Abstracts
Portal hypertension is the most common complication of chronic liver diseases, such as cirrhosis. The increased intrahepatic vascular resistance seen in hepatic disease is due to changes in cellular architecture and active contraction of stellate cells. In this article, we review the historical aspects of the kallikrein-kinin system, the role of bradykinin in the development of disease, and our main findings regarding the role of this nonapeptide in normal and experimentalmodels of hepatic injury using the isolated rat liver perfusion model (mono and bivascular) and isolated liver cells. We demonstrated that: 1) the increase in intrahepatic vascular resistance induced by bradykinin is mediated by B2 receptors, involving sinusoidal endothelial and stellate cells, and is preserved in the presence of inflammation, fibrosis, and cirrhosis; 2) the hepatic arterial hypertensive response to bradykinin is calcium-independent and mediated by eicosanoids; 3) bradykinin does not have vasodilating effect on the pre-constricted perfused rat liver; and, 4) after exertion of its hypertensive effect, bradykinin is degraded by angiotensin converting enzyme. In conclusion, the hypertensive response to BK is mediated by the B2 receptor in normal and pathological situations. The B1 receptor is expressed more strongly in regenerating and cirrhotic livers, and its role is currently under investigation.
bradykinin; hepatic metabolism; portal hypertension; bradykinin receptors
Hipertensão portal é a complicação mais comum das doenças crônicas do fígado, tais como cirrose. A resistência intravascular aumentada observada na doença hepática é devida a alterações na arquitetura celular e contração ativa das células estreladas. Neste trabalho revisamos aspectos históricos do estudo do sistema calicreína-cinina e os resultados de nossos estudos do papel deste nonapeptídeo no controle do tono vascular intra-hepático em condições normais e modelos experimentais de agressão hepática usando a perfusão de fígado isolado de rato (mono e bivascular) e células hepáticas isoladas. Nós demonstramos que: 1) o aumento da resistência vascular intrahepática induzido pela bradicinina é mediado por receptores B2, envolve a participação de células endoteliais sinusoidais e células estreladas e não é alterada pela presença de inflamação, fibrose ou cirrose; 2) a resposta hipertensiva induzida pela bradicinina no sistema arterial hepático é cálcio-independente emediada por eicosanóides; 3) bradicinina não tem efeito dilatador na circulação intra-hepática; 4) após exercer efeito vasoconstritor intra-hepático, a bradicinina é degradada pela enzima conversora de angiotensina. Em conclusão, a resposta hipertensiva à bradicinina é mediada pelo receptor B2 em condições normais e patológicas. Receptor B1 é expresso mais fortemente nos fígados em regeneração e cirróticos e seu papel está sob investigação.
bradicinina; metabolismo hepático; hipertensão porta; receptores de bradicinina
BIOMEDICAL AND MEDICAL SCIENCES
Portal hypertensive response to kinin
Maria KouyoumdjianI,II; Marcia R. NagaokaI,III; Mauricio R. Loureiro-SilvaI; Durval R. BorgesI,IV
ILaboratório de Hepatologia Experimental, UNIFESP, Rua Botucatu 862, Ed. J. L. Prado Vila Clementino, 04023-901 São Paulo, SP, Brasil
IIDepartamento de Bioquímica, UNIFESP, Rua Botucatu 862, Ed. J. L. Prado Vila Clementino, 04023-901 São Paulo, SP, Brasil
IIIDepartamento de Biociências, UNIFESP, Av. Ana Costa, 95, 4º andar, Vila Mathias, 11060-001 Santos, SP, Brasil
IVDepartamento de Medicina, UNIFESP, Rua Botucatu, 862, Ed. J. L. Prado, Vila Clementino, 04023-901 São Paulo, SP, Brasil
Correspondence to Correspondence to: Dr. Durval R. Borges E-mail: drborges@unifesp.br
ABSTRACT
Portal hypertension is the most common complication of chronic liver diseases, such as cirrhosis. The increased intrahepatic vascular resistance seen in hepatic disease is due to changes in cellular architecture and active contraction of stellate cells. In this article, we review the historical aspects of the kallikrein-kinin system, the role of bradykinin in the development of disease, and our main findings regarding the role of this nonapeptide in normal and experimentalmodels of hepatic injury using the isolated rat liver perfusion model (mono and bivascular) and isolated liver cells. We demonstrated that: 1) the increase in intrahepatic vascular resistance induced by bradykinin is mediated by B2 receptors, involving sinusoidal endothelial and stellate cells, and is preserved in the presence of inflammation, fibrosis, and cirrhosis; 2) the hepatic arterial hypertensive response to bradykinin is calcium-independent and mediated by eicosanoids; 3) bradykinin does not have vasodilating effect on the pre-constricted perfused rat liver; and, 4) after exertion of its hypertensive effect, bradykinin is degraded by angiotensin converting enzyme. In conclusion, the hypertensive response to BK is mediated by the B2 receptor in normal and pathological situations. The B1 receptor is expressed more strongly in regenerating and cirrhotic livers, and its role is currently under investigation.
Key words: bradykinin, hepatic metabolism, portal hypertension, bradykinin receptors.
RESUMO
Hipertensão portal é a complicação mais comum das doenças crônicas do fígado, tais como cirrose. A resistência intravascular aumentada observada na doença hepática é devida a alterações na arquitetura celular e contração ativa das células estreladas. Neste trabalho revisamos aspectos históricos do estudo do sistema calicreína-cinina e os resultados de nossos estudos do papel deste nonapeptídeo no controle do tono vascular intra-hepático em condições normais e modelos experimentais de agressão hepática usando a perfusão de fígado isolado de rato (mono e bivascular) e células hepáticas isoladas. Nós demonstramos que: 1) o aumento da resistência vascular intrahepática induzido pela bradicinina é mediado por receptores B2, envolve a participação de células endoteliais sinusoidais e células estreladas e não é alterada pela presença de inflamação, fibrose ou cirrose; 2) a resposta hipertensiva induzida pela bradicinina no sistema arterial hepático é cálcio-independente emediada por eicosanóides; 3) bradicinina não tem efeito dilatador na circulação intra-hepática; 4) após exercer efeito vasoconstritor intra-hepático, a bradicinina é degradada pela enzima conversora de angiotensina. Em conclusão, a resposta hipertensiva à bradicinina é mediada pelo receptor B2 em condições normais e patológicas. Receptor B1 é expresso mais fortemente nos fígados em regeneração e cirróticos e seu papel está sob investigação.
