Abstracts
Cell therapy for neurological disorders has advanced, and neural precursor cells (NPC) may become the ideal candidates for neural transplantation in a wide range of diseases. However, additional work has to be done to determine either the ideal culture environment for NPC expansion in vitro, without altering their plasticity, or the FGF-2 and EGF mechanisms of cell signaling in neurospheres growth, survival and differentiation. In this work we evaluated mouse neurospheres cultured with and without FGF-2 and EGF containing medium and showed that those growth factors are responsible for NPC proliferation. It is also demonstrated that endogenous production of growth factors shifts from FGF-2 to IGF-1/PDGFb upon EGF and FGF-2 withdrawal. Mouse NPC cultured in suspension showed different patterns of neuronal localization (core versus shell) for both EGF and FGF-2 withdrawal and control groups. Taken together, these results show that EGF and FGF-2 removal play an important role in NPC differentiation and may contribute to a better understanding of mechanisms of NPC differentiation. Our findings suggest that depriving NPC of growth factors prior to grafting might enhance their chance to effectively integrate into the host.
neural precursor cells; neurosphere; fibroblast growth factor 2; epidermal growth factor; differentiation
As terapias celulares para doenças neurológicas têm avançado e células precursoras neurais (NPC) surgem como candidatas ideais para o transplante de células neurais em muitas doenças. No entanto, trabalhos adicionais devem ser feitos para determinar o ambiente de cultivo ideal para a expansão in vitro das NPC, sem alterar sua plasticidade, e os mecanismos de sinalização celular do fator de crescimento epidérmico (EGF) e fator de crescimento de fibroblasto 2 (FGF-2) no crescimento, sobrevivência e diferenciação da neuroesfera. Nesse trabalho avaliamosNPCcultivadas na presença e na ausência de FGF-2 e EGF e mostramos que esses fatores de crescimento são responsáveis pela proliferação das NPC. Também foi demonstrado que a produção endógena de fatores de crescimento alterna de FGF-2 a fator de crescimento de insulina 1 (IGF-1) e fator de crescimento derivado de plaquetas b (PDGFb) após remoção de EGF e FGF-2. NPC de camundongo cultivadas em suspensão mostraram padrões de localização neuronal distintos (centro versus borda) tanto no grupo controle como no grupo sem EGF e FGF-2. Juntos, esses resultados mostram que a remoção de EGF e FGF-2 exerce importante ação na diferenciação de NPC e possivelmente contribui para melhor compreensão dos mecanismos envolvidos na diferenciação. Nossos achados sugerem que, privando as NPC de fatores de crescimento antes do transplante, talvez aumente as chances de que as células efetivamente se integrem ao hospedeiro.
células precursoras neurais; neuroesfera; fator de crescimento de fibroblasto 2; fator de crescimento epidérmico; diferenciação
BIOMEDICAL AND MEDICAL SCIENCES
Effects of FGF-2 and EGF removal on the differentiationof mouse neural precursor cells
Telma T. SchwindtI,* * These authors contributed equally to this work. ; Fabiana L. MottaI,* * These authors contributed equally to this work. ; Gabriela F. BarnabéI,* * These authors contributed equally to this work. ; Cristina G. MassantI; Alessander O. GuimarãesII; Maria Elisa CalcagnottoI; João B. PesqueroII; Luiz E. MelloI
IDepartamento de Fisiologia, Universidade Federal de São Paulo (UNIFESP), Edifício Antônio M. C. Paiva Rua Botucatu, 862, 04023-062 São Paulo, SP, Brasil
IIDepartamento de Biofísica, Universidade Federal de São Paulo (UNIFESP), Edifício Antônio M. C. Paiva Rua Botucatu, 862, 04023-062 São Paulo, SP, Brasil
Correspondence to Correspondence to: Dr. Luiz E. Mello E-mail: lemello@unifesp.br
ABSTRACT
Cell therapy for neurological disorders has advanced, and neural precursor cells (NPC) may become the ideal candidates for neural transplantation in a wide range of diseases. However, additional work has to be done to determine either the ideal culture environment for NPC expansion in vitro, without altering their plasticity, or the FGF-2 and EGF mechanisms of cell signaling in neurospheres growth, survival and differentiation. In this work we evaluated mouse neurospheres cultured with and without FGF-2 and EGF containing medium and showed that those growth factors are responsible for NPC proliferation. It is also demonstrated that endogenous production of growth factors shifts from FGF-2 to IGF-1/PDGFb upon EGF and FGF-2 withdrawal. Mouse NPC cultured in suspension showed different patterns of neuronal localization (core versus shell) for both EGF and FGF-2 withdrawal and control groups. Taken together, these results show that EGF and FGF-2 removal play an important role in NPC differentiation and may contribute to a better understanding of mechanisms of NPC differentiation. Our findings suggest that depriving NPC of growth factors prior to grafting might enhance their chance to effectively integrate into the host.
