Abstracts
Major modifications of immune system have been observed in African trypanosomiasis. These immune reactions do not lead to protection and are also involved in immunopathology disorders. The major surface component (variable surface glycoprotein,VSG) is associated with escape to immune reactions, cytokine network dysfunctions and autoantibody production. Most of our knowledge result from experimental trypanosomiasis. Innate resistance elements have been characterised. In infected mice, VSG preferentially stimulates a Th 1-cell subset. A response of <FONT FACE=Symbol>gd</FONT> and CD8 T cells to trypanosome antigens was observed in trypanotolerant cattle. An increase in CD5 B cells, responsible for most serum IgM and production of autoantibodies has been noted in infected cattle. Macrophages play important roles in trypanosomiasis, in synergy with antibodies (phagocytosis) and by secreting various molecules (radicals, cytokines, prostaglandins,...). Trypanosomes are highly sensitive to TNF-alpha, reactive oxygen and nitrogen intermediates. TNF-alpha is also involved in cachexia. IFN-gamma acts as a parasite growth factor. These various elements contribute to immunosuppression. Trypanosomes have learnt to use immune mechanisms to its own profit. Recent data show the importance of alternative macrophage activation, including arginase induction. L-ornithine produced by host arginase is essential to parasite growth. All these data reflect the deep insight into the immune system realised by trypanosomes and might suggest interference therapeutic approaches.
Trypanosome; African trypanosomiasis; immunology; macrophage; lymphocytes; nitric oxide; cytokine; autoantibodies
Modificações importantes no sistema imune são observadas na tripanosomíase Africana. Essas reações imunológicas não protegem e estão envolvidas em distúrbios imunopatológicos. O principal componente de superfície (glicoproteína variante de superfície, VSG) está associado à evasão das respostas imune, às disfunções da rede de citocinas e à produção de autoanticorpos. Muitos de nossos conhecimentos resultam da tripanosomíase experimental. Componentes da imunidade inata estão sendo caracterizados. Em camundongos infectados, a VSG estimula preferencialmente células Th1. Uma resposta de <FONT FACE=Symbol>gd</FONT> e células T CD8 aos antígenos do tripanossoma foi observada em gado tripanotolerante. Um aumento em células B CD5, responsável por IgM sérica e produção de autoanticorpos, foi observado no gado infectado. Os macrófagos desempenham importantes funções na tripanosomíase, em sinergismo com anticorpos (fagocitose) e pela secreção de várias moléculas (radicais, citocinas, prostaglandinas). Tripanossomas são altamente sensíveis ao TNF-alfa, espécies reativas de oxigênio e nitrogênio. O TNF-alfa também está envolvido em caquexia. O IFN-gama atua como um fator de crescimento do parasita. Esses vários componentes contribuem para a imunossupressão. Os tripanossomas usam os mecanismos imunes para seu próprio benefício. Dados recentes mostram a importância da ativação alternativa de macrófagos, incluindo a indução pela arginase. A L-ornitina produzida pela arginase do hospedeiro é essencial para o crescimento do parasita. Todos esses dados mostram o envolvimento no sistema imune realizado pelos tripanossomas e sugerem a interferência de métodos terapêuticos.
Tripanossoma; tripanosomíase Africana; imunologia; macrófago; linfócitos; óxido nítrico; citocina; autoanticorpos
BIOMEDICAL AND MEDICAL SCIENCES
Immunology and immunopathology of African trypanosomiasis
Philippe VincendeauI; Bernard BouteilleII
IEA 3677-Laboratoire de Parasitologie, Université de Bordeaux II, 146, rue Léo Saignat, 33076 Bordeaux Cedex, France
IIEA 3174-Institut d'épidémiologie neurologique et de neurologie tropicale, Faculté de médecine, Université de Limoges, 2, rue du Docteur Marcland, 87042 Limoges Cedex, France
ABSTRACT
Major modifications of immune system have been observed in African trypanosomiasis. These immune reactions do not lead to protection and are also involved in immunopathology disorders. The major surface component (variable surface glycoprotein,VSG) is associated with escape to immune reactions, cytokine network dysfunctions and autoantibody production. Most of our knowledge result from experimental trypanosomiasis. Innate resistance elements have been characterised. In infected mice, VSG preferentially stimulates a Th 1-cell subset. A response of gd and CD8 T cells to trypanosome antigens was observed in trypanotolerant cattle. An increase in CD5 B cells, responsible for most serum IgM and production of autoantibodies has been noted in infected cattle. Macrophages play important roles in trypanosomiasis, in synergy with antibodies (phagocytosis) and by secreting various molecules (radicals, cytokines, prostaglandins,...). Trypanosomes are highly sensitive to TNF-a, reactive oxygen and nitrogen intermediates. TNF-a is also involved in cachexia. IFN-g acts as a parasite growth factor. These various elements contribute to immunosuppression. Trypanosomes have learnt to use immune mechanisms to its own profit. Recent data show the importance of alternative macrophage activation, including arginase induction. L-ornithine produced by host arginase is essential to parasite growth. All these data reflect the deep insight into the immune system realised by trypanosomes and might suggest interference therapeutic approaches.
Key words: Trypanosome, African trypanosomiasis, immunology, macrophage, lymphocytes, nitric oxide, cytokine, autoantibodies.
RESUMO
Modificações importantes no sistema imune são observadas na tripanosomíase Africana. Essas reações imunológicas não protegem e estão envolvidas em distúrbios imunopatológicos. O principal componente de superfície (glicoproteína variante de superfície, VSG) está associado à evasão das respostas imune, às disfunções da rede de citocinas e à produção de autoanticorpos. Muitos de nossos conhecimentos resultam da tripanosomíase experimental. Componentes da imunidade inata estão sendo caracterizados. Em camundongos infectados, a VSG estimula preferencialmente células Th1. Uma resposta de gd e células T CD8 aos antígenos do tripanossoma foi observada em gado tripanotolerante. Um aumento em células B CD5, responsável por IgM sérica e produção de autoanticorpos, foi observado no gado infectado. Os macrófagos desempenham importantes funções na tripanosomíase, em sinergismo com anticorpos (fagocitose) e pela secreção de várias moléculas (radicais, citocinas, prostaglandinas). Tripanossomas são altamente sensíveis ao TNF-a, espécies reativas de oxigênio e nitrogênio. O TNF-a também está envolvido em caquexia. O IFN-g atua como um fator de crescimento do parasita. Esses vários componentes contribuem para a imunossupressão. Os tripanossomas usam os mecanismos imunes para seu próprio benefício. Dados recentes mostram a importância da ativação alternativa de macrófagos, incluindo a indução pela arginase. A L-ornitina produzida pela arginase do hospedeiro é essencial para o crescimento do parasita. Todos esses dados mostram o envolvimento no sistema imune realizado pelos tripanossomas e sugerem a interferência de métodos terapêuticos.
Palavras-chave: Tripanossoma, tripanosomíase Africana, imunologia, macrófago, linfócitos, óxido nítrico, citocina, autoanticorpos.
INTRODUCTION
Sleeping sickness or human African trypanosomiasis (HAT) is an endemic parasitic disease exclusively located in intertropical Africa where it is transmitted by the tsetse fly or Glossina, its unique vector (Vickerman 1985). The new taxonomy tools used in African trypanosomes (isoenzyme characterisation, DNA analysis) have allowed scientists to separate the Trypanosoma brucei clade in several subspecies. Two are infective for humans: T.b. gambiense, and T.b. rhodesiense. These extracellular parasites are injected into humans by the bite of infected tsetse fly..
The inoculation of trypanosomes into their mammalian hosts triggers a series of events involving, at first, innate immunity and, secondarily, specific immunity. The latter requires an efficient presentation of parasitic antigens, activation of T and B cells implying specific antigen receptor recognition, and the development of effector cells and molecules. These mechanisms are highly regulated by multiple signals delivered through a large number of receptors transduced across the plasma membrane and processed. During co-evolution with their hosts, trypanosomes have learnt to cope with host immune systems, by penetrating, diverting, and altering the numerous steps leading to the generation of an effective immune response. Major modifications of immune systems have been observed in trypanosomiasis: lymphadenopathy, splenomegaly (up to thirty times the normal size) with destruction of lymphatic tissue architecture and hypergammaglobulinemia. However, their effectiveness is limited as, most of the time, parasites cannot be eliminated and immunopathological phenomena, which induce tissular alterations, appear.
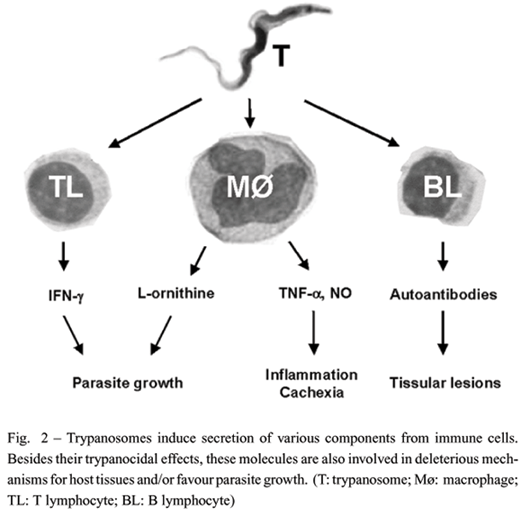
One of the major characteristics of trypanosomes is the presence of the Variant Surface Glycoprotein (VSG) which covers nearly all the membrane of trypanosomes in mammals and is the predominant surface antigen of African trypanosomes. VSG constitutes an important molecular interface between trypanosomes and the host immune system (Figure 1). VSG prevents trypanosome lysis by complement alternative pathway, and, above all, enables them to avoid the specific immune response via the phenomenon of antigenic variation (trypanosomes sequentially express antigenically distinct VSG). VSG also has several effects on immune elements such as induction of autoantibodies and cytokines, in particular tumour necrosis factor (TNF)-a (Tachado and Schofield 1994, Okomo-Assoumou et al. 1995a, Magez et al. 2002). Other trypanosome components and soluble factors, such as a trypanosome-released triggering factor (TLTF) which triggers interferon (IFN)-g production by T cells, are also involved in modulation of the immune system by acting on the synthesis of immune elements (Olsson et al. 1991, Vaidya et al. 1997) (Figure 2). Furthermore, increased levels of circulating endotoxins are a feature of human and experimental trypanosomiasis. These endotoxins, potent immunomodulatory molecules, participate to the immune disorders observed in trypanosomiasis. (Nyakundi et al. 2002). Elaboration of escape mechanisms to host immune defences and induction of parasite growth factor production are well developed by trypanosomes. In a recently discovered escape mechanism, host arginase induction, trypanosomes decrease immune response efficiency and increase the production of L-ornithine, an essential growth factor (Gobert et al. 2000, Vincendeau et al. 2003).
