetic tone (Hein et al. 1999). These knockout mice present cardiomyopathy induced by sympathetic hyperactivity and showed reduced exercise capacity, decreased maximal oxygen consumption, decreased cardiac contractility, and significant abnormalities in the ultrastructure of cardiac myocytes (Brum et al. 2002). Considering that most murine models of heart failure are based on the disruption or overexpression of genes for cardiac specific proteins, a2A and a2C knockout mice provide evidence that chronic elevation of sympathetic tone can lead to heart failure in the absence of genetically induced alterations in myocardial structural or functional proteins.
Even though a1-adrenergic receptors are expressed in a lower level in cardiac myocyte when compared with b adrenergic receptor (ratio of 10:1 for b and a1), genetic manipulation of a1-adrenergic receptors has also been demonstrated to affect cardiac structure and function (Simpson et al. 1991).The heart expresses all 3 subtypes of a1-adrenergic receptors, namely a1A, a1B, and a1D, being a1B-subtype more abundantly expressed in the heart. Transgenic mouse models with overexpression of a1B-adrenergic receptors demonstrated that this subtype might induce cardiac hypertrophy in some but not all transgenic strains (Akhter et al. 1997, Grupp et al. 1998, Lemire et al. 1998, Zuscik et al. 2001). In contrast, deletion of single a1-adrenergic receptor did not affect cardiac structure or function in resting mice (Cavalli et al. 1997,Rokosh and Simpson 2002, Tanoue et al. 2002). Of interest, mice lacking both a1A- and a1B-adrenergic receptors showed reduced cardiac growth after birth and functional alterations that partly resemble changes observed in heart failure (McCloskey et al. 2003, O'Connell et al. 2003, Turnbull et al. 2003).
G PROTEINS AND ADENYLYL CYCLASE
As previously mentioned, several G proteins are involved in adrenergic receptors signaling, including Gs and Gi, which modulate AC activity, and Gq that activates phospholipase C. Similar to that observed with adrenergic receptors, genetic manipulation of these downstream signaling components results in a variety of cardiac phenotypes.
Increased levels of inhibitory Gi are widely accepted as a contributor to b-adrenergic receptors dampened signaling in cardiac hypertrophy and failure. Indeed, mice genetically engineered to conditionally express a modified Gi coupled receptor (Ro1) developed lethal cardiomyopathy associated with a wide QRS complex arrhythmia, with a mortality rate greater than 90% at 16 weeks (Redfern et al. 2000). These results are the first to suggest the potential deleterious role for increased Gi expression in the development and progression of heart failure, which contrasts with the notion of Gi signaling being protectant due to its antiapoptotic effects. Thus, more research needs to be performed to elucidate the relative role of increased Gi signaling in heart failure.
In contrast to Gi, the role of increasing Gs signaling in heart failure has been described in the greatest detail. Transgenic mice overexpressing Gs show baseline tachycardia, enhanced chronotropic and inotropic response to isoproterenol, altered b-adrenergic receptor density, and increased frequency of cardiac arrhythmias (Iwase et al. 1996). Furthermore, overexpression of Gs is associated with myocyte hypertrophy, apoptosis and fibrosis (Iwase et al. 1996, Lader et al. 1998, Geng et al. 1999). These adverse effects seem to be related to enhanced L-type calcium currents in Gs transgenic mice, an effect independent of cAMP pathway.
Transgenic mice overexpressing Gq also exhibit a myopathic phenotype associated with cardiac hypertrophy and fibrosis, which recapitulates that observed in pressure overload hypertrophy (D'Angelo et al. 1997, Sakata et al. 1998). Likewise, transient cardiac expression of Gq led to hypertrophy and dilated cardiomyopathy (Mende et al. 1999). In addition, the decreased cardiac contractility observed in Gq transgenic mice seems to be related to a decreased calcium inflow by L-type calcium channels, and increased Gi. In these models, inhibition of Gi caused sudden death, which suggest that the increased Gi expression might be a compensatory mechanism to counteract other detrimental signaling caused by Gq pathway.
To date, overexpression of more distal components of Gs signaling pathways, specifically ACs' V and VI, does not induce any form of cardiomyopathy (Roth et al. 1999, Tepe and Liggett 1999). Agonist stimulated AC activity is higher in this model; however, this effect does not translate into markedly increased contractility. Furthermore, no evidence of cardiac structure injury was observed.
In summary, generation and characterization of genetically altered mouse models have greatly advanced our knowledge of the molecular mechanisms underlying the pathogenesis of heart failure and provided valuable insights into the identification of the molecular targets for therapeutic development.
b-ADRENERGIC RECEPTOR POLYMORPHISMS IN HEART FAILURE
The central role played by sympathetic nervous system and its receptors in heart failure makes polymorphisms in receptors genes attractive candidates for risk factor and/or predictors of response to treatment.