Key words: neural precursor cells, neurosphere, fibroblast growth factor 2, epidermal growth factor, differentiation.
RESUMO
As terapias celulares para doenças neurológicas têm avançado e células precursoras neurais (NPC) surgem como candidatas ideais para o transplante de células neurais em muitas doenças. No entanto, trabalhos adicionais devem ser feitos para determinar o ambiente de cultivo ideal para a expansão in vitro das NPC, sem alterar sua plasticidade, e os mecanismos de sinalização celular do fator de crescimento epidérmico (EGF) e fator de crescimento de fibroblasto 2 (FGF-2) no crescimento, sobrevivência e diferenciação da neuroesfera. Nesse trabalho avaliamosNPCcultivadas na presença e na ausência de FGF-2 e EGF e mostramos que esses fatores de crescimento são responsáveis pela proliferação das NPC. Também foi demonstrado que a produção endógena de fatores de crescimento alterna de FGF-2 a fator de crescimento de insulina 1 (IGF-1) e fator de crescimento derivado de plaquetas b (PDGFb) após remoção de EGF e FGF-2. NPC de camundongo cultivadas em suspensão mostraram padrões de localização neuronal distintos (centro versus borda) tanto no grupo controle como no grupo sem EGF e FGF-2. Juntos, esses resultados mostram que a remoção de EGF e FGF-2 exerce importante ação na diferenciação de NPC e possivelmente contribui para melhor compreensão dos mecanismos envolvidos na diferenciação. Nossos achados sugerem que, privando as NPC de fatores de crescimento antes do transplante, talvez aumente as chances de que as células efetivamente se integrem ao hospedeiro.
Palavras-chave: células precursoras neurais, neuroesfera, fator de crescimento de fibroblasto 2, fator de crescimento epidérmico, diferenciação.
INTRODUCTION
Evidence of neurogenesis in the adult brain of birds (Goldman and Nottebohm 1983), rodents and primates (Kuhn et al. 1996, Gould et al. 1999, Kuhn and Svendsen 1999), and the demonstration of stem cells in specific brain regions, such as the subventricular zone and the hippocampus (Eriksson et al. 1998, Van Praag et al. 2002), brought new perspectives for cell therapy and neural regeneration (Svendsen and Smith 1999). Although these cells in the adult brain have decreased plasticity and are not as easy to manipulate and to grow in culture as their embryonic stem cell counterparts (Gage 1998, Thomson et al. 1998), they do not involve ethical questions.
During the development of the central nervous system, there is extensive proliferation of neuroepithelial cells lining the ventricular walls which give rise to the neurons, astrocytes and oligodendrocytes of the mature brain (Jacobson 1991). An experimental model to study neural stem cells is the heterogeneous free floating aggregates of cells, termed neurospheres (Reynolds and Weiss 1996, McKay 1997, Gage 2000). Each neurosphere is derived from a single stem cell that, by asymmetrical division, gives rise to another stem cell and one progenitor cell. The progenitor cells, in turn, give rise only to other progenitor cells. In this way, only a small fraction of the neurosphere corresponds to real stem cells (Reynolds et al. 1992). Here we use the terminology neural precursor cells (NPC) to describe both cell types within the neurosphere (Svendsen et al. 1999, Svendsen and Caldwell 2000).