Understanding of the immune response was recently advanced by the discovery of the T and B subpopulations and, especially, of the T helper (Th) subsets, as well as the cytokines synthesised by each Th1 and Th2 subset. These factors control different aspects of the immune response, in peculiar the synthesis of nitric oxide, which is probably involved in several steps in the immune mechanisms. The role of gdT cells should also be taken into account as they have been implicated in other parasitic diseases such as malaria and leishmaniasis.
Most of the data concerning African trypanosomiasis have been obtained in animal diseases or experimental animal models. Few studies haveconcentrated only on the immunology of HAT. Results obtained from animal diseasesor experimental models can be investigated in human trypanosomiasis using adapted means. Genetic analysis of resistance and susceptibility to infection in inbred and congenic animal strains form the basis for research into equivalent genes in humans. Introduction of double-stranded RNA (ds RNA) into parasites induces potent and specific gene silencing a phenomenon called RNA interference (RNAi) and is a valuable tool to investigate trypanosome gene functions. The recent knowledge of the entire genome of T. brucei is an essential breakthrough to investigate immunology and immunopathology of HAT (Berriman et al. 2005).
CLINICAL SYMPTOMATOLOGY OF HAT
After a painful tsetse bite, the chancre (from several millimetres to centimetres) represents the initial lesion at the bite site, characterised by local erythema, oedema, heat, tenderness and a lack of any suppuration. Trypanosomes are present in the inflammatory tissues. The chancre disappears within 2 or 3 weeks. The disease evolves in two distinct successive phases determining its two pathological stages (Molyneux et al. 1996, Dumas and Bisser 1999). Within a few days after the tsetse bite, the patient enters the haemolymphatic stage I of the illness.
STAGE I, THE HAEMOLYMPHATIC STAGE
Clinical signs appear very early. Intermittent fever develops as a consequence of the successive waves of invasion of the blood by the trypanosomes. Adenopathies, splenomegaly, or even hepatological signs mark the invasion of the reticulo-endothelial system. Skin eruptions or trypanides are commonly observed. Severe pruritus with scratching skin lesions becomes unsupportable for the patient.
Cardiovascular alterations are less prominent, especially in the Gambian form. Irregular febrile episodes are accompanied by headaches, malaise, exhaustion, anorexia, extreme thirst, muscle and joint pains, pruritus, anaemia, rash and often deep hyperesthesia (the sign of the key of Kerandel). The lymph nodes are generally rubbery and mobile,painful at the beginning. Palpation of the subclavicular region (Winterbottom sign) is an important part of the diagnostic procedure in the Gambian form. Any adenopathy accompanied by fever should evoke the diagnosis of sleeping sickness in patients from endemic areas. Later, pruritus generalises. Oedema of the face and extremities appears early.
Few minor neurological and endocrine disorders may reveal the precocity of central nervoussystem (CNS) involvement, long before any detectable changes occur in the cerebrospinal fluid (CSF). Daytime somnolence or night-time insomnia may already be reported and electroencephalographic(EEG) tracings may reveal abnormalities. Psychiatric signs, with the alternation of irritability, changes in personality or mood affecting the daily and professional life of the patients, constitute often the first manifestation of the disease. The endocrine syndrome is marked by a permanent feeling of coldness, lack of appetite or in contrast hyperphagia, polydipsia and impotence, amenorrhea or infertility, indicative of vegetative and sexual disturbances.
STAGE II, THE MENINGOENCEPHALITIC STAGE
The meningoencephalitic stage appears slowlyand insidiously over a period of months or yearsdepending on the trypanosome. However, the clinical signs remain reversible for a long time withtreatment attesting to the predominance of potentially reversible inflammatory lesions over irreversible demyelinating lesions. The general signs ofthe haemolymphatic stage do not completely disappear: spikes of fever (but sometimes hypothermia), adenopathies and splenomegaly, cardiovascular manifestationsendocrine disturbances and typical pruritus.
The development of the neurological symptoms is progressive. As neurological signs occur already in stage I, biological criteria are the only means to confirm CNS invasion. The threshold criteria, which are commonly used, are based on CSF examination: more than 5 cells/µL and/or the presence of trypanosomes.
A wide variety of symptoms are encountered. The main symptoms from which sleeping sickness was named are daytime somnolence and nocturnal insomnia, the patients being "sleepy by day and restless by night". The sleep-wake cycle disturbances are accompanied with either or many of the following symptoms: headaches, sensory disturbances with diffuse superficial or deep sensations (muscle and bone hyperesthesia, either spontaneous or provoked; hyperpathia), presence of primitive reflexes (palm-mental reflex, sucking reflex), exaggerated deep tendon reflexes, psychiatric disorders (confusion, mood swings, agitation, aggressive behaviour, euphoria, absent gaze, mutism, indifference), and tremor (fine and diffuse without any myoclonic jerk at rest or during movement). Pyramidal alterations revealed by a Babinski sign can also be observed along with alterations in muscle tone, numbness or sensory deficit.
An abnormal number of monocytic cells is observed in the CSF. The early neurological symptoms correlate with the widespread meningeal inflammation, which occurs in both forms of HAT. The selective CNS locations explain in part the principal clinical neurological signs. Sleep-wake disturbances may result from invasion of the median eminence by parasites, which also accounts for neuro-endocrine dysfunctions with the involvement ofthe suprachiasmatic nuclei. Disorders of the sleep-wake cycle are accompanied by a state of apathy in the patient, the loss of muscle tone especially in the neck muscles and a drooping of the eyelids. Extrapyramidal symptoms signal the involvement of the striatum. Deep sensory disturbances with hyperpathia may result from the involvement of the thalamus and the early invasion of posterior spinal roots.
Apart from the disruptions of the circadian rhythm of the sleep-wake cycle, other biological rhythms are disturbed, such as body temperature, cortisol and prolactin or growth hormone secretion. The invasion of the subthalamic and hypophyseal regions account for the persistence of ad-endocrine disturbances such as impotence, amenorrhea or infertility and the development of disturbed sensations of hunger and thirst, with often hyperphagia and polydipsia in contrast to the poor general state of malnutrition of the patients. At the terminal phase of the disease, CNS demyelination and atrophy areaccompanied with disturbances in consciousnessand the development of dementia with incoherence, incontinence and epileptic fits. The patient dies in a state of cachexia and physiological misery.
GENETIC CONTROL OF TRYPANOSOMIASIS
The study of inbred and congeneic mouse strains has contributed greatly to our understanding of the genetic regulation of infectious diseases. A number of genes control infections of various pathogens by acting at the level of innate susceptibility, or at the level of acquired immunity. They may or may notbe linked to the major histocompatibility class(MHC) locus H2.
Inbred strains of mice differ in their susceptibility to infection with T. congolense, as judged by duration of survival following infection. Balb/c and A/J mice were the most susceptible and C57BL/6 the most resistant (Morrison et al. 1978). The existence of inbred susceptible and resistant strains of mice has made it possible to study the inheritance and mechanisms of host resistance. Resistance of mice to African trypanosomes is genetically determined. The control of resistance has been considered as dominant (Greenblatt et al. 1983) or recessive (De Gee et al. 1988). The use of different inbred mouse strains and trypanosome clones may explain this result (discrepancy).
In T. b. rhodesiense - infected mice, survivalwas not correlated with the height of the first peak of parasitemia, but a strong negative correlation between the second peak of parasitemia and survival time was noted (Seed and Sechelski 1995). However, in T. congolense-infected mice, the efficiency of clearance of the first peak of parasitemia was correlated with the survival period. Three loci influencing resistance of mice to T. congolense infection have been recently reported on chromosomes5, 17 and 1 (Kemp et al. 1997).
African cattle trypanosomiasis, mainly due to T. congolense and T. vivax, causes anaemia andweight loss, leading to death. Some African cattle breeds (N'Dama) are, however, able to live and be productive in endemic areas and are considered to be trypanotolerant. Trypanotolerance is genetically controlled and is an innate character, but can be increased by repeated infections. Trypanotolerant cattle are not refractory to trypanosome infections but limit proliferation of trypanosomes. Parasite counts are lower than in trypanosensitive cattle (Bos taurus, B. indicus). This resistance depends on the nutritional, physiological and stress conditions of the animal. Besides, studies of other factors in relation to host defence and survival, and especially cytokine production, have revealed that the ability to produce. IL-4 plays a role in the susceptibility to T. brucei infection (Bakhiet et al. 1996). Recently, mouse strain susceptibility to trypanosome infection has been correlated with an increase in host arginase production (Duleu et al. 2004).
Little is known about the effect of genetic polymorphism on infectious diseases in humans. Identification of human homologues for the murine genes controlling resistance and susceptibility to pathogens is in progress. Family studies should also be performed. Our knowledge of human trypanotolerance is based on reported cases (Lapeyssonnie 1960) and results from immunological screening (Lemesre et al. 1988, Authié et al. 1993). Subjects with a positive CATT (card agglutination trypanosomiasis test) were asymptomatic whereas the presence of blood parasites was observed. Moreover, although many Bantou people from Mbomo foci in the Congo were infected with T.b. gambiense, none of the pygmy population was infected. This effect, present before infection and unrelated to antibody production, is dependent on innate immunity factors. Moreover, in a recent study in the HAT focus of Sinfra (Côte d'Ivoire), single nucleotide polymorphisms within TNF-a and interleukin (IL)-10 promoters and genes were associated with susceptibility to HAT (Courtin et al. 2006).