Polymorphisms of many genes, including of adrenergic receptor signaling pathways and renin-angiotensin system together with environmental factors can markedly influence the progression of cardiac disease. Liggett et al. (Liggett et al. 1998, Liggett 2000b) have demonstrated that adrenergic polymorphism affects not only receptor signaling and sensitivity to pharmacological agents, but also influence clinical outcomes.
For human b1-adrenergic receptor, two major polymorphic loci have been identified. One of them is a Ser49Gly polymorphism in the extracellular N-terminus (Borjesson et al. 2000). The allelic distribution of Ser49Gly polymorphism has been associated with long-term survival (decreased mortality risk in subjects with Gly49) of patients with heart failure (Borjesson et al. 2000). This finding might be related to results form in vitro studies that demonstrated increased desensitization and down-regulation of the Gly49 variant (Levin et al. 2002, Rathz et al. 2002), consistent with the idea that b1-adrenergic receptor blockade or desensitization is protective in heart failure (Bristow 2000). However, contrasting results have been reported in the literature, since Podlowski et al. (Podlowski et al. 2000) found that Ser49Gly polymorphism is more frequent in patients with idiopathic dilated cardiomyopathy.
Another important polymorphism of b1-adrenergic receptor is an Arg389Gly polymorphism in the 4 intracellular loop, which participates in G-protein coupling. Arg389 variant is threefold more effective than is the Gly389 variant at activating adenylyl ciclase (Mason et al. 1999). Of interest, the Gly389 is considered to be the "wild type" allele since it was cloned first. However, its frequency in Caucasian population is ~ 0.27 (Liggett 2000b). In spite of data about this polymorphism, a few trends are apparent. Even though Tesson et al. (Tesson et al. 1999) have observed no direct correlation between Arg389Gly polymorphism and heart failure, more recently Arg389 variant, even when expressed at lower levels in mouse hearts, induced heart failure, whereas Gly389 variant did not (Mialet Perez et al. 2003). Indeed, responses to antagonists are greater in the Arg 389 than in Gly389 variant (Johnson et al. 2003, Mialet Perez et al. 2003, Sofowora et al. 2003).
a2C - adrenergic receptor polymorphism has uncovered a significant increased risk of progression to heart failure, particularly in African American subjects, when the gain of function Arg389b1-adrenergic receptor polymorphism is associated with the loss of function a2C-adrenergic deletion variant (decreased inhibition of noradrenaline release from sympathetic nerve terminals, which is consistent with findings of a2A/a2C adrenergic receptor knockout mice) (Small et al. 2002). Of interest, Arg389b1-adrenergic receptor polymorphism has not been associated with increased cardiovascular risk by itself; however it significantly increases the cardiovascular risk of a2C-adrenergic deletion variant. These results highlighted the importance of considering the combinations of individual polymorphisms, which results in several haplotypes that have not been investigated in detail.
Overall, as recently reviewed by Michel and Insel (Michel and Insel 2003), inconclusive results have been obtained regarding b1-adrenergic receptor polymorphisms. Further work is need to define the role of these polymorphisms play in heart disease and drug response, perhaps as haplotypes (Kirstein and Insel 2004, Lohse 2004).
Although b2-adrenergic receptors are expressed in the heart at lower concentrations than are b1-subtype, they are more numerous in many other sites, including vascular, bronchial, gastrointestinal smooth muscle, glands, leukocytes, and hepatocytes; and more importantly b2-adrenergic receptors are highly polymorphic. One rare and never homozygous Thr164Ile variant was associated with reduced survival and depressed exercise capacity in patients with heart failure (Liggett et al. 1998, Wagoner et al. 2000, Brodde et al. 2001). These results are consistent with findings in transgenic mice with Thr164Ile polymorphism targeted to the heart (Turki et al. 1996).
Another polymorphism associated with heart failure is Gln27Gly variant in b2-adrenergic receptor. It has recently been reported that heart failure patients homozygous for the Gln27 allele were less likely to respond to the b-blocker carvedilol compared with Glu27 allele (Kaye et al. 2003). This result suggests that Gln27 influences the responsiveness of heart failure patients to b-blocker therapy.In addition, we have recently reported that humans with polymorphism of b2-adrenergic receptor at codons 16 and 27, namely women who are homozygous for Arg16/Glu27 haplotype, have augmented muscle vasodilatory response to mental stress and exercise (Trombetta et al. 2005). Whether this result will be reproducible in heart failure patients is not known.
In summary, the contribution of b1 or b2 adrenergic receptor polymorphisms for progression of heart failure still need to be better investigated. Prospective studies of sufficient size are lacking, as well as, studies designed to consider the many complex haplotypes comprising a combination of individual polymorphisms.
ACKNOWLEDGMENTS
The authors acknowledge Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, processo 02/04588-8) for financial support and Dr.Carlos Eduardo Negrão for providing the microneurographic tracings of heart failure patients included in Figure 1.
Manuscript received on June 14, 2005; accepted for publication on November 4, 2005; presented by EDUARDO M. KRIEGER
Correspondence to: Patricia Chakur Brum
E-mail: pcbrum@usp.br
- AHLQUIST RP. 1948. A study of the adrenotropic receptors. Am J Physiol 153: 586-600.