Cell therapy for neurological disorders has advanced, and NPC may become the ideal candidates for neural transplantation in a wide range of diseases such as Parkinson (Dunnett and Bjorklund 1999). Previous studies have shown that mouse NPC can be pluripotent when exposed to the correct environment and produce hematopoietic cells in irradiated mice (Bjornson et al. 1999) and cells from distinct mesodermic lineages when injected in mouse blastocysts or chicken embryos (Clarke et al. 2000). Neurospheres are usually cultured in fibroblast growth factor 2 (FGF-2) and epidermal growth factor (EGF) containing medium in the first weeks and switched to EGF alone after 4 weeks. It was also shown that the acquisition of EGF responsiveness by neural precursor cells is promoted by FGF-2 in the early development in vitro. After several cell divisions, the same cell type may respond to both EGF and FGF- 2 (Ciccolini and Svendsen 1998). Yet, FGF-2 and EGF mechanisms of cell signaling in neurospheres growth, survival and differentiation are still unclear. It was previously reported that a method of neurospheres passaging, that maintain cell-cell contact, was also important for neurosphere growth (Svendsen et al. 1998).
Manipulation of environmental signals can make the cells differentiate in specific cell types and may help us to understand the mechanisms of neural and glial development to be assessed. Despite the advances in stem cell studies, additional work has to be done to determine the ideal culture medium forNPCexpansion in vitro without altering their plasticity. It is likely that on and off periods of growth factors might provide specific patterns of cell proliferation, migration and differentiation. In this work we evaluate mouse neurospheres cultured with and without FGF-2 and EGF containing medium and show that EGF and FGF-2 removal may influence their differentiation and expression of neurotrophins.
MATERIALS AND METHODS
ETHICAL ISSUES
This work was developed under the approval of the Ethics Committee of Universidade Federal de São Paulo (UNIFESP), file 0976-04.
HARVESTING AND CULTURING MOUSE NPC
mNPC were obtained from E14 (embryonic day 14) C57BL/6 mouse embryos. The fetuses were placed in a Petri dish containing PBS (phosphate buffered saline)/2% glucose, and the dissection was made under magnifying lens. The brains were sectioned and the tissue was incubated with Trypsin-EDTA solution (Invitrogen) for 15min at 37ºC. Trypsin was inactivated with fetal bovine serum, and, after cell sedimentation, the supernatant was removed and the cells were dissociated. Cell suspension was counted in a hemocytometer and the cells were seeded in 10 mL in a T25 flask at a density equivalent to 100,000 cells/mL. Each flask contains cells derived from a pool of 3 fetuses and all experiments were performed using three independent cultures of mNPC. The culture media is composed by 70% DMEM (Dulbecco's modified eagle medium; Invitrogen), 30% F12 (Invitrogen), 1% PSA (penicillin-streptomycin-amphotericin, Invitrogen), 2% B27 (Invitrogen), 20ng/mL EGF (Sigma), 20ng/mL FGF-2 (R&D), and 5µg/mL heparin (Sigma). Cells were maintained in an incubator at 37ºC under a 5% CO2 atmosphere. Every 3 days, half of the volume was replaced with fresh medium.
GROWTH FACTORS WITHDRAWAL
After neurosphere expansion (as described above), half of the population within one flask was kept in the complete media (control group - CTR) and the other half was cultured in the absence of growth factors (E/F less). The spheres were transferred to conical tubes and washed carefully 3 times with pre warmed DMEM. If necessary, the spheres were centrifuged at 130Xg. The spheres were put in growth factors free medium (DMEM/F12/B27) and kept in those conditions in suspension for 11 days. Every 3 days, half of the volume was replaced with fresh medium.