Palavras-chave: bradicinina, metabolismo hepático, hipertensão porta, receptores de bradicinina.
INTRODUCTION
The first description of isolated liver perfusion in the scientific literature was recorded in 1855 by Claude Bernard, when he was studying hepatic carbohydrate metabolism:
"J'ai choisi un chien adulte, vigoureux et bien portant, qui depuis plusiers jours était nourri exclusivement avec de la viande, et je l'ai sacrifié par la section du bulbe rachidien, sept heures aprés un repas copieux de tripes. Aussitôt l'abdomen fut ouvert, le foie fut enlevé en évitant de blesser son tissu, et cet organe encore tout chaud et avant que le sang êut le temps de se coaguler dans ses vaisseaux, fut soumis à un lavage à l'eau froide par la veine porte. Pour cela, je pris un tube de guttapercha, long de 1 mètre environ et portant à ses deux extrémités des ajustages en cuivre. Le tube étant préalablement rempli d'eau, une de ses extrémités fut solidement fixée au robinet de la fontaine du laboratoire de médicine du Collége de France. En ouvrant le robinet, l'eau traversa le foie avec une grande rapidité, car la force du courrant d'eau était capable, ainsi que cela fut mesuré, de soulever une colonne de mercure à 127 centimètres de hauteur (Bernard 1855)."
In 1948, Maurício Rocha e Silva, Wilson T. Beraldo, and Gastão Rosenfeld, working in the Biological Institute of São Paulo, described a substance that, when generated in the plasma by Bothrops jararaca venom or trypsin, showed an important arterial hypotensive action in cats and rabbits and stimulated guinea pig intestinal and uterine musculature contraction. They named the molecule bradykinin (BK, from Greek brady, slow and kinesia, movement) indicating a substance that produced a slow movement of the gut (Rocha e Silva et al. 1949). Their report follows: "In 1948, Beraldo, Rosenfeld, and I were interested to discover whether the venom of the B. jararaca liberated histamine from dog's liver as trypsin had been shown to do. The liver was perfused with defibrinated blood through the portal vein and the perfusates collected in the inferior vena cava just above the diaphragm. Since dog's blood practically does not contain histamine or any substance stimulating the guinea-pig gut, the perfusates were assayed directly upon the gut after the muscle had been made insensitive to the venom by desensitization. As soon as thes venom was injected in the perfusing cannula, the perfusates acquired enormous activity upon the guinea-pig ileum." (Rocha e Silva 1955).
The primary structure of bradykinin (Arg-Pro-Pro- Gly-Phe-Ser-Pro-Phe-Arg) was elucidated at the National Institute for Medical Research, Mill Hill, London (Elliott et al. 1960) and it was only in 1991 that the first kinin receptor cDNA was cloned (McEachern et al. 1991). In 1965, Ferreira found that the Bothrops jararaca venom contained a factor that potentiated several pharmacological actions of BK in vivo and in vitro. This factor was the basis for the development of the first generation of potent pharmaceuticals (captopril) for the treatment of arterial hypertension.
Angiotensin (hypertensin) was first described in 1939 in Argentina by Muñoz et al., and angiotensinconverting enzyme (ACE) was later characterized from horse plasma (Skeggs et al. 1956). The tissue conversion of angiotensin I (AI) to angiotensin II (AII) was demonstrated in Leal Prado's Laboratory at Escola Paulista de Medicina (Carlini et al. 1958) and this pioneering work was recognized later in an important review (Vane 1974).
In 1976, we demonstrated that BK and angiotensins I and II produced a portal hypertensive response in the liver, and that the liver efficiently inactivates BK (90% from an initial dose of 19µM) and converts AI to AII. We also verified that doses ranging from 0.17 to 0.34 nmols of AII were approximately 90% inactivated during a single passage through the liver (Borges et al. 1976). Later we verified that AI did not induce, per se, a portal hypertensive response, but its action was a result of its conversion to AII and subsequent action on theAT1 receptor, primarily within the periportal zone of the liver (Carvalho et al. 2007).
This review will summarize the hepatic response to bradykinin in normal and experimental models of liver disease.
CHARACTERISTICS OF THE KININ SYSTEM AND ITS ROLE IN DISEASES
The kinin system (KS) comprises several proteins and peptides, including precursors (kininogens), kinin-liberating enzymes (pro-kallikrein), biologically active kinins, and kinin-metabolizing enzymes. The kininogens (high molecular weight kininogen, HMWK, or low molecular weight kininogen, LMWK) are multifunctional glycoproteins synthesized by the liver (Borges and Gordon 1976) and other tissues (Figueroa et al. 1992); kininogens contain the kinin sequence in its mid portion and are the only precursors of the kinin peptides. Tissue and plasma kallikreins are serine proteases that differ from one another in their biochemical and functional characteristics (Bhoola et al. 1992). Kininogens and tissue kallikreins are expressed in many different tissues, whereas plasma kallikrein is predominantly produced by the liver. Plasma prokallikrein is a single chain glycoprotein synthesized and secreted by the liver; its cleavage by factor XIIa or its fragments generates plasma kallikrein. Its proteolytic activity is regulated by plasma inhibitors and hepatic clearance (Borges et al. 1981, 1986, Kouyoumdjian et al. 1989). Plasma kallikrein is primarily cleared by the liver (Borges et al. 1985); clearance is mediated by a galectin in a calcium-independent pathway (Nagaoka et al. 2003). The hepatic removal of tissue kallikreins (pig pancreatic and horse urinary) from circulation involves receptor-mediated endocytosis by C-type lectins, which were characterized as mannosyland galactosyl-specific, respectively (Kouyoumdjian et al. 1989).