- AKHTER SA, MILANO CA, SHOTWELL KF, CHO MC, ROCKMAN HA, LEFKOWITZ RJ AND KOCH WJ. 1997. Transgenic mice with cardiac overexpression of alpha1B-adrenergic receptors. In vivo alpha1-adrenergic receptor-mediated regulation of beta-adrenergic signaling. J Biol Chem 272: 21253-21259.
- ALBANESI FILHO FM. 2003. Epidemiologia da Insuficięncia Cardíaca. In Insuficięncia Cardíaca. BARRETTO ACP AND BOCCHI EA (Eds), Segmento, Săo Paulo, SP, Brasil, p. 13-22.
- ALTMAN JD, TRENDELENBURG AU, MACMILLAN, L, BERNSTEIN D, LIMBIRD L, STARKE K, KOBILKA BK AND HEIN L. 1999. Abnormal regulation of the sympathetic nervous system in alpha2A-adrenergic receptor knockout mice. Mol Pharmacol 56: 154-161.
- BISOGNANO JD ET AL. 2000. Myocardial-directed overexpression of the human beta(1)-adrenergic receptor in transgenic mice. J Mol Cell Cardiol 32: 817-830.
- BOHM M, REIGER B, SCHWINGER RH AND ERDMANN E. 1994. cAMP concentrations, cAMP dependent protein kinase activity, and phospholamban in non-failing and failing myocardium. Cardiovasc Res 28: 1713-1719.
- BONNEUX L, BARENDREGT JJ, MEETER K, BONSEL GJ AND VAN DER MAAS PJ. 1994. Estimating clinical morbidity due to ischemic heart disease and congestive heart failure: the future rise of heart failure. Am J Public Health 84: 20-28.
- BORJESSON M, MAGNUSSON Y, HJALMARSON A AND ANDERSSON B. 2000. A novel polymorphism in the gene coding for the beta(1)-adrenergic receptor associated with survival in patients with heart failure. Eur Heart J 21: 1853-1858.
- BRISTOW MR. 2000. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 101: 558-569.
- BRISTOW MR, GINSBURG R, MINOBE W, CUBICCIOTTI RS, SAGEMAN WS, LURIE K, BILLINGHAM ME, HARRISON DC AND STINSON EB. 1982. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing humanhearts. N Engl J Med 307: 205-211.
- BRODDE OE, BUSCHER R, TELLKAMP R, RADKE J, DHEIN S AND INSEL PA. 2001. Blunted cardiac responses to receptor activation in subjects with Thr164Ile beta(2)-adrenoceptors. Circulation 103: 1048-1050.
- BRUM PC, KOSEK J, PATTERSON A, BERNSTEIN D AND KOBILKA B. 2002. Abnormal cardiac function associated with sympathetic nervous system hyperactivity in mice. Am J Physiol Heart Circ Physiol 283: H1838-1845.
- BYLUND DB, EIKENBERG DC, HIEBLE JP, LANGER SZ, LEFKOWITZ RJ, MINNEMAN KP, MOLINOFF PB, RUFFOLO JR RR AND TRENDELENBURG U. 1994. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev 46: 121-136.
- CAVALLI A ET AL. 1997. Decreased blood pressure response in mice deficient of the alpha1b-adrenergic receptor. Proc Natl Acad Sci USA 94: 11589-11594.
- CHESLEY A, LUNDBERG MS, ASAI T, XIAO RP, OHTANI S, LAKATTA EG AND CROW MT. 2000. The beta(2)-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3'-kinase. Circ Res 87: 1172-1179.
- CHIDSEY CA AND BRAUNWALD E. 1966. Sympathetic activity and neurotransmitter depletion in congestive heart failure. Pharmacol Rev 18: 685-700.
- CHIEN KR. 1996. Molecular medicine in genetically engineered animals: series introduction. J Clin Invest 97: 2.
- CHO MC, RAO M, KOCH WJ, THOMAS SA, PALMITER RD AND ROCKMAN HA. 1999. Enhanced contractility and decreased beta-adrenergic receptor kinase-1 in mice lacking endogenous norepinephrine and epinephrine. Circulation 99: 2702-2707.
- CHRUSCINSKI AJ, ROHRER DK, SCHAUBLE E, DESAI KH, BERNSTEIN D AND KOBILKA BK. 1999. Targeted disruption of the beta2 adrenergic receptor gene. J Biol Chem 274: 16694-16700.
- CLELAND JG, KHAND A AND CLARK A. 2001. The heart failure epidemic: exactly how big is it? Eur Heart J 22: 623-626.
- COHN JN, LEVINE TB, OLIVARI MT, GARBERG V, LURA D, FRANCIS GS, SIMON AB AND RECTOR T. 1984. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 311: 819-823.