PREPARATION OF NEUROSPHERES SLICES
Whole neurospheres from the CTR and E/F-less groups were taken from the flasks, washed with PBS to remove the excess of culture medium and fixed in 4% PFA (paraformaldehyde) for 1h at room temperature. Neurospheres were washed three times with PBS and then transferred to PBS/10% sucrose solution for 1h at 4ºC, PBS/20% sucrose solution for 1h at 4ºC and, finally, PBS/30% sucrose solution at 4ºC overnight. Neurospheres were then mounted in Histo Prep (Fisher Scientific) and frozen in dry ice. Spheres were sliced at 10µm on a cryostat and placed on silanyzed slides (Superfrost slides, Fisher Scientific).
5-BROMO-2'DEOXYURIDINE (BRDU) INCORPORATION
BrdU (0.2µM) was added to the medium for 14h to the CTR and E/F less groups. To visually inspect the localization of proliferating cells in the neurosphere, whole neurospheres were sectioned in a cryostat and placed on silanyzed slides. For BrdU counting, spheres were dissociated using Trypsin and plated onto poly-lysin/laminin coated coverslips before immunocytochemistry. Sections or coverslips were incubated in HCl 1.5M for 30 min under gentle shaking, washed 3X 10 min in PBS and blocked in 5% normal goat serum and 0.1% TritonX-100 in PBS. The slides were incubated with anti-BrdU (Axyll/Accurate Chemical & Scientific Corporation, rat IgG, 1:200) for 2h and washed three times in PBS. Cells were incubated with the secondary antibody (Alexa 488 anti-rat IgG, Molecular Probes) for 1h and washed three times in PBS. DAPI solution (Sigma, 0.3µg/ml) was used as a nuclear stain. Cells were analyzed under a fluorescence microscope (Nikon, model Eclipse E600FM, Japan). The percentage of BrdU positive cells from the coverslips was statistically analyzed by using the ANOVA test with post hoc test Newman- Keuls with a significance level set at p < 0.05.
GROWTH CURVE
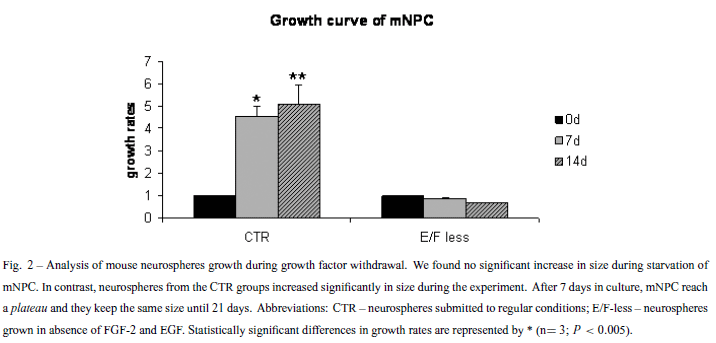
Isolated single neurospheres were placed in 96-multiwell plates to avoid neurospheres fusion. Multiwell plates were previously treated with Poly-(2-hydroxyethyl methacrylate) (Sigma) solution to avoid neurosphere adhesion on the bottom of the wells. Neurospheres were maintained in the presence (CTR) or in the absence (E/F-less) of growth factors for 11 days, and the diameter of the neurospheres were measured at every 3 days. We used the diameter measures to calculate the volume of the spheres during 11 days. For both CTR and E/F- less groups, 4 distinct neurospheres were measured in each triplicate. Growth curve results were submitted to the Student t test with a significance level set at p < 0.05.