INNATE IMMUNITY
NATURAL IMMUNITY
Normal human sera injected into T.b. brucei-infected mice caused a dramatic reduction in parasitemia (Laveran and Mesnil 1912). This phenomenon was not reproduced with the human trypanosome strains T.b. gambiense and T.b. rhodesiense. Trypanolytic factors (TLF) contained in normal human serum were identified as high-density lipoproteins (Rifkin 1978). Recently, two TLFs have been characterised in human serum. The first one (TLF1) belongs to a subclass of high-density lipoproteins and is inhibited by haptoglobin. In contrast, the second factor, TLF2, has a much higher molecular weight and does not appear to be a lipoprotein. Probably, the main trypanolytic effect is due to TLF2, which is not inhibited by haptoglobin (Raper et al. 1996). The trypanocidal effect of cape buffalo serum has been attributed to xanthine oxidase (Muranjan et al. 1997). Recently a trypanosome lysosomal protein (SRA) was found to be associated with resistanceto normal human serum. SRA is a truncated formof VSG and interacts with serum apolipoproteinL-I in the parasite lyzosome (Vanhamme and Pays 2004).
CHANCRE
The local response in the skin corresponds to the first protection developed by the host. Following inoculation of T. brucei into mammalian hosts, by the tsetse fly, a local skin reaction is induced by trypanosome proliferation and appears a few days after inoculation. In efferent lymphatic vessels, trypanosomes have been detected in lymph 1-2 days before the chancre. Their number declined during development of the chancre (6 days) and later increased. They are detected in the blood 5 days after inoculation. In T. congolense-infected sheep, neutrophils predominate in the early days and then T and B lymphocytes infiltrate the chancre. Later, T lymphocytes predominate, especially CD8+ T cells (Mwangi et al. 1990). An early response due to an increase in CD4+ and CD8+ T cells was revealed by flow cytometry in the afferent lymph draining the chancre. As the chancres regressed there was an increase in lymphoblasts and surface immunoglobulin bearing cells (Mwangi et al. 1996). During this first stage, trypanosomes expressed Variable Antigen Types (VATs) found characteristically in the tsetse fly, which changed after few days. An antibody response specific to these VATs appearedin the lymph and then in the plasma (Barry andEmergy 1984).
COMPLEMENT
Both in humans and animals, complement activation by two pathways is detected in HAT. The alternative pathway, independent of specific antibodies, was studied by the induction of trypanosome lysis (T. congolense and T.b. brucei) observed after the addition of fresh serum. Serum could induce trypanosome lysis only on uncoated VSG trypanosomes, as observed during the cycle of this parasite (procyclic forms). However, the appearance of VSG on parasites prevents trypanosome lysis by this alternative pathway (Ferrante and Allison 1983). For another strain of T.b. gambiense, it was demonstrated that the alternative pathway was incompletely activated without generation of the terminal complex (C5-C9) able to induce membrane lysis (Devine et al. 1986). The classical pathway, mediated by specific antibodies against trypanosomes, was also described and could be involved in parasite clearance by antibody-mediated lysis and/or opsonisation. The coated stages of T.b. brucei are lysed by antibodies with activation of complement by the classical pathway. Nevertheless, during these complement activations, the appearance of soluble fragments, including C3a and C5a anaphylatoxins and the C567 complex, could induce, on the one hand, the chemotactism of neutrophils and monocytes and, on the other hand, the release of amines involved in vasoconstriction and an increase in vascular permeability participating in the initial inflammatory response in the chancre.Immune complexes can also activate the complement. These immune complexes are constituted by antibodies specific to trypanosomes (e.g. anti-VSG antibody) leading to a rapid elimination of complement-fixing immune complexes (Russo et al. 1994) or by autoantibodies (see below), such as rheumatoid factor or anti-nucleic acid antibodies. These immune complexes with complement activation are also involved in some adverse effects, especially in tissue damage mediated by immune complex deposits (Nielsen 1985), such as thrombosis and glomerular involvement (Bruijn et al. 1988,van Velthuysen et al. 1994).
NATURAL KILLER CELLS
Natural killer (NK) cells have been identified asan important defence mechanism against tumourcells and intracellular pathogens, especially viruses. They are considered to belong to the lymphocyte lineage and have functions in both innate and acquired immune responses. NK cells lyse extracellular parasites. NK cells from T. cruzi-infected mice have been shown to exhibit significant activity against trypomastigotes of T. cruzi (Hatcher and Kuhn 1982).
NK cells secrete cytokines and especially IFN-g and TNF-a, which play major roles in trypanosomiasis and are regulated by cytokines which can activate or inhibit NK cell functions. NK cells also participate in the initiation of the inflammatory response, through the synthesis of chemokines.
In T. brucei-infected mice, NK activity was not modified in the early stages of infection, but was severely reduced from day 9 onwards (Askonas and Bancroft 1984). By contrast, NK cells were activated in mice infected with a natural extracellular trypanosome (T. musculi) and their critical role was demonstrated by the effects of their depletion byantiserum against asialo GM1 or their activation by polycytidylic copolymer (Albright et al. 1997).
T CELLS
Initial studies have evidenced alterations in T cell functions in trypanosomiasis, both in vivo and in vitro (Mansfield and Wallace 1974) (Figure 2). Histological examination revealed a massive B cellexpansion in the lymph nodes and spleen, which replaced the thymus-dependant area in T.b. brucei TREU 667-infected mice. These changes were seen within 7 days post-infection and persisted for at least 70 days. Moreover the role of T cells incontrolling infection was not clear (Askonas and Bancroft 1984).
Trypanosome antigen-specific T cell response was difficult to identify. In several studies, a transient proliferative T cell response to trypanosome antigens was noted in the first days of the infection followed by an absence of response (Gasbarre et al. 1980). The kinetoplastid membrane protein 11 of African trypanosomes is a potent stimulatorof T lymphocyte proliferation (Tolson et al. 1994).
In T. b. brucei - infected mice, an increasedproliferation of T cells was noted in the first days of infection in spleen and bone marrow, T blasts disappeared very rapidly. In T. congolense-infected cattle, antigen-specific proliferation of T cells was obtained with more or less difficulty according to the antigen, the T cell population and the time used. However, a strong trypanosome-specific T cell proliferation occurred in infected cattle following treatment (Emery et al. 1980, Lutje et al. 1995).
Most T cells in humans and mice bear Tabantigen receptors. These cells possess surface markers, which allow the discrimination of CD4+T cells (helper T cells) and CD8+ T cells (cytotoxic T cells). The knowledge of T cell subsets has been deeply modified by the discovery of two subsets of T helper cells, Th1 and Th2 cells. Th1 cells expressing a functional T cell response directed to VSG are generated in T.b. rhodesiense-infected mice. VSG specific T cells were found predominantly in the peritoneum. These cells did not pro-liferate but made a substantial IFN-g and IL-2 cytokine response (Schleifer et al. 1993). The cellular phenotype of VSG-responsive T cells (CD4+ CD3+) indicates that the VSG appear to preferentially stimulate a Th1 cell subset during infection.
Analysis of lymphocyte subsets in regionallymph nodes of T. congolense-infected N'Dama(trypanotolerant) and Boran (trypanosusceptible)were performed by flow cytometry. In both breeds, a significant decrease in the percentage of CD2+ and CD4+ T cells was observed, associated with an increase in the percentage of CD8+ T cells, B cells and gdT cells. VSG and two invariant antigens (33 kDa cysteine protease and 66 kDa antigen homologous to immunoglobulin heavy chain binding protein hsp70/Bip) induced in vitro proliferation and synthesis of IL-2 and IFN-g (Authié et al. 1992, Boulangé and Authié 1994, Lutje et al. 1995). No significant differences in the in vitro proliferation of lymph node cells to VSG, Concanavalin A (Con A) or hsp 70/Bip were observed between the two breeds. However, IFN-g production in response to Con A was higher in Boran at 35 days post infection.
Human and mouse immune systems contain few gdT cells, in marked contrast to those of ruminants (Hein and Mac Kay 1991). Functions of gdT cells remain largely unknown. Involvement of gdT cells in malaria and leishmaniasis has been observed (Rosat et al. 1995, Rzepczyk et al. 1997). A proliferative response of CD8+ T cells and gdT cells from trypanotolerant N'Dama to an antigen complex containing immunodominant epitopes was observed whereas a quasi absence of response was observed in trypanosusceptible Boran. The role of this gdT cell response in parasite resistance remains unclear. So, gdT cells, as CD4+ or CD8+, do not proliferate when stimulated with soluble VSG in vitro (Flynn and Sileghem 1994). It would be interesting to determine the role of cytokines synthesised by gdT cells.
Indeed, although specific T cells do not act on trypanosomes in the same way as the cytotoxic T cells in several infectious diseases such as viral infections, they markedly modify immune responses, especially by the secretion of cytokines. They greatly modify functions of B cells (antibody synthesis, isotype switch) and macrophages (antigen presentation, effector mechanisms).
B CELLS
In African trypanosomiasis, the main feature is a dramatic increase in immunoglobulin (Ig) levels(especially IgM), including trypanosome-specificantibodies and non-specific Ig production induced by cytokine activation of B cells. Some of these antibodies are also raised against autoantigens, corresponding to non-specific polyclonal activation of B-cells producing natural autoantibodies and also to antigen-driven antibodies induced by molecular mimicry (Figure 2). DNA from T.b. brucei stimulated B cell proliferation (Shoda et al. 2001). In T.b. brucei-infected mice, B lymphocytes display an aberrant activation phenotype (Sacco et al. 1994).