In humans, plasma kallikrein forms BK from HMWK, whereas tissue kallikreins form kallidin (Lys-BK) fromHMWKandLMWK.By contrast, both plasma and tissue kallikrein generate BK in rodents (Bhoola et al. 1992, Campbell 2001). The cleavage of Lys-BK by aminopeptidase generates BK, and cleavage by carboxypeptidase generates des-Arg9-BK and des-Arg9Lys- BK. Several enzymes are responsible for inactivation and metabolism of BK; for a long period of time, inactivation was attributed almost exclusively to angiotensinconverting enzyme (ACE) from the lung, although the liver plays an important role in BK inactivation (Prado et al. 1975). Hepatic modulation of KS, as demonstrated by the isolated and exanguinated perfused rat liver model, is shown in Figure 1.
In the rat liver, the kininase metalloendopeptidase EC 3.4.24.15 predominates over ACE and prolylendopeptidase (Molina et al. 1996). However, our evidence suggests that ACE, and not EC 3.4.24.15, is the main kininase involved in BK degradation by the liver (Gioli- Pereira et al. 2005). Hepatic EC 3.4.24.15 cleaves BK at the Phe5-Ser6 residue, releasing two peptides; the BK 1-5 fragment has a protective role against the deleterious effects of lipopolysaccharide in rats (Morinelli et al. 2001).
The KS has paracrine activity that is the release of BK from HMWK. Once released, BK has a potent effect on vascular tone: it relaxes the arterial vasculature, causing hypotension, and increases portal vascular tone, causing isolated hypertension in this area (Borges et al. 1976).
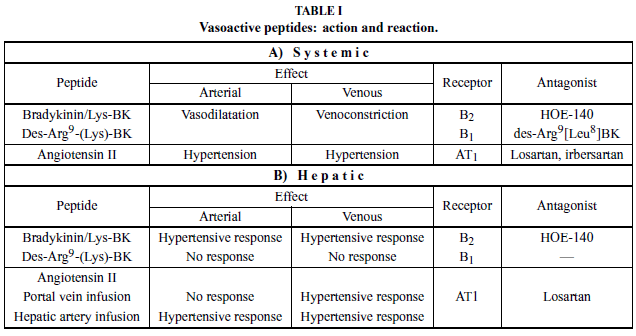
Bradykinin acts on two types of receptors: B1 (inducible) and B2 (constitutive; see Table I). Both mediate the same action, and their differentiation was based initially on a comparison of the potency of agonists and antagonists in several vascular preparations (Regoli et al. 1994). The B1 receptor (B1R) responds more efficiently to the octapeptide des-Arg9-BK than to BK itself whereas the B2 receptor (B2R) responds more efficiently to BK than to des-Arg9-BK. Antagonists of B1R and B2Rs are both available: des-Arg9[Leu8]-bradykinin and icatibant, a synthetic peptidomimetic (HOE-140), respectively (Hall 1997).
In addition to vascular effects, BK evokes the cardinal signs of inflammation (pain, swelling, redness, and heat) and is therefore characterized as a chemical mediator of the inflammatory response (Rocha e Silva 1964). BK acts near the top of the inflammatory cascade via interactions with B1R and B2R on target cell membranes. Activation of BK receptors is involved in the pathogenesis of multiple conditions (Table II).
B2R appears to be involved in the acute inflammatory response, whereas B1R has a role in chronic inflammatory processes (Perkins and Kelly 1993, Blais et al. 2000, Leeb-Lundberg et al. 2005, Costa-Neto et al. 2008). B1R may also play a significant role in the development of inflammatory diseases, particularly those with an immune etiology such as diabetes, asthma, rheumatoid arthritis, and multiple sclerosis (Couture et al. 2001). In streptozotocin-induced diabetes, B2R is overexpressed in peripheral tissues (Couture et al. 2001, Leeb- Lundberg et al. 2005) and, interestingly, the progression of diabetes mellituswas prevented by [Leu8]desArg9-BK (B1R antagonist), but not by icatibant (B2R antagonist) (Zuccollo et al. 1999).
There is direct and indirect evidence for the role of kinins in inflammatory airway diseases. Inhalation of BK or Lys-BK, but not des-Arg9-BK, causes bronchospasms in asthmatic person. Bronchoconstriction induced by BK was inhibited by HOE-140 (B2R antagonist), which confirms the involvement of B2R in BK-induced bronchospasm (Abraham et al. 2006).
In multiple sclerosis and septic shock, B1R has a protective role, while in other conditions it produces adverse effects, such as pain and inflammation (Calixto et al. 2004, Gabra et al. 2003). Although BK is capable of initiating the cascade of cytokine release that mediates hyperalgesic responses, antagonists of B1R and B2R fail to inhibit this response (Ferreira et al. 1993, Poole et al. 1999). In cardiovascular and renal diseases, these actions are mediated primarily by nitric oxide (NO) and prostaglandins.
Activation of the KS could prove beneficial due to its effects on angiogenesis, cardiac regeneration, and coronary blood flow; the KS also possesses anti-thrombotic, anti-inflammatory, and anti-apoptotic effects. It has been demonstrated that B2R is important in reducing the size of muscle infarction (Westerman et al. 2008).
KS activation is also related to basic pathological processes such as ischemia (Blais et al. 2000). In hypoxic situations, AII-induced cardiac angiogenesis is mediated by increased levels of BK and B2R activation (Munk et al. 2007). In the ischemic heart, des-Arg9-BK and BK promote protective effects, via B1R and B2R, that are due to the involvement of other mediators such as NO, prostanoid, and endothelium-derived hyperpolarizing factor (EDHF) (Lagneux et al. 2003).
In renal transplantation, the incidence of primary dysfunction and poor initial graft function appears to depend on the length of cold storage and has thus been ascribed to injury from harvesting, cold storage, and warm reperfusion after blood vessel reconnection; i.e., cold ischemia-warm reperfusion injury (IRI). Kakoki et al. (2007) showed that both B1R and B2R protect the kidney from damage caused by IRI, including reducing DNA damage, apoptosis, morphological and functional kidney changes, and mortality. Recently, Wang et al. (2008) showed that B1R antagonism promoted down regulation of pro-inflammatory and upregulation of antiinflammatory molecules, representing a new therapeutic strategy for the prevention and treatment of renal IRI.
Souza et al. (2004) demonstrated an increase in tissue kallikrein activity and activation of B2R after intestinal IRI, demonstrating the role ofB2Rin the development of inflammation and tissue loss. Therefore, B2R antagonists may be useful adjunct therapy for the treatment of the severe inflammatory injuries that follow ischemia and reperfusion of the superior mesenteric artery. B2R, but not B1R, also protects against cardiac IRI by inhibiting apoptosis and limiting ventricular remodeling (Yin et al. 2007). The role of kinins in hepatic IRI is now under investigation in our laboratory.