IMMUNOCYTOCHEMISTRY FOR β-TUBULIN III, GFAP (GLIAL FIBRILLARY ACIDIC PROTEIN) AND GAL-C (GALACTOCEREBROSIDE C)
The slides were blocked and permeabilized in 5% normal goat serum and 0.1% Triton X-100 in PBS for 30 min (except for Gal-C, in which detergent was not added). Primary antibodies for β-tubulin III (Sigma, mouse IgG, 1:300), GFAP (DAKO, rabbit IgG, 1:500), Gal-C (Chemicon, mouse IgG3, 1:200) were added, and cells were incubated overnight at 4ºC. The slides were washed in PBS and incubated with the secondary antibodies (Alexa 546 anti-mouse IgG, Alexa 488 antimouse IgG or Alexa 488 anti-rabbit IgG, Molecular Probes, 1:250). After washing with PBS, DAPI solution (Sigma, 0.3µg/ml)was used as a nuclear stain. Cells were analyzed under a fluorescence microscope. β-Tubulin III was considered a reliable marker for neuronal differentiation because it was shown previously that, besides being expressed by both mature neurons and neural progenitors, β-tubulin III+ migrating cells from neurospheres adopt a neuronal phenotype and express neurochemical markers (GABA, glutamate) and mature neuronal markers (Neu-N, MAP-2ab) (Ostenfeld and Svendsen 2004). GFAP is used as an astrocytic marker, but it can be also expressed by immature neural progenitor cells.
TERMINAL DEOXYNUCLEOTIDYL TRANSFERASEMEDIATED BIOTINYLATED UTP NICK END LABELING (TUNEL)
In situ cell death detection kit (Roche) labels apoptotic cells, based on labelling of DNA strand breaks, with fluorescein (green fluorescence). Coverslips containing dissociated cells were fixed with 4% paraformaldehyde in PBS for 10 min, washed 3 times with PBS and blocked and permeabilized in 5% normal goat serum and 0.2% Triton X-100 in PBS for 15 min at room temperature. Cells were washed one time with PBS and the enzyme and nucleotide mix were added (Enzyme-TdT and Label-nucleotide) and incubated for 30 min at 37ºC. Cells were washed 3 times with PBS and the nuclei were labelled with DAPI for 5 min. Cells were washed with PBS, mounted and analyzed under a fluorescence microscope. TUNEL data were analyzed using the Student t test (significance set at p < 0.05).
RNA EXTRACTION AND CDNA SYNTHESIS
Total RNA was isolated by using TRizol (Invitrogen) reagent according to manufacturers' protocol. Total RNA concentration and integrity were determined by spectrophotometer readings at absorbance 260nm and 280nm and by observation of the Ribosomal RNA bands in a 1% agarose gel electrophoresis respectively. To avoid DNA contamination, the RNA was previously treated for 30 min at 37ºC with 1 U RQ1 RNase-Free DNase (Promega) in presence of 20 U RNAseOUTTM (Invitrogen) RNAse Inhibitor and the DNase was inactivated by a 95ºC incubation for 15 min and immediately chilled on ice. First strand cDNA synthesis was performed using SuperScriptTM II Reverse Transcriptase (Invitrogen) as suggested by the manufacturer using 5µg of total RNA. Resultant cDNA was then used for PCR as described below.