Antibodies specific to trypanosomes are induced by several parasite antigens, including variant and invariant VSG epitopes, as well as membrane, cytoplasmic and nuclear antigens, through T-dependent and T-independent pathways (Reinitzand Mansfield 1990). Antibodies directed against trypanosome VSG components appeared in seraand their binding to the surface coat of the trypanosomes was able to induce a decrease in parasitemia, both in the blood and extravascular spaces, specifically by immune lysis of parasites and their destruction by the Kupffer cells in the liver. Only heterologous antigenic variants ( < 0.1%) remain to repopulate the blood and tissues. Parasites are eliminated due to VSG-specific IgM (appearing at high levels, 3-4 days after infection). In contrast, VSG-specific IgG does not seem to be involved in the destruction of trypanosomes, as they appeared after the disappearance of this VAT population. Another induction of antibodies, linked to the new VSG epitopes, appeared in sera and also contributed to decrease the new VAT-specific population. The VAT-specific antibodies therefore decreased to low levels, whereas antibodies, belonging predominantly to the IgM class specific to invariant epitopes, remained at high levels. During infection, B cell non-specific stimulation was enhanced as T-independent B cell responses to the VSG successive parasitemias. In contrast, specific trypanosome B cell response, depending on T cell regulation, was depressed. Several factors may contribute to this immunosuppression. Macrophages may become unable to present antigens to T cells (by defects in antigen processing and association of epitopes with MHC Class II) and produce immunosuppressive factors as nitric oxide (NO), prostaglandins (PG), and cytokines. An increase in immunosuppressive cytokines, such as INF-g and transforming growth factor (TGF)-b, was also detected during infection. However, TGF-b is known to inhibit the production of IL-4, IL-5, IL-6, the major cytokines implied in B cell proliferation and differentiation (Fargeas et al. 1992).
Several autoantibodies are detected duringAfrican trypanosomiasis. High levels of polyclonal Igs were a marked feature of HAT. The specificity of these Igs is frequently characterised against a large range of autoantigens. Autoantibodies were directed against red blood cells (Kobayakawa et al. 1979), liver and cardiolipids (MacKenzie and Boreham 1974), nucleic acids: DNA and RNA (Kobayakawa et al. 1979, Hunter et al. 1992b), intermediate filaments (Anthoons et al. 1986) and rheumatoid factors (Kazyumba et al. 1986). Autoantibodies directed against components of CNS myelin have also been reported. They are specific for the major glycosphingolipids of myelin, the galactocerebrosides, and were detected in sera from both experimentally infected animals (Jauberteau et al. 1991) and patients from the Ivory Coast (Amevigbe et al. 1992). Other autoantibodies directed against notyet characterised proteins have been described in HAT patients (Asonganyi et al. 1989) as well as antibodies directed at myelin basic protein in experimentally infected animals (Hunter et al. 1992a). Other antibodies were raised against an epitope containing L-tryptophan, a precursor to the neurotransmetter serotonin, (Okomo-Assoumou et al. 1995b) or recognised some neuronal components of the cytoskeleton, neurofilament proteins. In some cases, these autoantibodies (anti-galactocerebrosides and anti-neurofilaments) are associated with the neurological stage of the disease and their detection in sera and CSF could contribute towards defining the neurological involvement of HAT (Courtioux et al. 2005). In vivo demyelinisation has been produced by purified antibodies to galactocerebroside (Saida et al. 1979). There are several hypotheses for the origin of these antibodies. They may be induced by a non-specific stimulation of B cells producing natural autoantibodies (Arneborn et al. 1983, Mortazavi-Milani et al. 1984). In other cases, antigen-driven autoantibodies are specific to epitopes of the causative infecting agent with molecular mimicry to self antigens, inducing a cross-reactivity to intermediate filaments (Dales et al. 1983, Fujinami et al. 1983, Davies 1997) as demonstrated for anti-neurofilament and anti-galactocerebroside antibodies which recognised respectively a flagellar component and a proteolipidic epitope of trypanosomes, and epitopes expressed by neurones (Ayed et al. 1997, Girard et al. 2000).
A subpopulation of B cells, identified by the expression of high levels of surface Igs and of CD5 in humans and Ly-1 in mice is responsible for most serum IgM (Kipps 1990). These CD5 cells produce autoantibodies, and antibodies to thymus-independent antigens. In cattle infected with T. congolense, a dramatic increase in these cells (more than four times the control value in blood) was measured and correlated with increases in serum Igs and in theabsolute number of B cells (Naessens and Williams 1992). An induction of these CD5 B cells (directly by parasite products or indirectly through the cytokine network) could account for the alteration in immunoglobulin synthesis and antibody production observed in trypanosomiasis.
MACROPHAGES
Mononuclear phagocytes play a key role in all steps of immune response in the inflammatory phase, as antigen presenting cells, in specific immunity, in synergy with antibodies and cytokines. They also can be involved in immunosuppressive and immunopathological phenomena. Quantitative, biochemical and functional changes of mononuclear phagocytes are observed in trypanosomiasis. In T. brucei-infected mice, histological examination showed a marked expansion in macrophages of the liver, spleen and bone marrow. The Kupffer cells in the liver increased in number and were often found in mitosis. The cells contained abundant phagolysosomes (vacuolated cytoplasm). An increase uptake of intravenously injected sheep red blood cells was also noted.
Macrophages are highly sensitive to environmental factors, especially microorganisms, microorganism-derived products and cytokines. A clear reduction in mannose receptors, Fc receptors, C3bi receptors (Mac-1) and F4/80 occurs by day 4 post T. b. brucei infection. The expression of MHC Class II molecules (Ia antigens) was reported to haveincreased in T. b. brucei-infected mice and decreased in T.b. rhodesiense-infected mice. The antigen-presenting function was reported to be unmodified in T. b. brucei-infected mice and defective in T. b. rhodesiense-infected mice. Macrophages react to stimuli by adapted response. They secrete many factors with various functions. They synthesise cytokines and effector mediators.
Macrophages may play an important role in protection against trypanosomes, particularly in the presence of homologous antiserum. The immunological clearance of [75Se]-methionine-labelled T. brucei in mice has been conducted to investigate the respective roles of antibodies, macrophage activation and complement in the removal of circulating parasites. The clearance was largely accomplished by antibody-mediated hepatic phagocytosis. C3 is necessary for the full opsonic activity present in murine clearance in passively immunised mice (MacAskill et al. 1980). These in vivo studies extend previous studies on the in vitro phagocytic function of macrophages in the presence of immune serum (Takayanagi et al. 1992).
As the existence of receptors for the Fc region of IgM on the macrophage membrane is still controversial, the role of IgM antibodies on trypanosome phagocytosis in the absence of complement remains unlikely. Receptor-mediated phagocytosis is enhanced during infection. It is possible that trypanosomes phagocytosed through receptors (C3b receptors, Fc receptors, etc.) or after destruction by complement-mediated lysis trigger macrophage suppressor activity, although the participation of soluble factors or another cell types cannot be ruled out.
Furthermore, macrophages from T.b. brucei-infected mice are able to synthesise reactive oxygen intermediates (ROI) after triggering by phorbol myristate acetate. Oxygen-derived species are among the most toxic products produced by macrophages. Trypanosomes are highly sensitive to these species, and in peculiar to hydrogen peroxide and hypochlorous acid, synthesised during phagocytosis.
Macrophages from trypanosome-infected mice also synthesise reactive nitrogen intermediates(RNI). Trypanosomes are highly sensitive to the cytostatic / cytotoxic effects of these compounds (Vincendeau and Daulouède 1991, Vincendeau et al. 1992). They are highly reactive radicals with short half-lives, which can react together to form potent and more stable effector molecules able to act on distant targets such as extracellular parasites. We have recently shown that T.b. gambiense are highly sensitive to S nitroso-compounds, which arenew effector molecules synthesised by activatedhuman macrophages in vitro (Mnaimneh et al.1997). Nitrosylated compounds could representnew effector molecules with a potent effect on targets distant from macrophages. In a recent study DNA from T.b. brucei have increased macrophage production of IL-12, TNF-a and NO (Shoda et al. 2001).
Macrophages are also active in secreting PGs which modulate lymphocyte and macrophage functions. During a T. b. brucei infection, the ratio of PGE2/PGF1a is reversed, with an overproduction of PGE2 (Fierer et al. 1984). Macrophages are involved in immunosuppressive mechanism, and VSG can also inhibits macrophage functions (Flynn and Sileghem 1991, Schleifer and Mansfield 1993, Coller et al. 2003).
Macrophages respond to, and synthesise, a large number of cytokines. The production of IL-1 is increased in T.b. brucei-infected mice, but this increase may be due to release rather than synthesis (Sileghem et al. 1989). In murine macrophages, VSG induces IL-1 and TNF-a synthesis. Human monocytes can also be induced by trypanosomes and secreted factors from trypanosomes to express TNF-a RNA transcripts and secrete TNF-a in culture supernatants (Daulouède et al. 2001).
Classical and alternative states of macrophage activation are observed in trypanosomiasis (Figure 2). Classical activation precedes alternative activation in murine trypanosomiasis. However, both activation states are expressed in these mice. By inducing alternative macrophage activation, trypanosomes induce host arginase which both decrease trypanocidal nitrosylated compound synthesis and increases L-ornithine production (Gobert et al. 2000). L-ornithine is the first step of polyamine synthesis, essential for parasite growth and trypanothione synthesis (Vincendeau et al. 2003).
CYTOKINES AND CHEMOKINES
A profound dysregulation of the cytokine network is observed in trypanosomiasis. The first evidence of overproduction of TNF-a/cachectin was shown in T.b. brucei-infected rabbits. (Rouzer and Cerami 1980). TNF-a is known to induce fever, asthenia, cachexia and hypertriglyceridemia. High levels of TNF-a are associated with the presence of patent inflammatory signs in the early phase of human trypanosomiasis and of major neurologic signs in the late phase (Okomo-Assoumou et al. 1995a). A persistently increased serum TNF-a level could contribute to the hypergammaglobulinemia observed in trypanosomiasis because the role of TNF-a on activation, proliferation and differentiation of B cells has already been shown (Roldan et al. 1992). Nevertheless, TNF-a participates in the mechanisms leading to trypanosome elimination: TNF-a acts indirectly in a cascade of events leading to cell activation or directly on parasites due to its cytotoxic properties (Lucas et al. 1994). Initial control of parasitemia in T.b. brucei-infected mice was diminished by the injection of anti-TNF-a antibodies (Lucas et al. 1993). VSG can trigger TNF-a production by macrophages, which are the cells, which produce the most of this molecule. Moreover, TNF-a production can be stimulated by IFN-g. IFN-g and TGF-b can be produced by CD8 T cells activated by TLTF released by T.b. brucei (Vaidya et al. 1997). TGF-b has immunosuppressive effects. An interesting fact is that IFN-g stimulates parasite growth (Olsson et al. 1991). The binding of epidermal growth factor (EGF) on T.b. brucei receptors favoured parasite growth and was one of the first cytokine-parasite interactions noted (Hide et al. 1989). All these data show that by interfering with the cytokine network and by using cytokines as growth factor, trypanosomes can completely modify the effector functions of the immune system. The effects of cytokines could also be completely different according to the presence of co-stimulators and the time period during which they are produced in trypanosome infected animals.