Although the KS has been related to thrombotic risk, it was observed that B2R knock-out mice were protected against arterial thrombosis, and that overexpression of the AT2 receptor had a compensatory effect (Schmaier 2008).
KS, via B2R activation, is associated with suppression of reactive oxygen species production and consequent prevention of renal tissue damage (Chao et al. 2007). The antifibrotic effect of B2R within the kidney was also demonstrated by Schanstra and co-workers (2002), supporting the potential role of BK in the antifibrotic effects of ACE inhibitors. Recently, in the unilateral ureteral obstruction model of renal fibrosis, B1R has been found to be overexpressed, and delayed treatment with an orally active nonpeptide B1R antagonist which blocked macrophage infiltration, leading to reversal of renal fibrosis (Klein et al. 2009).
The participation of kinin receptors during sepsis remains unclear. It is well known that B1R is induced and overexpressed during tissue injury, following inflammatory processes, and after LPS administration.
LIVER AND THE KININ SYSTEM
The liver has a very sophisticated vascular system that plays an active role in the maintenance of hepatic function. As a key organ in the control of the internal milieu, the liver relies on appropriate tissue perfusion to control the composition of the blood that drains into the systemic circulation. Excluding the hepatic artery and its branches, which are responsible for only 20% to 40% of the blood entering the liver, the intra-hepatic circulation is maintained at very lowpressure in physiological conditions. The vascular tone in portal venules, sinusoids, and hepatic venules is actively controlled by several relaxing and contracting substances, including BK (Borges et al. 1976, Loureiro-Silva et al. 1999, 2001, Gioli-Pereira et al. 2005, Nagaoka et al. 2006).
BK plays an important role in the control of vascular tone in physiological and pathological conditions. Both a widespread distribution throughout the vascular system and its very short half-life in circulation (Pellacani et al. 1992) indicate that BK participates in vascular tone control in an autocrine/paracrine way. However, since it can be detected in the circulation, an endocrine mechanism of action may also be involved, particularly in pathologic conditions such as sepsis (Weipert et al. 1988). Interestingly, BK has a dual role in the vascular tone control: it is a potent endothelium-dependent vasodilator but can also cause vasoconstriction by acting directly on vascular smooth muscle cells.
The vasodilatory effect of BK depends on endothelial cells that, after stimulation, release at least 3 compounds: nitric oxide, prostacyclin, and EDHF (Busse et al. 1994, Blatter et al. 1995, Nakashima et al. 1993). Briefly, activation of B2R located on endothelial cells initiates a sequence of events that results in the production of nitric oxide and prostacyclin. These two compounds migrate to adjacent smooth muscle cells and, by activating guanylate and adenylate cyclases, increase the intra-cellular concentration of the second messengers cGMP and cAMP, respectively (Holzmann et al. 1980, Waldman and Murad 1987). These cyclic nucleotides activate different mechanisms that reduce intracellular calcium concentration and, ultimately, cause smooth muscle cell relaxation. The vasodilatory effect of EDHF was originally attributed to the activation of smooth muscle potassium channels. More recent evidence suggests that EDHF release from endothelial cells occurs following the opening of potassium channels in stimulated endothelial cells (Busse et al. 2002). Although several putative candidates have been proposed, the identity of EDHF remains uncertain (Villar et al. 2008). In humans, the vasodilatory effect of BK has been demonstrated in both arteries and veins (Bönner et al. 1992).
Most of the compounds and pathways that mediate this BK effect are expressed in the liver and are likely to participate in intrahepatic vascular tone control. This is most likely to be true of intra-hepatic vessels that are structurally similar to the vessels where the BK vasodilatory effect has been demonstrated, i.e., the portal vein, hepatic artery, and their branches (Loureiro-Silva et al. 1999).
The vasoconstrictive effect of BK is caused by the direct action of this peptide on vascular smooth muscle. It is endothelium-independent and can, in fact, be blunted by the presence of endothelial tissue. Although most of studies have shown that this BK effect is mediated by B2R, the participation of B1R has also been demonstrated (Felipe et al. 2007). It seems clear that the pivotal event in BK-induced constriction of the vascular smooth muscle cell is an increase in intracellular Ca2+ concentration, which is primarily due to increased Ca2+ influx from the extracellular space (Bkaily et al. 1997, Dong et al. 2005, Eguchi et al. 1997). Using a rabbit saphenous vein preparation, Eguchi et al. (1997) demonstrated the involvement of Ca2+ release from intracellular thapsigargin-sensitive storages sites and a Gprotein- mediated increase in the Ca2+ sensitivity of the contractile apparatus. Most of the BK vasoconstriction effect observed in different preparations has been shown to be mediated by the release of eicosanoids (Wong et al. 1977, Barabé et al. 1979, Gioli-Pereira et al. 2005).
To study the portal hypertensive response (PHR) to BK, we used the isolated rat liver perfusion (IRLP) model in both monovascular (via portal vein) and bivascular (via portal vein and hepatic artery) conditions, in a hemoglobin-free medium enriched with oxygen. The IRLP is an elegant experimental model, since the liver integrity is maintained and changes in the intrahepatic vascular tone can be easily detected by measuring the perfusion pressure; perfusion pressure can be monitored by the connection of a vertical, open, graduated (cm) column to the portal cannula between the peristaltic pump and the liver in the monovascular perfusion (Borges et al. 1976). In the bivascular perfusion technique, the portal perfusion pressure is measured as described, and the hepatic artery pressure is measured by the pressure transducer. Using these experimental models, we have studied the intrahepatic vascular response to several vasoactive compounds, including BK, des-Arg9-BK, AI, and AII, in both normal and pathologic conditions. Additionally, we have carried out in vitro studies using isolated liver cells. Our findings are summarized in the following paragraphs.