- COMMUNAL C, SINGH K, PIMENTEL DR AND COLUCCI WS. 1998. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activationof the beta-adrenergic pathway. Circulation 98:1329-1334.
- COMMUNAL C, SINGH K, SAWYER DB AND COLUCCI WS. 1999. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation 100: 2210-2212.
- D'ANGELO DD, SAKATA Y, LORENZ JN, BOIVIN GP, WALSH RA, LIGGETT SB AND DORN II GW. 1997. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA 94: 8121-8126.
- DEVIC E, XIANG Y, GOULD D AND KOBILKA B. 2001. Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharmacol 60: 577-583.
- ENGELHARDT S, HEIN L, WIESMANN F AND LOHSE MJ. 1999. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci USA 96: 7059-7064.
- ENGELHARDT S, BOKNIK P, KELLER U, NEUMANN J, LOHSE MJ AND HEIN L. 2001. Early impairment of calcium handling and altered expression of junctin in hearts of mice overexpressing the beta1-adrenergic receptor. Faseb J 15: 2718-2720.
- ENGELHARDT S, HEIN L, KELLER U, KLAMBT K AND LOHSE MJ. 2002. Inhibition of Na(+)-H(+) exchange prevents hypertrophy, fibrosis, and heart failure in beta(1)-adrenergic receptor transgenic mice. Circ Res 90: 814-819.
- ENGELHARDT S, HEIN L, DYACHENKOW V, KRANIAS EG, ISENBERG G AND LOHSE MJ. 2004. Altered calcium handling is critically involved in the cardiotoxic effects of chronic beta-adrenergic stimulation. Circulation 109: 1154-1160.
- ESCHENHAGEN T, MENDE U, DIEDERICH M, NOSE M, SCHMITZ W, SCHOLZ H, SCHULTE AM, ESCH J, WARNHOLTZ A AND SCHAFER H. 1992. Long term beta-adrenoceptor-mediated up-regulation of Gi alpha and G(o) alpha mRNA levels and pertussis toxin-sensitive guanine nucleotide-binding proteins in rat heart. Mol Pharmacol 42: 773-783.
- FELDMAN AM, CATES AE, VEAZEY WB, HERSHBERGER RE, BRISTOW MR, BAUGHMAN KL, BAUMGARTNER WA AND VAN DOP C. 1988. Increase of the 40,000-mol wt pertussis toxin substrate (G protein) in the failing human heart. J Clin Invest 82: 189-197.
- FRANCIS GS, GOLDSMITH SR, LEVINE TB, OLIVARI MT AND COHN JN. 1984. The neurohumoral axis in congestive heart failure. Ann Intern Med 101: 370-377.
- GENG YJ, ISHIKAWA Y, VATNER DE, WAGNER TE, BISHOP SP, VATNER SF AND HOMCY CJ. 1999. Apoptosis of cardiac myocytes in Gs alpha transgenic mice. Circ Res 84: 34-42.
- GERHARDSTEIN BL, PURI TS, CHIEN AJ AND HOSEY MM. 1999. Identification of the sites phosphorylated by cyclic AMP-dependent protein kinase on the beta 2 subunit of L-type voltage-dependent calcium channels. Biochemistry 38: 10361-10370.
- GRUPP IL, LORENZ JN, WALSH RA, BOIVIN GP AND RINDT H. 1998. Overexpression of alpha1B-adrenergic receptor induces left ventricular dysfunction in the absence of hypertrophy. Am J Physiol 275: H1338-1350.
- HAJJAR RJ, MULLER FU, SCHMITZ W, SCHNABEL P AND BOHM M. 1998. Molecular aspects of adrenergic signal transduction in cardiac failure. J Mol Med 76: 747-755.
- HALL RA AND LEFKOWITZ RJ. 2002. Regulation of G protein-coupled receptor signaling by scaffold proteins. Circ Res 91: 672-680.
- HARDING VB, JONES LR, LEFKOWITZ RJ, KOCH WJ AND ROCKMAN HA. 2001. Cardiac beta ARK1 inhibition prolongs survival and augments beta blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci USA 98: 5809-5814.
- HAUSDORFF WP, LOHSE MJ, BOUVIER M, LIGGETT SB, CARON MG AND LEFKOWITZ RJ. 1990. Two kinases mediate agonist-dependent phosphorylation and desensitization of the beta 2-adrenergic receptor. Symp Soc Exp Biol 44: 225-240.
- HEIN L, LIMBIRD LE, EGLEN RM AND KOBILKA BK. 1999. Gene substitution/knockout to delineate the role of alpha 2-adrenoceptor subtypes in mediating central effects of catecholamines and imidazolines. Ann N Y Acad Sci 881: 265-271.
- ISHIKAWA Y, SOROTA S, KIUCHI K, SHANNON RP, KOMAMURA K, KATSUSHIKA S, VATNER DE, VATNER SF AND HOMCY CJ. 1994. Downregulation of adenylylcyclase types V and VI mRNA levels in pacing-induced heart failure in dogs. J Clin Invest 93: 2224-2229.