Chemokines also play essential roles in infectious disease control. They induce cell recruitment and activation. They induce adhesion molecules on cells of the immune system, which can bind to various cells, mainly endothelial cells, which express adapted ligands (Hickey 1999). Cytokines and chemokines can also be involved in neurological disorders (Sorensen et al. 1999). So, TNF-a has been reported to contribute to the pathophysiology of cerebral malaria. Mice chronically infected with T.b. brucei develop inflammatory lesions ofthe CNS after treatment with subcurative doses ofa trypanocidal agent (Hunter et al. 1991). Chemokines favour macrophage and lymphocyte recruitment in CNS of T.b. brucei-infected animals. The activity of these cells in precise and selective areas of CNS might induce alterations leading to various disorders, such as sleep and endocrine disorders (Lundkvist et al. 2004, Buguet et al. 1993, 2001).
The presence of TNF-a RNA transcripts inthe CNS of these mice suggests that TNF-a production could play a role in these lesions. Also, TNF-a and other cytokines contribute to the generation of somnogenic molecules such as IL-1 (Pentreath 1994). In a recent study an intra cerebral infusion of soluble type I TNF-a receptor reduced trypanosome-induced neurodegeneration (Ning et al. 2003). High levels of plasmatic IL-10 are also found in human trypanosomiasis. A number of aspects deserve further investigation: the study of all the various cytokines and soluble cytokine receptors, the possible existence of membrane or soluble cytokine receptors synthesised by the parasite, and the interaction and modulation of all these elements. Cytokines have been shown to play an essential role in the synthesis of NO, whose effects on several features of immune response have been observed over the past few years.
NITRIC OXIDE
Nitric oxide is a short-lived diatomic free radical synthesised from L-arginine by NO synthase (NOS). Calcium-dependent constitutive NOS (cNOS) release small amounts (picomoles) of NO within a short time, whereas calcium-independent inducible NOS (iNOS) release high levels (nanomoles) ofNO for a long time. Expression of iNOS in macrophages, neutrophils, hepatocytes, endothelial cells and epithelial cells is regulated at transcription level by a number of agents, including microbial products and cytokines. In vitro, murine cells produce large amounts of NO after exposure to a combination of stimuli: lipopolysaccharide (LPS), IFN-g, IL-1, TNF-a etc. Human monocytes treated by IL-4 express CD23 antigen. The crosslink of CD23 induces iNOS expression, the release of NO and various other molecules (IL-6, TNF-a, oxygen radicals, lipid mediators).
Nitric oxide is involved in the inflammatoryresponse mediated by endotoxin, cytokines or physiochemical stress. NO produced by cytokine-activated macrophages is important in host defenceand plays a crucial role in controlling infections in vivo. The role of NO and cytokines has been studied in detail in mice infected by intracellular parasitesas Leishmania. In this murine model, IFN-g synthesised by Th1 cells leads to iNOS activity, whereas IL-4 and IL-10 synthesised by Th2 cells have a suppressive effect.
NO or other nitrogen intermediates can alsoreact with the oxygen intermediates and form peroxynitrite and hydrogen radicals. Moreover, NO can form nitrosylated compounds which is able to transport and liberate NO on targets distant fromNO producing cells. Nitrosylated compounds can not only act on extracellular parasites, but also modify parasite antigens and host cell function. These compounds may have various effects (parasite killing, alteration of tissue functions such as neurotransmission, etc.) according to their localisation (spleen, liver, peritoneum, CNS, etc.). By selective inhibition of Th1 cells, NO exerts a negative feedback effect. The altered production of NO, induced by dysregulation of the cytokine network,may lead to alteration of immune response andmay also be involved in pathophysiological mechanisms. Nitrotyrosine, a marker of peroxynitrite formation, and iNOS are immunodetected in the brains of T.b. brucei-infected mice. Nitrotyrosine staining is associated with the appearance of neurological signs (Keita et al. 2000).
In HAT, nitrite production is increased at first. NO can also be stored as nitroso compounds. These NO-adducts are indirectly detected, as they induce the appearance of antibodies directed to nitrosylated antigens (Semballa et al. 2004). However, in trypanosome-infected mice, a decrease in plasmatic L-arginine leads to a decreased NO production. L-arginine is consumed by arginase, which synthetics L-ornithine and urea. L-ornithine is the precursor of polyamines and trypanothione. By inducing arginase, trypanosomes bypass NO production and benefit growth factor production. Arginase induction by parasites might be considered as a new strategy elaborated by parasites to escape host defence and benefit growth factors.
IMMUNOSUPPRESSION
The increased susceptibility of T. gambiense-infected patients to secondary infections was pointed out in the initial observations and reports of the Sleeping Sickness Commission (Low and Castellani 1903). Cellular immunity (skin tests to PPD, Candida or streptococcal antigens and sensitisation with DNCB) and humoral immunity (response to the H antigen of Salmonella typhi) were depressed in patients with HAT (Greenwood et al. 1973). In a recent study, no statistical difference was found between the prevalence of HIV infection in HAT patients and controls (Meda et al. 1995).
Immunosuppression was also observed and investigated in experimental trypanosomiasis and trypanosome - infected cattle (Ilemobade et al. 1982). In these models, immunosuppression was attributed to polyclonal B cell activation as well as the generation of suppressor T cells and suppressor macrophages. General B cell activation was noted in trypanosomiasis (hypergammaglobulinemia and a large increase of B cells in the spleen, as well as the presence of numerous Mott cells in cerebral spinal fluid and plasma cells in perivascular infiltrates), whereas specific antibody response to trypanosome antigens were reduced (Sacks and Askonas 1980).
A marked suppression of antibody responseto Brucella abortus was reported in cattle infected with T. congolense (Rurangirwa et al. 1983). Trypanosoma evansi infection in sheep delayed and depressed the increase in total cell and lymphoblast output from a lymph node draining the site of a Pasteurella haemolytica vaccine administration. These reduced outputs may limit the dissemination ofantigenic specific cells (Onah et al. 1997).
Cells, cytokines and prostaglandins have been studied in order to know their contribution, aloneor in synergy, and with or without parasitic elements, to immunosuppression mechanisms. Trypanosome membrane fragments have been foundto mimic the immunosuppressive effects of living parasites (Clayton et al. 1979). A deficient production of IL-2 and of IL-2 receptor expression has been shown in several models (Alcino and Fresno 1985, Sileghem et al. 1989). The roles of macrophage-derived factors, especially prostaglandinsand IFN-g secreted by CD8+ T cells in the suppression of IL-2 receptor expression on CD4+ and CD8+ T cells, were also shown (Darji et al. 1993). Besides its action on the Th1 subset, rather than the Th 2 subset, NO also acts on other elements, favouring immunosupression.
IMMUNOINTERVENTION
The resistance of mice to African trypanosomescan be increased non-specifically by immunostimulants such as Calmette-Guérin bacilli and Propionibacterium acnes (Murray and Morrison1979, Black et al. 1989). These immunostimulants are considered to activate macrophages. P. acnes-treated macrophages inhibited T. brucei growth in vitro (Black et al. 1989).
An acquired resistance has been observedin trypanocide-treated cattle. In a cohort study in Zaire during the 10 year observation period of adults previously diagnosed and treated for HAT,the risk of a second episode of HAT was greatly reduced compared to the risk of a first episode in previously undiagnosed adults (Khonde et al. 1995). Induction of protective immunity by vaccination is an important goal to control infectious diseases.However, a vaccine must be very effective and not only delay development of the disease, but also include a large number of antigenic variants. A major aim is to identify antigen(s) that elicit a protective immune response in trypanosomiasis. Identifying molecules inducing durable protection may lead to their production as recombinant antigens. Nucleic acid vaccines represent a new promising approach. They are able to induce all the elements of the specific immune response unlike killed microorganisms or defined protein. Studies using dead or living trypanosomes, soluble-released antigens, purified VSG and irradiated parasites have shown that protection is restricted to the VSG-specific epitopes (Pays 1995). Strategies using invariant antigens (particularly those in the flagellar pocket) may be very worthwhile (Olecnick et al. 1988). Furthermore, in experimental murine trypanosomiasis, vaccination based on glycosylphosphatidyl (GPI)anchor of VSG can prevent TNF-a associatedimmunopathology and decrease disease severity(Magez et al. 2002). TNF-a associated immunopathology may also be prevented by selective inhibitors of macrophage functions (Mamani-Matsuda et al. 2004).
CONCLUSION
The knowledge of host and parasite genomes andall immune response elements can help in understanding immune mechanisms (natural and acquired) developed in trypanosomiasis. These mechanisms are triggered starting with the first contact of hosts with trypanosomes and chancre formations. One of the initial questions which needs to be addressed is the influence of parasite inoculation, and especially of Glossina saliva elements, on the modulation of both innate and specific immune response of hosts. Studies have shown that tick saliva contains innate immunity inhibitors (complement, NK cells, etc.), reduces macrophage cytokine elaboration, and impairs the earliest stages of specific immunity (Wikel 1996). At the onset of specific response, IL-12 plays a major role in T cell shift towards a Th1 response. It would be interesting to investigate its role, as well as all the elements composing the cytokine network. Indeed, the presence of parasite molecules interacting with these elements, which indicates that a parasite can deeply penetrate the host immune system, might lead to paradoxical and perverse effects on immune response.