QUANTITATIVE REAL-TIME PCR
Expression analysis of mRNA was performed in the ABI PRISM 7700 sequence detection system (Applied Biosystem, EUA) using SYBR®-Green amplification detection system. Each reaction was performed in a final volume of 20µL using cDNA reversed transcribed from 25ng of the RNA, 10µL of the SYBR®-Green Universal PCR Master Mix and 1µL of each forward and reverse primers (10µM each) shown below. We conducted the Real-Time PCR reactions separately using the following temperature protocol: 50ºC-2 min, 95ºC-10 min, and 50 cycles of 95ºC-15s and 60ºC-1 min, followed by a dissociation curve protocol to check the specificity of the amplicon produced in each reaction. To check reaction efficiency, we previously run standard curves for each primer set and cDNA sample. As the efficiency of all reactions was approximately 100% (> 95%), we were able to use 2-ΔCt parameter to express relative expression data, taking TBP (TATA binding protein) as endogenous control. Real-Time PCR experiments were submitted to the Student t test with a significance level set at p<0.05. Primers, forward and reverse, respectively, are represented in the 50  30 direction:
30 direction:
β-tubulin III: AGACCTACTGCATCGACAATGAAG
and GCTCATGGTAGCAGACACAAGG;
GFAP: AAGAGTGGTATCGGTCCAAGTTTG
and CAGTTGGCGGCGATAGTCAT;
EGF: CCAAACGCCGAAGACTTATCC
and CTTATTACCGATGGGATAGCCC;
FGF2: CCAACCGGTACCTTGCTATGA
and TTCGTTTCAGTGCCACATACCA;
IGF1: GCCACACTGACATGCCCAAG and TGCACTTCCTCTACTTGTGTTCTTC;
NT3: TTACAGGTGAACAAGGTGATGTCC
and CCGGCAAACTCCTTTGATCC;
PDGFa: CATTCGCAGGAAGAGAAGTATTG
and CTGGTCTTGCAAACTGCGGG;
PDGFb: GAAAGCTCATCTCGAGGGAGG
and GCGTCTTGCACTCGGCG;
Nestin: TGACCATTTAGATGCTCCCCAG
and GTCCATTCTCCATTTTCCCATTC;
TBP: GAATCTTGGCTGTAAACTTGACCT
and TCTTATTCTCATGATGACTGCAGCA.
RESULTS
To assess if there was a significant difference in growth and in the expression of growth factors and neural specific proteins between the CTR and E/F-less groups, three independent cultures of mNPC derived from E14 embryos were subjected to growth factors deprivation for 11 days, followed by analyses of proliferation, cell death and differentiation.
To confirm if removal of EGF and FGF-2 would have an effect in the proliferation rates of neurospheres in suspension, BrdU incorporation and growth rates of isolated neurospheres were evaluated. BrdU labeling decreased 50% after growth factor withdrawal (Fig. 1). The low proliferation rates could implicate in a lack of size increment for the E/F-less group, as observed in the growth curves (Fig. 2). However, the percentage of TUNEL positive cells did not change significantly during the growth factors starvation process, when compared to the CTR group (Fig. 3).
Given that growth factors removal leads to a decrease in cell proliferation, we expected to find cells undergoing differentiation, even in suspension. Our results show that staining of mNPC for neuronal and astrocytic markers (β-tubulin III and GFAP respectively) yielded a different distribution and localization of neurons in the E/F-less and CTR groups. GFAP was found across the whole neurosphere whereas β-tubulin III positive cells were concentrated in the neurosphere core (Fig. 4). In order to check if there is an increase in the transcripts of β-tubulin III, gfap and nestin, we performed quantitative PCR. The results showed a decreased expression of nestin and gfap, but a tendency of increase in β-tubulin III expression in the E/F-less group when compared to CTR (Fig. 5).
Our results showed important differences in the proliferation and distribution of cell populations within the neurosphere after growth factors withdrawal. Then, we asked if growth factors produced by the NPC could be differentially expressed and play a role in cell survival and differentiation after EGF and FGF-2 removal. The expression of a number of growth factors that are relevant for cell proliferation and survival (egf - Epidermal Growth Factor, fgf-2 - Fibroblast Growth Factor 2, igf-1 - Insulin-like Growth Factor 1, nt3 - Neurotrophin 3, pdgfa and pdgfb - Plateled Derived Growth Factor α and β) was measured in the CTR and the E/F-less groups by using the Real-Time PCR technique. Our Real-Time PCR results showed that, after growth factors withdrawal, mNPC decreased the expression of fgf-2 and increased the expression of igf-1 and pdgfb (Fig. 5).