- IWASE M ET AL. 1996. Adverse effects of chronic endogenous sympathetic drive induced by cardiac GS alpha overexpression. Circ Res 78: 517-524.
- JABER M ET AL. 1996. Essential role of beta-adrenergic receptor kinase 1 in cardiac development and function. Proc Natl Acad Sci USA 12: 12974-12979.
- JANSSEN PM ET AL. 2002. Intracellular beta-blockade: overexpression of Galpha(i2) depresses the betaadrenergic response in intact myocardium. Cardiovasc Res 55: 300-308.
- JOHNSON JA, ZINEH I, PUCKETT BJ, MCGORRAY SP, YARANDI HN AND PAULY DF. 2003. Beta 1- adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin Pharmacol Ther 74: 44-52.
- JOSUE O. 1907. Hypertrophie cardioque causée par l'adrenaline et la toxine typhique. C R Soc Biol 63: 285-290.
- KATZ AM. 2001. Crossovers between functional and proliferative signaling: key to understanding the pathophysiology and management of heart failure. J Cell Mol Med 5: 125-131.
- KATZ AM. 2002. Proliferative signaling and disease progression in heart failure. Circ J 66: 225-231.
- KATZ AM. 2003. Heart failure: a hemodynamic disorder complicated by maladaptive proliferative responses. J Cell Mol Med 7: 1-10.
- KAUMANN AJ. 1991. Some aspects of heart beta adrenoceptor function. Cardiovasc Drugs Ther 5: 549-560.
- KAYE DM, SMIRK B, WILLIAMS C, JENNINGS G, ESLER M AND HOLST D. 2003. Beta-adrenoceptor genotype influences the response to carvedilol in patients with congestive heart failure. Pharmacogenetics 13: 379-382.
- KIRCHHEFER U, SCHMITZ W, SCHOLZ H AND NEUMANN J. 1999. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res 42: 254-261.
- KIRSTEIN SL AND INSEL PA. 2004. Autonomic nervous system pharmacogenomics: a progress report. Pharmacol Rev 56: 31-52.
- KOCH WJ, ROCKMAN HA, SAMAMA P, HAMILTON RA, BOND RA, MILANO CA AND LEFKOWITZ RJ. 1995. Cardiac function in mice overexpressing the beta-adrenergic receptor kinase or a beta ARK inhibitor. Science 268: 1350-1353.
- KUNST G, KRESS KR, GRUEN M, UTTENWEILER D, GAUTEL M AND FINK RH. 2000. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res 86: 51-58.
- KUSCHEL M, ZHOU YY, CHENG H, ZHANG SJ, CHEN Y, LAKATTA EG AND XIAO RP. 1999. G(i) protein-mediated functional compartmentalization of cardiac beta(2)-adrenergic signaling. J Biol Chem 274: 22048-22052.
- LADER AS, XIAO YF, ISHIKAWA Y, CUI Y, VATNER DE, VATNER SF, HOMCY CJ AND CANTIELLO HF. 1998. Cardiac Gsalpha overexpression enhances L-type calcium channels through an adenylyl cyclase independent pathway. Proc Natl Acad Sci USA 95: 9669-9674.
- LANDS AM, ARNOLD A, MCAULIFF JP, LUDUENA FP AND BROWN JR TG. 1967. Differentiation of receptor systems activated by sympathomimetic amines. Nature 214: 597-598.
- LEFKOWITZ RJ, PIERCE KL AND LUTTRELL LM. 2002. Dancing with different partners: protein kinase a phosphorylation of seven membrane-spanning receptors regulates their G protein-coupling speci- ficity. Mol Pharmacol 62: 971-974.
- LEMIRE I, ALLEN BG, RINDT H AND HEBERT TE. 1998. Cardiac - specific overexpression of alpha1- BAR regulates betaAR activity via molecular crosstalk. J Mol Cell Cardiol 30: 1827-1839.
- LEVIN MC, MARULLO S, MUNTANER O, ANDERSSON B AND MAGNUSSON Y. 2002. The myocardium- protective Gly-49 variant of the beta 1-adrenergic receptor exhibits constitutive activity and increased desensitization and down-regulation. J Biol Chem 277: 30429-30435.
- LIGGETT SB. 2000a. beta(2)-adrenergic receptor pharmacogenetics. Am J Respir Crit Care Med 161: S197-201.
- LIGGETT SB. 2000b. Pharmacogenetics of beta-1- and beta-2-adrenergic receptors. Pharmacology 61: 167-173.
- LIGGETT SB, WAGONER LE, CRAFT LL, HORNUNG RW, HOIT BD, MCINTOSH TC AND WALSH RA. 1998. The Ile164 beta2-adrenergic receptor polymorphism adversely affects the outcome of congestive heart failure. J Clin Invest 102: 1534-1539.
- LOHSE MJ. 2004. Beta-adrenoceptor polymorphisms and heart failure. Trends Mol Med 10: 55-58.