In recirculating monovascular liver perfusion, BK induces a PHR that is mediated by B2R and modulated by the NO system in normal livers as well as in livers affected by inflammation, fibrosis, and cirrhosis (Loureiro- Silva et al. 2001, Nagaoka et al. 2006). During liver regeneration, the PHR to BK is present, although it is decreased from day 2 until 7 when compared to days 0 and 1 (Teixeira et al. 1999). The PHR was abolished in the presence of 6µM icatibant and, interestingly, when this B2R antagonist was used at higher concentration (> 120µM), portal pressure continuously increased until this antagonist was removed from the circuit (Gioli- Pereira et al. 2005).
Molar excess of captopril (ACE inhibitor) and JA-2 (EC 3.4.24.15 specific inhibitor) did not modify the PHR to BK. However, ACE, but not EC 3.4.24.15 inhibitor, increased the BK half-life in the liver perfusion model. On the other hand, HOE-140 inhibited the hypertensive action of BK but had no effect on its hydrolysis (Gioli- Pereira et al. 2005).
The hypertensive effects on the hepatic microcirculation were also studied in a bivascular rat liver perfusion, performed in the anterograde mode. When BK was injected into the arterial bed, it produced a double hemodynamic effect, including both hepatic arterial and portal hypertensive responses. The arterial hypertensive response was calcium-independent and mediated by eicosanoids; the PHR, occurring through a distinct pathway, was not inhibited by naproxen. On the other hand, BK injected into the portal vein produced a portal, but not arterial, hypertensive response. This response was calcium-dependent and was not affected by the presence of naproxen (Gioli-Pereira et al. 2005).
The hemodynamic effects were associated with metabolic effects when BK was injected via the arterial route; these effects included a decrease in oxygen consumption and increase in glucose release. The metabolic effects on oxygen consumption (in the absence of Ca2+) and on glucose release were not altered by naproxen. When BK reaches the liver by the venous route, the hemodynamic effect was also associated with the metabolic effects of decreased oxygen consumption and increased glucose release. The effect on oxygen consumption was calcium-dependent and was not affected by naproxen. The effect on glucose release was neither calcium- dependent nor affected by naproxen.
The hypertensive effect of BK on sinusoidal cells of the periportal region is followed by ACE hydrolysis of the peptide, primarily in the perivenous region. Therefore, we suggested that ACE is the main kininase involved in hepatic BK degradation (Gioli-Pereira et al. 2005).
Using the indicator dilution technique to study distribution volumes and transport phenomena in the liver, we observed that, in the steady state, BK is predominantly distributed in the extracellular space. The mean hepatic extraction of BK (200 nmol) was 16 nmol (8%), which is compatible with the fact that BK is rapidly hydrolyzed by ACE after its action on sinusoidal B2R in the periportal region (Gioli-Pereira et al. 2005).
Due to their anatomic location and ability to contract or relax in response to vasoactive mediators, endothelins, AII or TXA2, and NO or CO, respectively, stellate cells seem to be involved in the regulation of portal flow (Rockey 2003). Nevertheless, Kupffer cells and endothelial cells also contain filaments, tubules, and proteins that confer upon these cells the ability to regulate sinusoidal diameter (McCuskey and Reilly 1993). Using isolated liver cells, we demonstrated that sinusoidal cells (quiescent stellate and the fraction containing Kupffer and endothelial cells), but not hepatocytes, respond to BK, causing a basal extracellular acidification (microphysiometer model). This response to BK was specific because it was blocked by HOE-140, a B2R antagonist (Gioli-Pereira et al. 2005).
As discussed before, B1R is generally absent from normal tissue, but its expression can be induced in a variety of cells following an inflammatory stimulus. We extensively pursued the effect of des-Arg9-BK on the intrahepatic vascular response. Among the experimental models that we have studied were: lipopolysaccharide (classical model of induction of B1R), acute phase inflammation (turpentine oil injection), chronic inflammation (i.p. CCl4 injection up to 56 days), fibrosis (i.p. porcine serum injection), cirrhosis (bile duct ligation, BDL), and partial hepatectomy (70% resection). des- Arg9-BK or des-Arg9-Lys-BK did not elicit change in the portal pressure in any of these models. In a methoxamine pre-constricted liver, the hypertensive effect of BK was maintained, but no intrahepatic vascular response to B1R agonists was observed in normal, LPStreated, or BDL rats (Nagaoka et al. 2006). On isolated cells, we also observed that des-Arg9-BK did not elicit a response in hepatocyte or sinusoidal hepatic cells (Gioli-Pereira et al. 2005).
Interestingly, in spite of the absence of intrahepatic vascular response, B1R agonists were metabolized by the liver of normal and LPS-treated rats at the same rate, suggesting an efficient clearance machinery for these kinins (Nagaoka et al. 2006).
The induction of B1R was confirmed by Western blot in some experimental groups which were studied, such as BDL and partial hepatectomy (Nagaoka et al. 2006). In the CCl4-ip model, there was a slight band at all different times of treatment (10, 21, and 56 days). Interestingly, in the BDL group, B1R expression increased in relation to the time of progression of cirrhosis, suggesting a possible role of B1R in the development of fibrosis; this hypothesis is now under investigation. In the partial hepatectomy group, B1R expressionwas observed on days 1 and 2 and disappeared on day 7, suggesting a role in tissue remodeling (Nagaoka et al. 2006).
These results were confirmed by Sancho-Bru and co-workers (2007), who showed that rat normal liver expresses both B1R and B2R and, in addition, cirrhotic livers (CCl4 induction) have an overexpression of these receptors. ContinuousBKinfusion had a hepato-protective and anti-apoptotic role, via reduction in hepatocellular injury and attenuation of the progression of experimental fibrosis. They suggested that the anti-fibrotic effect of BK infusion was probably associated with the decrease of activated hepatic stellate cells and reduction of collagen and TGF-β1 expression (Sancho-Bru et al. 2007).
In summary, BK induces an increase in the intrahepatic vascular resistance that is mediated by B2R and preserved in the presence of inflammation, fibrosis, and cirrhosis. The hepatic arterial hypertensive response to BK is calcium-independent and mediated by eicosanoids, and BK does not have a vasodilatation effect on the pre-constricted perfused rat liver. Following its hypertensive effect, BK is degraded by ACE. In cirrhosis and hepatic regeneration, B1R is overexpressed, suggesting a role in fibrogenesis and the tissue remodeling that now is under investigation.