Studies done on transgenic knock-in and knock-out mice have produced many major findings in the understanding of infectious diseases. It has been shown, for instance, that the disruption of IL-4 gene in T.b. brucei-infected B 10.Q mice alters the control of parasitemia and the production of anti-VSG antibodies, though shortening their lifeexpectancy (Bakhiet et al. 1996). The use of different gene promoters/enhancers may contribute to defining the role of a transgene in every type of tissue. The various immune effector mechanisms found to be efficient in controlling trypanosomiasis (antibodies, complement, phagocytosis, TNF-a, NO, etc.) might be effective on one organ, but ineffective elsewhere. Moreover, the chronology of each state of activation of a cell might be tightly regulated to allow an efficient immune response and avoid immune disorders. New data from genome, transcriptome and proteome analysis might help in understanding the sequence of all events involvedin resistance/susceptibilityy to trypanosome and in disease development. Further research on appropriate immunological means, associated with chemotherapeutic agents, might be useful in curing chemotherapy-resistant trypanosomiasis.
Manuscript received on September 27, 2005; accepted for publication on October 5, 2005; presented by LUCIA MENDONÇA PREVIATO
Correspondence to: Philippe Vincendeau
- ALBRIGHT JW, JIANG D AND ALBRIGHT JF. 1997. Innate control of the early course of infection in mice inoculated with Trypanosoma musculi Cell Immunol 176: 146-152.
- ALCINO O AND FRESNO M. 1985. Suppressor factor of T-cell activation and decreased interleukin 2 activity in experimental African trypanosomiasis. Infect Immun 50: 382-387.
- AMEVIGBE M, JAUBERTEAU-MARCHAN MO, BOUTEILLE B, DOUA F, BRETON JC, NICOLAS JA AND DUMAS M. 1992. Human African trypanosomiasis: presence of antibodies to galactocerebrosides. Am J Trop Med Hyg 45: 652-662.
- ANTHOONS JAMS, VAN MARCK EAE AND GIGASE PLJ. 1986. Autoantibodies to intermediate filaments in experimental infections with Trypanosoma brucei gambiense Z Parasitenkd 72: 443-452.
- ARNEBORN P, BIBERFELD G, FORSGREN M AND VON STEDINGK LV. 1983. Specific and non specific B-cell activation in measles and varicella. Clin Exp Immunol 51: 165-172.
- ASKONAS BA AND BANCROFT GJ. 1984. Interaction of African trypanosomes with the immune system. Philos Trans R Soc Lond [Biol] 307: 41-50.
- ASONGANYI T, LANDO G AND NGU JL. 1989. Serum antibodies against human brain myelin proteins in Gambian trypanosomiasis. Ann Soc Belge Med Trop 69: 213-221.
- AUTHIÉ E, MUTETI DK, MBAWA ZR, LONSDALE-ECCLES JD, WEBSTER P AND WELLS CW. 1992. Identification of a 33-kilodalton immunodominant antigen of Trypanosoma congolense as a cysteine protease. Mol Biochem Parasitol 56: 103-116.
- AUTHIÉ E ET AL. 1993. Some new prospects in epidemiology and fight against human African trypanosomiasis. Res Rev Parasitol 51: 29-46.
- AYED Z, BRINDEL I, BOUTEILLE B, VAN MEIRVENNE N, DOUA F, HOUINATO D, DUMAS M AND JAUBERTEAU MO. 1997. Detection and characterization of autoantibodies directed against neurofilament proteins in human African trypanosomiasis. Am J Trop Med Hyg 57: 1-6.
- BAKHIET M, JANSSON L, BÜSCHER P, HOLMDAHL R, KRISTENSSON K AND OLSSON T. 1996. Control of parasitemia and survival during Trypanosoma brucei brucei infection is related to strain-dependent ability to produce IL-4. J Immunol 157: 3518-3526.
- BARRY JD AND EMERGY DL. 1984. Parasite development and host responses during the establishment of Trypanosoma brucei infection transmitted by tsetse fly. Parasitology 88: 67-84.
- BERRIMAN M, GHEDIN E, HERTZ-FOWLER C, BLANDIN G, RENAULD H, BARTHOLOMEU DC, LENNARD NJ, CALER EL AND HAMLIN NE. 2005. The genome of the African trypanosome Trypanosoma brucei Science 309: 416-422.
- BLACK SJ, MURRAY M, SHAPIRO SZ, KAMINSKY R, BOROWY NK, MUSANGA R AND OTIENO-OMONDI F. 1989. Analysis of Propionibacterium acnes-induced non-specific immunity to Trypanosoma brucei in mice. Parasite Immunol 11: 371-383.
- BOULANGÉ A AND AUTHIÉ E. 1994. A 69 kDa immunodominant antigen of Trypanosoma (Nannomonas) congolense is homologous to immunoglobulin heavy chain binding protein (BiP). Parasitology 109: 163-173.
- BRUIJN JA, OEMAR BS, EHRICH JH AND FLEUREN GJ. 1988. Immune complex formation in the kidney: recent observations in experimental trypanosomiasis. Ann Soc Belge Med Trop 68: 11-14.
- BUGUET A, BERT J, TAPIE P, TABARAUD F, DOUA F, LONSDORFER J, BOGUI P AND DUMAS M. 1993. Sleep-wake cycle in human African trypanosomiasis. J Clin Neurophysiol 10: 190-196.
- BUGUET A, BOURDON L, BOUTEILLE B, CESPUGLIO R, VINCENDEAU P, RADOMSKI MW AND DUMAS M. 2001. The duality of sleeping sickness: focusing on sleep. Sleep Med Rev 5: 139-153.
- CLAYTON CE, SACKS DL, OGILVIE BM AND ASKONAS BA. 1979. Membrane fractions of trypanosomes mimic the immunosuppressive and mitogenic effects of living parasites on the host. Parasite Immunol 1: 241-249.
- COLLER S, MANSFIELD P, PAULNOCK JM AND DONNA M. 2003. Glycosylinositolphosphate soluble variant surface glycoprotein inhibits IFN-gamma-induced nitric oxide production via reduction inSTAT1 phosphorylation in African trypanosomiasis. J Immunol 171: 1466-1472.
- COURTIN D, ARGIRO L, JAMONNEAU V, N'DRI L, N'GUESSAN P, ABEL L, DESSEIN A, COT M, LAVEISSIÈRE C AND GARCIA A. 2006. Interest of tumor necrosis factor-alpha -308 G/A and interleukin-10 -592 C/A polymorphisms in human African trypanosomiasis. Infect Genet Evol 6: 123-129.
- COURTIOUX B, BISSER S, M'BELESSO P, NGOUNGOU E, GIRARD M, NANGOUMA A, JOSENANDO T, JAUBERTEAU-MARCHAN MO AND BOUTEILLE B. 2005. Dot-ELISA for a more reliable human African trypanosomiasis staging. J Clin Microbiol 43: 4789-4795.
- DALES S, FUJINAMI RS AND OLDSTONE MBA. 1983. Infection with vaccinia favors the selection of hybridomas synthesizing autoantibodies against intermediate filaments, one of them cross-reacting with the virus hemagglutinin. J Immunol 131: 1546-1553.
- DARJI A, SILEGHEM M, HEREMANS H, BRYS L AND DE BAETSELIER P. 1993. Inhibition of T-cell responsiveness during experimental infections withTrypanosoma brucei: active involvement of endogenous IFN-g. Infect Immun 61: 3098-3102.
- DAULOUÈDE S, BOUTEILLE B, MOYNET D, DE BAETSELIER P, COURTOIS P, LEMESRE JL, BUGUET A, CESPUGLIO R AND VINCENDEAU P.2001. Human macrophage tumor necrosis factor(TNF)-alpha production induced by Trypanosomabrucei gambiense and the role of TNF-alpha in parasite control. J Infect Dis 183: 988-991.
- DAVIES JM. 1997. Molecular mimicry: can epitope mimicry induce autoimmune disease? Immunol Cell Biol 75: 113-126.
- DE GEE AL, LEVINE RF AND MANSFIELD JM. 1988. Genetics of resistance to the African trypanosomes. VI. Heredity of resistance and variable surface glycoprotein-specific immune responses. J Immunol 140: 283-288.
- DEVINE DV, FALK RJ AND BALBER AE. 1986. Restriction of the alternative pathway of human complement by intact Trypanosoma brucei subsp. gambiense Infect Immun 52: 223-229.
- DULEU S, VINCENDEAU P, COURTOIS P, SEMBALLA S, LAGROYE I, DAULOUÈDE S, BOUCHER JL,WILSON KT, VEYRET B AND GOBERT PA. 2004. Mouse strain susceptibility to trypanosome infection: an arginase-dependent effect. J Immunol 172: 6298-6303.
- DUMAS M AND BISSER S. 1999. Clinical aspects of human African trypanosomiasis. In: DUMAS M, BOUTEILLE B AND BUGUET A (Eds), Progress in human African trypanosomiasis, sleeping sickness, Paris: Springer Verlag, p. 215-233.
- EMERY DL, WELLS PW AND TENYWA T. 1980. Trypanosoma congolense: specific transformation invitro of leukocytes from infected or immunised cattle. Exp Parasitol 50: 358-368.
- FARGEAS C, WU CY, NAKAJIMA T, COX D, NUTMAN T AND DELESPESSE G. 1992. Differential effects of transforming growth factor b on the synthesis of Th1 and Th2-like lymphokines by human T lymphocytes. Eur J Immunol 22: 2173-2176.
- FERRANTE A AND ALLISON AC. 1983. Alternative pathway activation of complement by African trypanosomes lacking a glycoprotein coat. Parasite Immunol 5: 491-498.
- FIERER J, SALMON JA AND ASKONAS BA. 1984. African trypanosomiasis alters prostaglandin production by murine peritoneal macrophages. Clin Exp Immunol 58: 548-556.
- FLYNN JN AND SILEGHEM M. 1991. The role of the macrophage in induction of immunosuppression in Trypanosoma congolense-infected cattle. Immunology 74: 310-316.
- FLYNN JN AND SILEGHEM M. 1994. Involvement of gdT cells in immunity to trypanosomiasis. Immunology 83: 86-92.
- FUJINAMI RS, OLDSTONE MBA, WROBLEWSKA Z, FRANKEK ME AND KOPROWSKI H. 1983. Molecular mimicry in virus infection: cross-reaction of measles virus phosphoprotein or herpes simplex virus protein with human intermediate filaments. Proc Natl Acad Sci USA 80: 2346-2350.
- GASBARRE LC, HUG K AND LOUIS JA. 1980. Murine T lymphocyte specificity for African trypanosomes. I. Induction of a T lymphocyte dependent proliferative response to Trypanosoma brucei Clin Exp Immunol 41: 97-106.