DISCUSSION
This work demonstrates that mNPC can survive and differentiate in suspension after EGF and FGF-2 removal, but have reduced its proliferation capability. Only very few BrdU positive cells were found in the E/F-less groups (Fig. 1). The decreased BrdU incorporation after starvation suggests that EGF and FGF-2 have a crucial role for the maintenance of mNPC cell cycling. Measurement of growth rates also showed that neurospheres subjected to growth factor removal did not grow during the period of growth factor absence (Fig. 2). However, cell death percentage is around 5% in both CTR and E/F-less groups, indicating that cells can survive in this condition. The same cell survival under growth factor deprivation must be due to the differential expression of growth factors and cytokines in the neurosphere. Immunocytochemistry assays showed that mNPC cultured in suspension differentiated into the three basic neural cell types: neurons, astrocytes and a few oligodendrocytes (data not shown), in both the E/F-less and CTR groups. This reproduces in vitro the development of the cerebral cortex in vivo (Wright et al. 2006).
Campos and colleagues (2004) have recently proposed a three-dimensional model of mouse neurospheres based on the expression of specific proteins and BrdU labeling. This model shows the presence of immature (Nestin+) and cycling (BrdU+) cells in the outer layer of the neurosphere, as well as laminin α2 and integrin β1. Yet, the astrocytic marker GFAP is expressed across the whole neurosphere, while some extracellular matrix proteins and the neuronal marker β-tubulin III are preferably expressed in the neurosphere core. Our findings are in agreement with the mouse model forGFAPand β-tubulin III of mNPC cultured in suspension in the presence of EGF and FGF-2 (CTR group) (Fig. 4). We hypothesize that this distribution of neurons is caused by a decreased gradient of growth factors from the outer layer to the center of neurospheres. Since the concentration of EGF and FGF-2 inside the sphere might be lower than on the outside in the CTR condition, cells in the neurosphere core are able to stop proliferation and start differentiation even in suspension.
On the other hand, in the E/F-less group, with the absence of growth factors in the culture medium, neurons concentrated on the border of the neurospheres, whereas GFAP positive cells had the same localization (Fig. 4). We hypothesize that, in the CTR group, cells from the neurosphere core are exposed to a lower concentration of growth factors than are the cells from the borders, because of the gradient. This condition induces the cells from the core to initiate differentiation. After growth factor removal, cells on the borders of the neurosphere (that lack growth factors) initiate differentiation, while cells from the core produce growth factors, maintaining these cells in an undifferentiated state. In the core of the growth factor deprived neurospheres, the smaller concentrations of such autocrine production of growth factors, as compared to exogenous administration in the medium, would be sufficient to maintain an undifferentiated state while insufficient to trigger mitotic activity, as shown by BrdU uptake experiments and growth rates of neurospheres (Figs. 1, 2). This represents a reasonable explanation for understanding the altered distribution of neuronal phenotypes (core versus shell) as a consequence of growth factor withdrawal.
Mouse neurospheres are known to produce a variety of growth factors when they are subjected to growth factor deprivation (Einstein et al. 2006). Our Real-Time PCR results showed that mNPC from the E/F-less group decreased the expression of fgf-2, gfap and nestin, and increased the expression of igf-1 and pdgfb (Fig. 5). Taken together, these results suggest that, during EGF and FGF-2 removal, there is an alteration in the expression of growth factors and of neural specific proteins (gfap, β-tubulin III and nestin). When we removed growth factors from the culture media, cells stopped proliferating and started differentiating. Therefore, the decreased expression of nestin, a marker for stem cells or immature neural progenitors, was expected. Also, given that we encountered a tendency of increased transcripts of β-tubulin III, we suggest that growth factor removal preferentially lead to neuronal differentiation rather than glial differentiation, when compared to the standard culture media. In addition, as previously mentioned, gfap can be expressed by immature neural progenitors. In this way, if growth factor withdrawal induces differentiation of cells in suspension, the number of neural stem/immature cells should be expected to decrease. This, in part, could explain the difference in the level of differentiation and in the neuronal distribution in the neurospheres before and after growth factor removal.