ACKNOWLEDGMENTS
Several studies from the Laboratory of Experimental Hepatology have been supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Desenvolvimento Científico eTecnológico (CNPq).
Manuscript received on July 14, 2008; accepted for publication on November 3, 2008; presented by LUIZ R. TRAVASSOS
In commemoration of the 75th anniversary of Escola Paulista de Medicina/Universidade Federal de São Paulo.
- ABRAHAM WM, SCURI M AND FARMER SG. 2006. Peptide and non-peptide bradykinin receptor antagonists: role in allergic airway disease. Eur J Pharmacol 533: 215-221.
- BARABÉ J, MARCEAU F, THÉRIAULT B, DROUIN JN AND REGOLI D. 1979. Cardiovascular actions of kinins in the rabbit. Can J Physiol Pharmacol 57: 78-91.
- BERNARD C. 1855. Sur le mécanisme de la formation du sucre dans le foie. Compte Rendu des Séances de L'Académie des Sciences XLI: 461-469.
- BHOOLA KD, FIGUEROA CD AND WORTHY K. 1992. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev 44: 1-80.
- BKAILY G, JAALOUK D, JACQUES D, ECONOMOS D, HASSAN G, SIMAAN M, REGOLI D AND POTHIER P. 1997. Bradykinin activates R-, T-, and L-type Ca2+ channels and induces a sustained increase of nuclear Ca2+ in aortic vascular smooth muscle cells. Can J Physiol Pharmacol 75: 652-660.
- BLAIS C JR, MARCEAU F, ROULEAU JL AND ADAM A. 2000. The kallikrein-kininogen-kinin system: lessons from the quantification of endogenous kinins. Peptides. 21: 1903-1940.
- BLATTER LA, TAHA Z, MESAROS S, SHACKLOCK PS, WIER WG AND MALINSKI T. 1995. Simultaneous measurement of Ca++ and nitric oxide in bradykinin-stimulated vascular endothelial cells. Circ Res 76: 922-924.
- BÖNNER G, PREIS S, SCHUNK U, WAGMANN M, CHROSCH R AND TOUSSAINT C. 1992. Effect of bradykinin on arteries and veins in systemic and pulmonary circulation. J Cardiovasc Pharmacol 20 (Suppl 9): S21-S27.
- BORGES DR AND GORDON AH. 1976. Kininogen and kininogenase synthesis by the liver of normal and injured rats. J Pharm Pharmacol 28: 44-48.
- BORGES DR, PRADO JL, LIMÃOS EA AND CAMARGO ACM. 1976. Catabolism of vasoactive polypeptides by perfused rat liver. Naunyn-Schmiedeberg's Arch Pharmacol 295: 33-40.
- BORGES DR, WEBSTER ME, GUIMARÃES JA AND PRADO JL. 1981. Synthesis of prekallikrein and metabolism of plasma kallikrein by perfused rat liver. Biochem Pharmacol 30: 1065-1069.
- BORGES DR, GORDON AH, GUIMARÃES JA AND PRADO JL. 1985. Rat plasma kallikrein clearance by perfused rat liver. Braz J Med Biol Res 18: 187-194.
- BORGES DR, SAMPAIO CAM, DE LA LLOSA P AND PRADO JL. 1986. The liver is the main organ to clear plasma and tissue kallikreins from plasma in vivo Adv Exp Biol Res 198A: 229-233.
- BUSSE R, HECKER M AND FLEMING I. 1994. Control of nitric oxide and prostacyclin synthesis in endothelial cell. Arnemittelforschung/Drug Res 44: 392-396.
- BUSSE R, EDWARDS G, FÉLÉTOU M, VANHOUTTE PM AND WESTON AH. 2002. EDHF: bringing the concepts together. Trends Pharmacol Sci 23: 374-380.
- CALIXTO JB, MEDEIROS R, FERNANDES ES, FERREIRA J, CABRINI DA AND CAMPOS MM. 2004. Kinin B1 receptors: key G-protein-coupled receptors and their role in inflammatory and painful processes. Br J Pharmacol 143: 803-818.
- CAMPBELL DJ. 2001. The kallikrein-kinin system in humans, Clin. Exp. Pharmacol. Physiol. 28: 1060-1065.
- CARLINI EA, PICARELLI Z AND PRADO JL. 1958. Pharmacological activity of hypertensin I and its conversion into hypertensin II. Bull Soc Chim Biol 40: 1825-1834.
- CARVALHO LT, NASCIMENTO EA, TEIXEIRA FO, NAGAOKA MR, BORGES DR AND KOUYOUMDJIAN M. 2007. The hepatic conversion of angiotensin I and the portal hypertensive response to angiotensin II in normal and regenerating liver. J Gastroenterol Hepatol 22: 1543-1548.
- COSTA-NETO CM, DILLENBURG-PILLA P. HEINRICH TA, PARIERAS-E-SILVA LT, PEREIRA MG, REIS RI AND SOUZA PP. 2008. Participation of kallikrein-kinin system in different pathologies. Int Immunopharmacol 8: 135-142.
- COUTURE R, HARRISSON M, VIANNA RM AND CLOUTIER F. 2001. Kinin receptors in pain and inflammation. Eur J Pharmacol 429: 161-167.
- CHAO J, LI HJ, YAO YY, SHEN B, GAO L, BLEDSOE G AND CHAO L. 2007. Kinin infusion prevents renal inflammation, apoptosis, and fibrosis via inhibition of oxidative stress and mitogen-activated protein kinase activity. Hypertension 49: 490-497.
- DONG Y-L, VEGIRAJU S AND YALLAMPALLI C. 2005. Ca2+ signaling in human fetoplacental vasculature: effect of CGRP in umbilical vein smooth muscle cytosolic Ca2+ concentration. Am J Physiol Hearth Circ Physiol 289: 960-967.
- EGUCHI D, NISHIMURA J, KOBAYASHI S, KOMORI K, SUGIMACHI AND KANAIDE H. 1997. Mechanism of contraction induced by bradykinin in the rabbit saphenous vein. Brit J Pharmacom 120: 371-378.
- ELLIOTT DF, LEWIS GP AND HORTON EW. 1960. The structure of bradykinin - A plasma kinin from ox blood. Biochem Biophys Res Comm 3: 87-91.