- GIRARD M, BISSER S, BÜSCHER P, BOUTEILLE B, PREUD'HOMME JL AND JAUBERTEAU MO. 2000. Cross-reactivity of galactocerebroside autoantibodies with a Trypanosoma brucei proteolipidic epitope. Clin Exp Immunol 119: 516-522.
- GOBERT A, DAULOUÈDE S, LEPOIVRE M, BOUCHER JL, CESPUGLIO R, BUGUET A, BOUTEILLE B, VEYRET B AND VINCENDEAU P. 2000. L-arginine availability modulates local NO production and parasite killing in experimental trypanosomiasis. Infect Immun 68: 4653-4657.
- GREENBLATT HC, DIGGS CL AND AIKAWA M. 1983. Antibody-dependent phagocytosis of Trypanosoma rhodesiense by murine macrophages. Am J Trop Med Hyg 32: 34-45.
- GREENWOOD BM, WHITTLE HC AND MOLYNEUX DH. 1973. Immunosuppression in Gambian trypanosomiasis. Trans R Soc Trop Med Hyg 67: 846-850.
- HATCHER FM AND KUHN RE. 1982. Destruction of T. cruzi by natural killer cells. Science 218: 295-296.
- HEIN WR AND MAC KAY CR. 1991. Prominence of gdT cells in the ruminant immune system. Immunol Today 12: 30-31.
- HICKEY WF. 1999. Leukocyte traffic in the central nervous system: the participants and their roles. Semin Immunol 11: 125-137.
- HIDE G, GRAY A, HARRISON CM AND TAIT A. 1989. Identification of an epidermal growth factor receptor homologue in trypanosomes. Mol Biochem Parasitol 36: 51-59.
- HUNTER CA, COW JW, KENNEDY PGE, JENNINGS FW AND MURRAY M. 1991. Immunopathology of experimental African sleeping sickness: detection of cytokine mRNA in the brains of Trypanosoma brucei brucei-infected mice. Infect Immun 59: 4636-4640.
- HUNTER CA, JENNINGS FW, KENNEDY PGE AND MURRAY M. 1992a. Astrocyte activation correlates with cytokine production in central nervous system of Trypanosoma brucei brucei-infected mice. Lab Invest 67: 635-642.
- HUNTER CA, JENNINGS FW, TIERNEY JF, MURRAY M AND KENNEDY PGE. 1992b. Correlation of autoantibody titres with central nervous system pathology in experimental African trypanosomiasis. J Neuroimmunol 41: 143-148.
- ILEMOBADE AA, ADEGBOYE DS, ONOVIRAN O AND CHIMA JC. 1982. Immunodepressive effects of trypanosomal infection in cattle immunized against contagious bovine pleuropneumonia. Parasite Immunol 4: 273-282.
- JAUBERTEAU MO, BEN YOUNES-CHENOUFI A, AMEVIGBE M, BOUTEILLE B, DUMAS M, BRETON JC AND BAUMANN N. 1991. Galactocerebrosides are antigens for immunoglobulins in sera of an experimental model of trypanosomiasis in sheep. J Neurol Sci 101: 82-86.
- KAZYUMBA G, BERNEY M, BRIGHOUSE G, CRUCHAUD A AND LAMBERT PH. 1986. Expression of B cell repertoire and autoantibodies in human African trypanosomiasis. Clin Exp Immunol 65: 10-18.
- KEITA M, VINCENDEAU P, BUGUET A, CESPUGLIO R, VALLAT JM, DUMAS M AND BOUTEILLE B. 2000. Inducible nitric oxide synthase and nitrotyrosine in the central nervous system of mice chronically infected with Trypanosoma brucei brucei Exp Parasitol 95: 19-27.
- KEMP SJ, IRAQI F, DARVASI A, SOLLER M AND TEALE AJ. 1997. Localization of genes controlling resistance to trypanosomiasis in mice. Nat Genet 16: 194-196.
- KHONDE N, PEPIN J, NIYONSENGA T, MILORD F AND DE WALS P. 1995. Epidemiological evidence for immunity following Trypanosoma brucei gambiense sleeping sickness. Trans R Soc Trop Med Hyg 89: 607-611.
- KIPPS TJ. 1990. The CD5 B cell. Adv Immunol 47: 117-185.
- KOBAYAKAWA T, LOUIS J, SHOZO I AND LAMBERT PH. 1979. Autoimmune response to DNA, red cells, and thymocyte antigens in association with polyclonal antibody synthesis during experimental African trypanosomiasis. J Immunol 122: 296-301.
- LAPEYSSONNIE L. 1960. Seconde note sur un cas exceptionnel de trypanosomiase. Présence de parasites pendant 21 ans, sans signes cliniques appréciables, chez une femme traitée sans succčs pendant 10 ans. Bull Soc Path Exot 53: 20-32.
- LAVERAN A AND MESNIL F. 1912. Trypanosomes et trypanosomiases. Paris: Masson, p. 118-154.
- LEMESRE JL, NOIREAU F, MAKOUNDOU ML, LOUEMBET MT AND FRÉZIL JL. 1988. Apport des techniques sérologiques dans l'analyse du liquide céphalo-rachidien de patients congolais atteints de la maladie du sommeil. Bull Soc Path Exot 81: 506-510.
- LOW GC AND CASTELLANI A. 1903. Report of the Sleeping Sickness Commission. Proc R Soc London 2: 14.
- LUCAS R, MAGEZ S, SONGA B, DARJI A, HAMERS R AND DE BAETSELIER P. 1993. A role for TNF during African trypanosomiasis: involvement in parasite control, immunosuppression and pathology. Res Immunol 144: 370-375.
- LUCAS R, MAGEZ S, DE LEYS R, FRANSEN L, SSHEERLINCK JP, RAMPELBERG M, SABLON E AND DE BAETSELIER P. 1994. Mapping the lectin-like activity of tumor necrosis factor. Science 263: 814-817.
- LUNDKVIST GB, KRISTENSSON K AND BENTIVOGLIO M. 2004. Why trypanosomes cause sleeping sickness? Physiology 19: 198-206.
- LUTJE V, MERTENS B, BOULANGÉ A, WILLIAMS DJ AND AUTHIÈ E. 1995. Trypanosoma congolense: proliferative responses and interleukin production in lymph node cells of infected cattle. Exp Parasitol 81: 154-164.
- MACASKILL JA, HOLMES PH, WHITELAW DD, MCCONNEL JM, JENNINGS EW AND URQUHART GM. 1980. Immunological clearance of 75Se-labelled Trypanosoma brucei in mice. II. Mechanisms in immune animals. Immunology 40: 629-635.
- MACKENZIE AR AND BOREHAM PLF. 1974. Autoimmunity in trypanosome infections. I. Tissue autoantibodies in Trypanosoma (Trypanozoon) brucei infections of the rabbit. Immunology 26: 1225-1238.
- MAGEZ S, STIJLEMANS B, BARAL TOYA AND DE BAETSELIER P. 2002. DNA from protozoan parasites Babesia bovis, Trypanosoma cruzi, and T. brucei is mitogenic for B lymphocytes and stimulates macrophage expression of interleukin-12, tumor necrosis factor-alpha, and nitric oxide. Microbes Infect 4: 999-1006.
- MAMANI-MATSUDA M, RAMBERT J, LEJOLY-BOISSEAU H, DAULOUÈDE S, MALVY D, THIOLAT D, COVES S, COURTOIS P, VINCENDEAU P AND MOSSALAYI MD. 2004. Quercetin induces theapoptosis of Trypanosoma brucei gambiense anddecreases the pro-inflammatory response of human macrophages. Antimicrob Agents Chemother 48: 924-929.
- MANSFIELD JM AND WALLACE JH. 1974. Suppression of cell-mediated immunity in experimental African trypanosomiasis. Infect Immun 10: 335-339.
- MEDA HA, DONA F, LAVEISSIÈRE C, MIEZAN TW, GAENS F, BRATTEGAARD K, DE MUYRCK A AND DE COCK KM. 1995. Human immunodeficiency virus infection and human African trypanosomiasis: a case control study in Côte d'Ivoire. Trans R Soc Trop Med Hyg 89: 639-643.
- MNAIMNEH S, GEFFARD M, VEYRET B AND VINCENDEAU P. 1997. Albumin nitrosylated by activated macrophages possesses antiparasitic effectsneutralized by anti-NO-acetylated-cysteine antibodies. J Immunol 158: 308-314.
- MOLYNEUX DH, PENTREATH V AND DOUA F. 1996. African trypanosomiasis in man. In: COOK GC (Ed), Manson's tropical Diseases, 20th ed., London (UK): WB Saunders Company Ltd, p. 1183-1196.
- MORRISON WI, ROELANTS GE, MAYOR-WITHEY KS and MURRAY M. 1978. Susceptibility of inbred strains of mice to Trypanosoma congolense: correlation with changes in spleen lymphocyte populations. Clin Exp Immunol 32: 25-40.
- MORTAZAVI-MILANI SM, FACER CA AND HOLBOROW EJ. 1984. Induction of anti-intermediate filament antibody in rabbit experimentally infected with Trypanosoma brucei brucei Immunology 52: 423-426.
- MURANJAN M, WANG Q, LI YL, HAMILTON E, OTIENO-OMONDI FP, WANG J, VAN PRAAGH A, GROOTENHUS JG AND BLACK SJ. 1997. The trypanocidal cape buffalo serum protein is xanthineoxidase. Infect Immun 65: 3806-3814.
- MURRAY M AND MORRISON WI. 1979. Non-specific induction of increased resistance in mice to Trypanosoma congolense and T. brucei by immunostimulants. Parasitology 79: 349-366.
- MWANGI DM, HOPKINS J AND LUCKINS AG. 1990. Cellular phenotypes in Trypanosoma congolense infected sleep: the local skin reaction. Parasite Immunol 12: 647-658.
- MWANGI DM, HOPKINS J AND LUCKINS AG. 1996. Trypanosoma congolense infection in sheep: cellular phenotypes in lymph and lymph nodes associated with skin reactions. J Comp Path 114: 51-61.