- LOHSE MJ, ENGELHARDT S AND ESCHENHAGEN T. 2003a. What is the role of beta-adrenergic signaling in heart failure? Circ Res 93: 896-906.
- LOHSE MJ, VILARDAGA JP AND BUNEMANN M. 2003b. Direct optical recording of intrinsic efficacy at a G protein-coupled receptor. Life Sci 74:397-404.
- LOWES BD ET AL. 2002. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med 346: 1357-1365.
- MACMILLAN LB, HEIN L, SMITH MS, PIASCIK MT AND LIMBIRD LE. 1996. Central hypotensive effects of the alpha2a-adrenergic receptor subtype. Science 273: 801-803.
- MANN DL, KENT RL, PARSONS B AND COOPER GT. 1992. Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation 85: 790-804.
- MARTIN NP, WHALEN EJ, ZAMAH MA, PIERCE KL AND LEFKOWITZ RJ. 2004. PKA-mediated phosphorylation of the beta1-adrenergic receptor promotes Gs/Gi switching. Cell Signal 16: 1397-1403.
- MARX SO, REIKEN S, HISAMATSU Y, JAYARAMAN T, BURKHOFF D, ROSEMBLIT N AND MARKS AR. 2000. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101: 365-376.
- MASON DA, MOORE JD, GREEN SA AND LIGGETT SB. 1999. A gain-of-function polymorphism in a G-protein coupling domain of the human beta1-adrenergic receptor. J Biol Chem 274: 12670-12674.
- MATSUDA JJ, LEE H AND SHIBATA EF. 1992. Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circ Res 70: 199-207.
- MCCLOSKEY DT, TURNBULL L, SWIGART P,O'CONNELL TD, SIMPSON PC AND BAKER AJ. 2003. Abnormal myocardial contraction in alpha- (1A)- and alpha(1B)-adrenoceptor double-knockout mice. J Mol Cell Cardiol 35: 1207-1216.
- MENDE U, KAGEN A, MEISTER M AND NEER EJ. 1999. Signal transduction in atria and ventricles of mice with transient cardiac expression of activated G protein alpha(q). Circ Res 85: 1085-1091.
- MIALET PEREZ J, RATHZ DA, PETRASHEVSKAYA NN, HAHN HS, WAGONER LE, SCHWARTZ A, DORN GW AND LIGGETT SB. 2003. Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med 9: 1300-1305.
- MICHEL MC AND INSEL PA. 2003. Receptor gene polymorphisms: lessons on functional relevance from the beta 1-adrenoceptor. Br J Pharmacol 138: 279-282.
- MILANO CA, ALLEN LF, ROCKMAN HA, DOLBER PC, MCMINN TR, CHIEN KR, JOHNSON TD, BOND RA AND LEFKOWITZ RJ. 1994. Enhanced myocardial function in transgenic mice overexpressing the beta 2-adrenergic receptor. Science 264: 582-586.
- MORISCO C, ZEBROWSKI DC, VATNER DE, VATNER SF AND SADOSHIMA J. 2001. Beta-adrenergic cardiac hypertrophy is mediated primarily by the beta(1)-subtype in the rat heart. J Mol Cell Cardiol 33: 561-573.
- MORRIS AJ AND MALBON CC. 1999. Physiological regulation ofGprotein-linked signaling. Physiol Rev 79: 1373-1430.
- NEGRÃO CE ET AL. 2001. Abnormal neurovascular control during exercise is linked to heart failure severity. Am J Physiol Heart Circ Physiol 280: H1286- 1292.
- NEUMANN J, SCHMITZ W, SCHOLZ H, VON MEYERINCK L, DORING V AND KALMAR P. 1988. Increase in myocardial Gi-proteins in heart failure. Lancet 2: 936-937.
- O'CONNELL TD ET AL. 2003. The alpha(1A/C)- and alpha(1B)-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J Clin Invest 111: 1783-1791.
- PACKER M. 1992. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol 20: 248-254.
- PATTERSON AJ, ZHU W, CHOW A, AGRAWAL R, KOSEK J, XIAO RP AND KOBILKA B. 2004. Protecting the myocardium: a role for the beta2 adrenergic receptor in the heart. Crit Care Med 32: 1041- 1048.
- PING P AND HAMMOND HK. 1994. Diverse G protein and beta-adrenergic receptor mRNA expression in normal and failing porcine hearts. Am J Physiol 267: H2079-2085.
- PODLOWSKI S ET AL. 2000. Beta1-adrenoceptor gene variations: a role in idiopathic dilated cardiomyopathy? J Mol Med 78: 87-93.
- PORT JD AND BRISTOW MR. 2001. Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol 33: 887-905.
- POST SR, HAMMOND HK AND INSEL PA. 1999. Betaadrenergic receptors and receptor signaling in heart failure. Annu Rev Pharmacol Toxicol 39: 343-360.