- FELIPE SA, RODRIGUES ES, MARTIN RP, PAIVA AC, PESQUERO JB AND SHIMUTA SI. 2007. Functional expression of kinin B1 and B2 receptors in mouse abdominal aorta. Braz J Med Biol Res 40: 649-655.
- FERREIRA SH. 1965. A bradykinin-potentiating factor (BPF) present in the venom of Bothrops jararaca Brit J Pharmacol 24: 163-169.
- FERREIRA SH, LORENZETTI BB AND POOLE S. 1993. Bradykinin initiates cytokine-mediated inflammatory hyperalgesia. Br J Pharmacol 110: 1227-1231.
- FIGUEROA CD, GONZALEZ CB, MULLER-ESTERL W AND BHOOLA KD. 1992. Cellular localization of human kininogens. Agents and Actions Suppl 38: 617-626.
- GABRA BH, COUTURE R AND SIROIS P. 2003. Functional duality of kinin receptors in pathophysiology. Med Sci (Paris) 19: 1101-1110.
- GIOLI-PEREIRA L, NASCIMENTO EA, SANTOS EL, BRACHT A, JULIANO MA, PESQUERO JB, BORGES DR AND KOUYOUMDJIAN M. 2005. Fate of bradykinin on the rat liver when administered by the venous or arterial route. J Gastroenterol Hepatol 20: 463-473.
- HALL JM. 1997. Bradykinin receptors: pharmacological properties and biological roles. Pharmacol Ther 56: 131-190.
- HOLZMANN S, KUKOVETZ WR AND SCHMIDT K. 1980. Mode of action of coronary arterial relaxation by prostacyclin J Cyclic Nucleotide Res 6: 451-460.
- IMAMURA T, KOBAYASHI H, KHAN R, NITTA H AND OKAMOTO K. 2006. Induction of vascular leakage and blood pressure lowering through kinin release by a serine proteinase from Aeromonas sobria J Immunol 177: 8723-8729.
- KAKOKI M, MCGARRAH RW, KIM HS AND SMITHIES O. 2007. Bradykinin B1 and B2 receptors both have protective roles in renal ischemia/reperfusion injury 2007. Proc Natl Acad Sci USA 104: 7576-7581.
- KLEIN J ET AL. 2009. Delayed blockade of the kinin B1 receptor reduces renal inflammation and fibrosis in obstructive nephropathy. DOI: 10.1096/fj.08-115600. Faseb J 23: 134-142.
- KOUYOUMDJIAN M, BORGES DR AND PRADO JL. 1984. Kinin-inactivating endopeptidase from rat liver. Int J Biochem 16: 733-739.
- KOUYOUMDJIAN M, BORGES DR, PRADO ES AND PRADO JL. 1989. Identification of receptors in the liver that mediate endocytosis of circulating tissue kallikreins. Biochim Biophys Acta 980: 299-304.
- LAGNEUX C, ADAM A AND LAMONTAGNE D. 2003Astudy of the mediators involved in the protection induced by exogenous kinins in the isolated rat heart. Intern Immunopharmacol 3: 1511-1518.
- LEEB-LUNDBERG LM, MARCEAU F, MÜLLER-ESTERL W, PETTIBONE DJ AND ZURAW BL. 2005. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev 57: 27-77.
- LOUREIRO-SILVA MR, MOLINA HM AND BORGES DR. 1999. Substâncias vasoativas e a modulação do sistema microvascular hepático. Rev Ass Med Brasil 45: 206-216.
- LOUREIRO-SILVA MR, MOLINA HM AND BORGES DR. 2001. The portal hypertensive response to bradykinin in inflamed or cirrhotic rats is mediated by the B2-type receptors. J Gastroenterol Hepatol 16: 41-45.
- MCCUSKEY RS AND REILLY FD. 1993. Hepatic microvasculature: dynamic structure and its regulation. Semin Liver Dis 13: 1-12.
- MCEACHERN AE, SHELTON ER, BHAKTA S, OBERNOLTE R, BACH C, ZUPPAN P, FUJISAKI J, ALDRICH RW AND JARNAGIN K. 1991. Expression cloning of a rat B2 bradykinin receptor. Proc Natl Acad Sci USA 88: 7724-7728.
- MOLINA HM, CARMONA AK, KOUYOUMDJIAN M, BORGES DR AND JULIANO L. 1996. Liver bradykinin-inactivating- endopeptidase is similar to the metalloendopeptidase. (EC 3.4.24.15) Immunopharmacol 32: 176-179.
- MOLINA HM, CARMONA AK, KOUYOUMDJIAN M AND BORGES DR. 2000. Thimet oligopeptidase EC 3.4.24.15 is a major liver kininase. Life Sci 67: 509-520.
- MORINELLI TA, WEBB JG, JAFFA AA, PRIVITERA PJ AND MARGOLIUS HS. 2001. A metabolic fragment of bradykinin, Arg-Pro-Pro-Gly-Phe, protects against the deleterious effects of lipopolysaccharide in rats. J Pharmacol Exp Therapy 296: 71-76.
- MUNK VC, SANCHEZ DE MIGUEL L, PETRIMPOL M, BUTZ N, ERIKSSON U, HEIN L, HUMAR R AND BATTEGAY EJ. 2007. Angiotensin II induces angiogenesis in the hypoxic adult mouse heart in vitro through an AT2-B2 receptor pathway. Hypertension 49: 1178-1185.
- MUÑOZ JM, BRAUN-MENENDEZ E, FASCIOLO JC AND LELOIR LF. 1939. Hypertensin: the substance causing renal hypertension. Nature 144: 980.
- NAGAOKA MR, STRITAL E, KOUYOUMDJIAN M AND BORGES DR. 2003. Participation of a galectin-dependent mechanism in the hepatic clearance of tissue-type plasminogen activator and plasma kallikkrein. Thromb Res 108: 257-262.
- NAGAOKA MR, GOMIERO L, TEIXEIRA FO, AGOSTINO FG, POUZA JEP, MINARY P, BORGES DR AND KOUYOUMDJIAN M. 2006. Is the expression of kinin B1 receptor related to intrahepatic vascular response? Biochim Biophys Acta 1760: 1831-1836.