- NAESSENS J AND WILLIAMS DJL. 1992. Characterization and measurement of CD5+ B cells in normal and Trypanosoma congolense-infected cattle. Eur J Immunol 22: 1713-1718.
- NIELSEN KH. 1985. Complement in trypanosomiasis. In: TIZARD I (Ed), Immunology and pathogenesis of trypanosomiasis, Boca Raton (Florida, USA): CRC Press, p. 133-144.
- NING Q, LINGLI H AND WENMIN L. 2003. Intraventricular infusion of antagonists of IL-1 and TNF-a attenuates neurodegeneration induced by the infection of Trypanosoma brucei J Neuroimmunol 138: 92-98.
- NYAKUNDI JN, CRAWLEY B, SMITH RA AND PENTREATH VW. 2002. The relationships between intestinal damage and circulating endotoxins in experimental Trypanosoma brucei brucei infections. Parasitology 124: 589-595.
- OKOMO-ASSOUMOU MC, DAULOUÈDE S, LEMESRE JL, N'ZILA-MOUANDA A AND VINCENDEAU P. 1995a. Correlation of high serum levels of tumor necrosis factor-a with disease severity in human African trypanosomiasis. Am J Trop Med Hyg 53: 539-543.
- OKOMO-ASSOUMOU MC, GEFFARD M, DAULOUÈDE S, CHAUGIER C, LEMESRE JL AND VINCENDEAU P. 1995b. Circulating antibodies directed against tryptophan-like epitopes in sera of patients with human African trypanosomiasis. Am J Trop Med Hyg 52: 461-467.
- OLECNICK JG, WOLFE R, NAYMAN RK AND MCLAUGHLIN J. 1988. A flagellar pocket membrane fraction from Trypanosoma brucei rhodesiense Immunogold localization and nonvariant immunoprotection. Infect Immun 56: 92-98.
- OLSSON T, BAKHIET M, EDLUND C, HOJEBERG B, VAN DER MEIDE PH AND KRISTENSSON K. 1991. Bidirectional activating signals between Trypanosoma brucei and CD8+ T cells: a trypanosome-released factor triggers interferon-g production that stimulates parasite growth. Eur J Immunol 21: 2447-2454.
- ONAH DN, HOPKINS J AND LUCKINS AG. 1997. Effects of Trypanosoma evansi on the output of cells from a lymph node draining the site of Pasteurella haemolytica vaccine administration. J Comp Pathol 117: 73-82.
- PAYS E. 1995. La variation antigénique et le problčme du vaccin contre les trypanosomes africains. Bull Mem Acad Roy Med Belg 150: 123-135.
- PENTREATH VW. 1994. Trypanosomiasis and the nervous system. Pathology and immunology. Trans R Soc Trop Med Hyg 89: 9-15.
- RAPER J, NUSSENZWEIG V AND TOMLINSON S. 1996. The main lytic factor of Trypanosoma brucei brucei in normal human serum is not high density lipoprotein. J Exp Med 183: 1023-1029.
- REINITZ DM AND MANSFIELD JM. 1990. T-cell-independent and T-cell-dependent B-cell responses toexposed variant surface glycoprotein epitopes intrypanosome - infected mice. Infect Immun 58:2337-2342.
- RIKFIN MR. 1978. Identification of the trypanocidal factor in normal human serum/high-density lipoprotein. Proc Natl Acad Sci USA 75: 3450-3454.
- ROLDAN E, RODRIGUEZ C, NAVAS G, PARRA C AND BRIEVA JE. 1992. Cytokine network regulating terminal maturation of human bone narrow B cells capable of spontaneous and high rate of Ig secretion in vitro J Immunol 149: 2367-2371.
- ROSAT JP, CONCEIÇÃO-SILVA F, WAANDERS GA, BEERMANN F, WILSON A, OWEN MJ, HAYDAY AC, HUANG S, AGUET M AND MACDONALD HR. 1995. Expansion of gamma delta+ T cells in BALB/c mice infected with Leishmania major is dependent upon Th2-type CD4+ T cells. Infect Immun 63: 3000-3004.
- ROUZER CA AND CERAMI A. 1980. Hypertriglyceridemia associated with Trypanosoma brucei brucei infection in rabbits: role of defective triglyceride removal. Mol Biochem Parasitol 2: 31-38.
- RURANGIRWA FR, MUSOKE AJ, NANTULYA VM AND TABEL H. 1983. Immune depression in bovine trypanosomiasis effects of acute and chronic Trypanosoma congolense and chronic Trypanosoma vivax infections on antibody response to Brucella abortus vaccine. Parasite Immunol 5: 207-276.
- RUSSO DC, WILLIAMS DJ AND GRAB DJ. 1994. Mechanisms for the elimination of potentially lytic complement-fixing variable surface glycoprotein antibody-complexes in Trypanosoma brucei Parasitol Res 80: 487-492.
- RZEPCZYK CM, ANDERSON K, STAMATIOU S, TOWNSEND E, ALLWORTH A, MCCORMACK J AND WHITBY M. 1997. gd T cells: their immunobiology and role in malaria infections. Int J Parasitol 27: 191-200.
- SACCO RE, HAGEN M, DONELSON JE AND LYNCH RG. 1994. B lymphocytes of mice display an aberrant activation phenotype and are cell cycle arrested in G0/G1A during acute infection with Trypanosoma brucei J Immunol 153: 1714-1723.
- SACKS DL AND ASKONAS BA. 1980. Trypanosome-induced suppression of antiparasite responses during experimental African trypanosomiasis. Eur J Immunol 10: 971-974.
- SAIDA K, SAIDA T, ALVINGL GR, BROWN MJ, SILBERGERG DH AND ASBURY AK. 1979. In vivo demyelinisation produced by purified antibodies to galactocerebroside. J Neuropathol Exp Neurol 38: 338-348.
- SCHLEIFER KW AND MANSFIELD JM. 1993. Suppressor macrophages in African trypanosomiasis inhibitT cell proliferative responses by nitric oxide andprostaglandins. J Immunol 151: 5492-5503.
- SCHLEIFER KW, FILUTOWICZ LR, SCHOPF LR AND MANSFIELD JM. 1993. Characterization of T helper cell responses to the trypanosome variant surface glycoprotein. J Immunol 150: 2910-2919.
- SEED JR AND SECHELSKI J. 1995. The inheritance of factors controlling resistance in mice infected with Trypanosoma brucei rhodesiense J Parasitol 81: 653-657.
- SEMBALLA S, GEFFARD M, DAULOUÈDE S, VEYRET B, LEMESRE JL, HOLZMULER P, MALVY D AND VINCENDEAU P. 2004. Antibodies directed against nitrosylated neoepitopes in sera of patients with human African trypanosomiasis. Trop Med Int Hlth 9: 1104-1110.
- SHODA LK, KEGERREIS KA, SUAREZ CE, RODITI I, CORRAL RS, BERTOT GM, NORIMINE J AND BROWN WC. 2001. DNA from protozoan parasites Babesia bovis, Trypanosoma cruzi, and T. brucei is mitogenic for B lymphocytes and stimulates macrophage expression of interleukin-12, tumor necrosis factor alpha, and nitric oxide. Infect Immun 69: 2162-2171.
- SILEGHEM MR, DARJI A, HAMERS R AND DE BAETSELIER P. 1989. Modulation of IL-1 production and IL-1 release during experimental trypanosome infections. Immunology 68: 137-139.
- SORENSEN TL ET AL. 1999. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest 103: 807-815.
- TACHADO DS AND SCHOFIELD L. 1994. Glycosylphosphatidylinositol toxin of Trypanosoma bruceiregulates Il-1a and TNF-a expression in macrophages by protein tyrosine kinase mediated signal transduction. Biochem Biophys Res Comm 205: 984-991.
- TAKAYANAGI T, KAWAGUCHI H, YABU Y, ITOH M AND YANO K. 1992. Inhibition of IgM antibody-mediated aggregation of Trypanosoma gambiense in the presence of complement. Experientia 48: 1002-1006.
- TOLSON DL, JARDIM A, SCHNUR LF, STEBECK C, TUCKEY C, BEECROFT RP, TEH HS, OLAFSON RW AND PEARSON TW. 1994. The kinetoplastid membrane protein 11 of Leishmania donovani and African trypanosomes is a potent stimulator of T-lymphocyte proliferation. Infect Immun 62: 4893-4899.
- VAIDYA T, BAKHIET M, HILL KL, OLSSON T, KRISTENSSON K AND DONELSON JE. 1997. The gene for a lymphocyte triggering factor from African trypanosomes. J Exp Med 186: 433-438.
- VAN VELTHUYSEN ML, MAYEN AE, PRINS FA, DE HEER E, BRUIJN JA AND FLEUREN GJ. 1994.Phagocytosis by glomerular endothelial cells in infection-related glomerulopathy. Nephrol Dial Transplant 9: 1077-1083.
- VANHAMME L AND PAYS E. 2004. The trypanosome lytic factor of human serum and the molecular basis of sleeping sickness. Int J Parasitol 34: 887-898.
- VICKERMAN K. 1985. Developmental cycles and biology of pathogenic trypanosomes. Brit Med Bull 41: 105-114.
- VINCENDEAU P AND DAULOUÈDE S. 1991. Macrophage cytostatic effect on Trypanosoma musculiinvolves an L-arginine-dependent mechanism. J Immunol 146: 4338-4343.
- VINCENDEAU P, DAULOUÈDE S, VEYRET B, DARDÉ ML, BOUTEILLE B AND LEMESRE JL. 1992. Nitric oxide-mediated cytostatic activity on Trypanosoma brucei gambiense and Trypanosoma bruceibrucei Exp Parasitol 75: 353-360.
- VINCENDEAU P, GOBERT AP, DAULOUÈDE S, MOYNET D AND MOSSALAYI MD. 2003. Arginases in parasitic diseases. Trends Parasitol 19: 9-12.
- WIKEL SK. 1996. Tick modulation of host cytokines. Exp Parasitol 84: 304-309.
Publication Dates
-
Publication in this collection
28 Nov 2006 -
Date of issue
Dec 2006
History
-
Accepted
05 Oct 2005 -
Received
27 Sept 2005