- RATHZ DA, BROWN KM, KRAMER LA AND LIGGETT SB. 2002. Amino acid 49 polymorphisms of the human beta1-adrenergic receptor affect agonist-promoted trafficking. J Cardiovasc Pharmacol 39: 155-160.
- RAU T ET AL. 2003. Overexpression of wild-type G alpha(i)-2 suppresses beta-adrenergic signaling in cardiac myocytes. FASEB J 17: 523-525.
- REDFERN CH ET AL. 2000. Conditional expression of a Gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc Natl Acad Sci USA 97: 4826-4831.
- REIKEN S, GABURJAKOVA M, GUATIMOSIM S, GOMEZ AM, D'ARMIENTO J, BURKHOFF D, WANG J, VASSORT G, LEDERER WJ AND MARKS AR. 2003. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J Biol Chem 278: 444-453.
- REITER M. 1988. Calcium mobilization and cardiac inotropic mechanisms. Pharmacol Rev 40: 189-217.
- ROCKMAN HA, CHIEN KR, CHOI DJ, IACCARINO G, HUNTER JJ, ROSS J JR, LEFKOWITZ RJ AND KOCH WJ. 1998. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci USA 95: 7000-7005.
- ROHRER DK, DESAI KH, JASPER JR, STEVENS ME, REGULA JR DP, BARSH GS, BERNSTEIN D AND KOBILKA BK. 1996. Targeted disruption of the mouse beta1-adrenergic receptor gene: developmental and cardiovascular effects. Proc Natl Acad Sci USA 93: 7375-7380.
- ROKOSH DG AND SIMPSON PC. 2002. Knockout of the alpha 1A/C-adrenergic receptor subtype: the alpha 1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc Natl Acad Sci USA 99: 9474-9479.
- RONA G. 1985. Catecholamine cardiotoxicity. J Mol Cell Cardiol 17: 291-306.
- ROTH DM, GAO MH, LAI NC, DRUMM J, DALTON N, ZHOU JY, ZHU J, ENTRIKIN D AND HAMMOND HK. 1999. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation 99: 3099-3102.
- SAKATA Y, HOIT BD, LIGGETT SB, WALSH RA AND DORN 2ND GW. 1998. Decompensation of pressure-overload hypertrophy in G alpha q-overexpressing mice. Circulation 97: 1488-1495.
- SCHWINGER RH, MUNCH G, BOLCK B, KARCZEWSKI P, KRAUSE EG AND ERDMANN E. 1999. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J Mol Cell Cardiol 31: 479-491.
- SIBLEY DR, STRASSER RH, BENOVIC JL, DANIEL K AND LEFKOWITZ RJ. 1986. Phosphorylation/dephosphorylation of the beta-adrenergic receptor regulates its functional coupling to adenylate cyclase and subcellular distribution. Proc Natl Acad Sci USA 83: 9408-9412.
- SIMMERMAN HK AND JONES LR. 1998. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev 78: 921-947.
- SIMPSON PC, KARIYA K, KARNS LR, LONG CS AND KARLINER JS. 1991. Adrenergic hormones and control of cardiac myocyte growth. Mol Cell Biochem 104: 35-43.
- SMALL KM, WAGONER LE, LEVIN AM, KARDIA SL AND LIGGETT SB. 2002. Synergistic polymorphisms of beta1- and alpha2C-adrenergic receptors and the risk of congestive heart failure. N Engl J Med 347: 1135-1142.
- SOFOWORA GG ET AL. 2003. A common beta1-adrenergic receptor polymorphism (Arg389Gly) affects blood pressure response to beta-blockade. Clin Pharmacol Ther 73: 366-371.
- STARLING EH. 1897. The Arris and Bale Lectures on some points in the pathology of heart disease. Lancet 149: 569-572.
- STEINBERG SF. 1999. The molecular basis for distinct beta-adrenergic receptor subtype actions in cardiomyocytes. Circ Res 85: 1101-1111.
- SULAKHE PV AND VO XT. 1995. Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization. Mol Cell Biochem: 149-150: 103-126.
- TACHIBANA H, NAGA PRASAD SV, LEFKOWITZ RJ, KOCH WJ AND ROCKMAN HA. 2005. Level of beta-adrenergic receptor kinase 1 inhibition determines degree of cardiac dysfunction after chronic pressure overload-induced heart failure. Circulation 111: 591-597.
- TANOUE A, KOSHIMIZU TA AND TSUJIMOTO G. 2002. Transgenic studies of alpha(1)-adrenergic receptor subtype function. Life Sci 71: 2207-2215.
- TENDERA M. 2004. The epidemiology of heart failure. J Renin Angiotensin Aldosterone Syst 5 (Suppl 1): S2-6.
- TEPE NM AND LIGGETT SB. 1999. Transgenic replacement of type V adenylyl cyclase identifies a critical mechanism of beta-adrenergic receptor dysfunction in the G alpha q overexpressing mouse. FEBS Lett 458: 236-240.