- NAKASHIMA M, MOMBOULI J-V, TAYLOR AA AND VANHOUTTE PM. 1993. Endothelium-dependent hyperpolarization caused by bradykinin in human coronary arteries. J Clin Invest 92: 2867-2871.
- PELLACANI A, BRUNNER HR AND NUSSBERGER J. 1992. Antagonizing and measurement: approaches to understanding of hemodynamic effects of kinins. J Cardiov Pharmacol 20(Suppl 9): S28-S34.
- PERKINS MN AND KELLY D. 1993. Induction of bradykinin B1 receptors in vivo in a model of ultra-violet irradiationinduced thermal hyperalgesia in the rat. Br J Pharmacol 110: 1441-1444.
- PEROSA SR ET AL. 2007. Kinin B1 and B2 receptors are overexpressed in the hippocampus of humans with temporal lobe epilepsy. Hippocampus 17: 26-33.
- POOLE S, LORENZETTI BB, CUNHA JM, CUNHA FQ AND FERREIRA SH. 1999. Bradykinin B1 and B2 receptors, tumour necrosis factor alpha and inflammatory hyperalgesia. Br J Pharmacol 126: 649-656.
- PRADO JL, LIMÃOS EA, ROBLERO J, FREITAS JO, PRADO ES AND PAIVA AC. 1975. Recovery and conversion of kinins in exsanguinated rat preparations. Naunyn Schmiedebergs Arch Pharmacol 290: 191-205.
- PREDIGER RDS, MEDEIROS R, PANDOLFO P, DUARTE FS, PASSOS GF, PESQUERO JB, CAMPOS MM, CALIXTO JB AND TAKAHASHI RN. 2008. Genetic deletion or antagonism of kinin b1 and b2 receptors improves cognitive deficits in a mouse model of Alzheimer's disease. Neuroscience 151: 631-643.
- REGOLI D, GOBEIL F, NGUYEN QT, JURIK D, SEOANE PR, SALVINO JM AND SAWUTZ DG. 1994. Bradykinin receptor types and B2 subtypes. Life Sciences 55: 735-749.
- ROCHA E SILVA M. 1955. Bradykinin: occurrence and properties. In: GADDUM JH (Ed), Polypeptides which stimulate plain muscle. E&S Livingstone Ltd, Edinburgh and London, p. 45-57.
- ROCHA E SILVA M. 1964. Chemical mediators of the acute inflammatory reaction. Ann NY Acad Sci 116: 899-911.
- ROCHA E SILVA M, BERALDO WT AND ROSENFELD G. 1949. Bradykinin, a hypotensive and smooth muscle stimulation factor released from plasma globulin by snake venoms and by trypsin. Am J Physiol 156: 261-273.
- ROCKEY DC. 2003. Vascular mediators in the injured liver. Hepatology 37: 4-12.
- SANCHO-BRU P ET AL. 2007. Bradykinin attenuates hepatocellular damage and fibrosis in rats with chronic liver injury. Gastroenterol 133: 2019-2028.
- SCHANSTRA JP ET AL. 2002. In vivo bradykinin B2 receptor activation reduces renal fibrosis. J Clin Invest 110: 371- 379.
- SCHMAIER AH. 2008. Assembly, activation, and physiologic influence of the plasma kallikrein/kinin system. Int Immunopharmacol 8: 161-165.
- SKEGGS LT, KAHN JR AND SHUMWAY NP. 1956. The preparation and function of the hypertensin-converting enzyme. J Exp Med 103: 295-299.
- SOUZA DG, LOMEZ ESL, PINHO V, PESQUERO JB, BADER M, PESQUERO JL AND TEIXEIRA MM. 2004. Role of Bradykinin B2 and B1 Receptors in the Local, Remote, and Systemic Inflammatory Responses That Follow Intestinal Ischemia and Reperfusion Injury. The Journal of Immunology 172: 2542-2548.
- TEIXEIRA FO, KOUYOUMDJIAN M AND BORGES DR. 1999. Reatividade portal após hepatectomia parcial. GED 18: S53.
- VANE JR. 1974. The fate of angiotensin I. Handbook of Experimental Pharmacology XXXVII: 17-40.
- VILLAR IC, HOBBS AJ AND AHLUWALIA A. 2008. Sex difference in vascular function: implications of endotheliumderived hyperpolarizing factor. J Endocrinol 197: 447-462.
- WALDMAN SA AND MURAD F. 1987. Cyclic GMP synthesis and function. Pharmacol Rev 39: 163-169.
- WANG PH, CAMPANHOLLE G, CENEDEZE MA, FEITOZA CQ, GONÇALVES GM, LANDGRAF RG, JANCAR S, PESQUERO JB, PACHECO-SILVA A AND CÂMARA NO. 2008. Bradykinin B1 receptor antagonism is beneficial in renal ischemia-reperfusion injury. PLoS ONE 3: e3050.
- WEIPERT J, HOFFMANN H, SIEBERG M AND WHALLEY ET. 1988. Attenuation of arterial blood pressure fall in endotoxin sock in the rat using the competitive bradykinin antagonist Lys-Lys-[Hyp2, Thi5,8, DPhe7]-BK (B4148). Br J Pharmacol 94: 282-284.
- WESTERMANN D, SCHULTHEISS HP AND TSCHÖPE C. 2008. New perspective on the tissue kallikrein-kinin system in myocardial infarction: Role of angiogenesis and cardiac regeneration. Intern Immunopharmacol 8: 148-154.
- WONG PY, TERRAGNO DA, TERRAGNO NA AND MCGIFF JC. 1977. Dual effect of bradykinin and prostaglandin metabolism: relationship to the dissimilar vascular actions of kinins. Prostaglandins 13: 1113-1125.
- YIN H, CHAO J, BADER M AND CHAO L. 2007. Differential role of kinin B1 and B2 receptors in ischemia-induced apoptosis and ventricular remodeling. Peptides 28: 1383-1389.
- ZUCCOLO A, NAVARRO M, FRONTERA M, CUEVA F, CARATTINO M AND CATANZARO OL. 1999. The involvement of kallikrein-kinin system in diabetes type I (insulitis). Immunopharmacol 45: 69-74.
Publication Dates
-
Publication in this collection
19 Aug 2009 -
Date of issue
Sept 2009
History
-
Accepted
03 Nov 2008 -
Received
14 July 2008