- TESSON F ET AL. 1999. Characterization of a unique genetic variant in the beta1-adrenoceptor gene and evaluation of its role in idiopathic dilated cardiomyopathy. CARDIGENE Group. J Mol Cell Cardiol 31: 1025-1032.
- TROMBETTA IC ET AL. 2005. Gly16 + Glu27 beta2-adrenoceptor polymorphisms cause increased forearm blood flow responses to mental stress and handgrip in humans. J Appl Physiol 98: 787-794.
- TURKI J, LORENZ JN, GREEN SA, DONNELLY ET, JACINTO M AND LIGGETT SB. 1996. Myocardial signaling defects and impaired cardiac function of a human beta 2-adrenergic receptor polymorphism expressed in transgenic mice. Proc Natl Acad Sci USA 93: 10483-10488.
- TURNBULL L, MCCLOSKEY DT, O'CONNELL TD, SIMPSON PC AND BAKER AJ. 2003. Alpha 1-adrenergic receptor responses in alpha 1AB-AR knockout mouse hearts suggest the presence of alpha 1D-AR. Am J Physiol Heart Circ Physiol 284: H1104-1109.
- UNGERER M, BOHM M, ELCE JS, ERDMANN E AND LOHSE MJ. 1993. Altered expression of beta-adrenergic receptor kinase and beta 1- adrenergic receptors in the failing human heart. Circulation 87: 454-463.
- XIANG Y AND KOBILKA B. 2003a. The PDZ-binding motif of the beta2-adrenoceptor is essential for physiologic signaling and trafficking in cardiac myocytes. Proc Natl Acad Sci USA 100: 10776-10781.
- XIANG Y AND KOBILKA BK. 2003b. Myocyte adrenoceptor signaling pathways. Science 300: 1530-1532.
- XIANG Y, DEVIC E AND KOBILKA B. 2002. The PDZ binding motif of the beta 1 adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. J Biol Chem 277: 33783-33790.
- XIAO RP. 2000. Cell logic for dual coupling of a single class of receptors to G(s) and G(i) proteins. Circ Res 87: 635-637.
- XIAO RP. 2001. Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE:15.
- XIAO RP, ZHANG SJ, CHAKIR K, AVDONIN P, ZHU W, BOND RA, BALKE CW, LAKATTA EG AND CHENG H. 2003. Enhanced G(i) signaling selectively negates beta2-adrenergic receptor (AR)-but not beta1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation 108: 1633-1639.
- XIAO RP, ZHU W, ZHENG M, CHAKIR K, BOND R, LAKATTA EG AND CHENG H. 2004. Subtype-specific beta-adrenoceptor signaling pathways in the heart and their potential clinical implications. Trends Pharmacol Sci 25: 358-365.
- WAGONER LE, CRAFT LL, SINGH B, SURESH DP, ZENGEL PW, MCGUIRE N, ABRAHAM WT, CHENIER TC, DORN GW 2ND AND LIGGETT SB. 2000. Polymorphisms of the beta(2)-adrenergic receptor determine exercise capacity in patients with heart failure. Circ Res 86: 834-840.
- WALLUKAT G. 2002. The beta-adrenergic receptors. Herz 27: 683-690.
- WALLUKAT G, PODLOWSKI S, NISSEN E, MORWINSKI R, CSONKA C, TOSAKI A AND BLASIG IE. 2003. Functional and structural characterization of anti-beta1-adrenoceptor autoantibodies of spontaneously hypertensive rats. Mol Cell Biochem 251: 67-75.
- WEHRENS XH AND MARKS AR. 2003. Altered function and regulation of cardiac ryanodine receptors in cardiac disease. Trends Biochem Sci 28: 671-678.
- WEHRENS XH, LEHNART SE AND MARKS AR. 2005. Intracellular calcium release and cardiac disease. Annu Rev Physiol 67: 69-98.
- WONG W AND SCOTT JD. 2004. AKAPsignalling complexes: focal points in space and time. Nat Rev Mol Cell Biol 5: 959-970.
- ZAUGG M, XU W, LUCCHINETTI E, SHAFIQ SA, JAMALI NZ AND SIDDIQUI MA. 2000. Beta-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation 102: 344-350.
- ZHAO XL, GUTIERREZ LM, CHANG CF AND HOSEY MM. 1994. The alpha 1-subunit of skeletal muscle L-type Ca channels is the key target for regulation by A-kinase and protein phosphatase-1C. Biochem Biophys Res Commun 198: 166-173.
- ZHU WZ, ZHENG M, KOCH WJ, LEFKOWITZ RJ, KOBILKA BK AND XIAO RP. 2001. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci USA 98: 1607-1612.
- ZUSCIK MJ ET AL. 2001. Hypotension, autonomic failure, and cardiac hypertrophy in transgenic mice overexpressing the alpha 1B-adrenergic receptor. J Biol Chem 276: 13738-13743.
Publication Dates
-
Publication in this collection
17 Aug 2006 -
Date of issue
Sept 2006
History
-
Accepted
04 Nov 2005 -
Received
14 June 2005