Real-Time PCR experiments (Fig. 5) clearly demonstrate that endogenous production of growth factors shifts from FGF-2 to IGF-1/PDGFb upon growth factor withdrawal. Erickson and co-workers (2008) showed that the addition of low concentrations of IGF-1 was necessary for continued passaging and that NPC survived for long periods in culture without EGF or FGF-2 when IGF-1 was added to the media. It has been suggested that IGF-1 effects on NPC lifespan were mediated by means of its own tyrosine kinase receptor. The increased expression of PDGFb can be related to cell survival upon growth factor withdrawal (Ishii et al. 2008), since the PDGFb receptor signaling in neurospheres can prevent apoptosis. The hypothesis that the differential expression of igf-1 and pdgfb in the E/F-less group is important to maintain cell survival is in agreement with the proportion of TUNEL positive cells (only 5%, for both groups). It is also known that PDGF influences the generation of neurons and glia during embryogenesis and in early postnatal life (Forsberg-Nilsson et al. 2003). Again, it can be related to the decreased expression of gfap and nestin. Thus, the increased expression of igf-1 and pdgfb, induced by the absence of EGF and FGF-2 in the culture media, may play a role in cell death and differentiation mechanisms in mNPC.
To conclude, the main findings of this work are that, under EGF and FGF-2 starvation, mNPC: i) stop growing; ii) show increased expression of igf-1 and pdgfb that can promote cell survival; iii) have changes in the distribution of β-tubulin III+ cells according to the availability of growth factors; iv) induce differentiation of cells in suspension. These results may contribute to the understanding of EGF and FGF-2 mechanisms on NPC differentiation and are relevant for defining the ideal culture medium for neural progenitors expansion in vitro. In addition, these findings could be useful for therapies using NPC aiming at improved lifespan and integration of pre-differentiated cells after transplantation. Our findings suggest that depriving NPC of growth factors prior to grafting might enhance their chance to effectively integrate into the host.
ACKNOWLEDGMENTS
TT Schwindt, GF Barnabé and ME Calcagnotto are Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) fellows; AO Guimarães is Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) fellow; supported by grants from Institutos do Milênio de Bioengenharia Tecidual and CNPq (Brazil); FGF-2 was kindly gifted by Prof. Hugo Aguirre Armelin, from Instituto de Química, Universidade de São Paulo.
JACOBSON M. 1991. The germinal cell, histogenesis, and lineages of nerve cells. Plenum Press, London.
Manuscript received on June 27, 2008; accepted for publication on December 16, 2008; presented by LUIZ R. TRAVASSOS
In commemoration of the 75th anniversary of Escola Paulista de Medicina/ Universidade Federal de São Paulo.
- BJORNSON CR, RIETZE RL, REYNOLDS BA, MAGLI MC AND VESCOVI AL. 1999. Turning brain into blood: a hematopoietic fate adopted by adult neural stem cells in vivo Science 283: 534-537.
- CAMPOS LS, LEONE DP, RELVAS JB, BRAKEBUSCH C, FASSLER R, SUTER U AND FFRENCH-CONSTANT C. 2004. Beta1 integrins activate a MAPK signalling pathway in neural stem cells that contributes to their maintenance. Development 131: 3433-3444.
- CICCOLINI F AND SVENDSEN CN. 1998. Fibroblast growth factor 2 (FGF-2) promotes acquisition of epidermal growth factor (EGF) responsiveness in mouse striatal precursor cells: identification of neural precursors responding to both EGF and FGF-2. J Neurosci 18: 7869-7880.
- CLARKE DL, JOHANSSON CB, WILBERTZ J, VERESS B, NILSSON E, KARLSTROM H, LENDAHL U AND FRISEN J. 2000. Generalized potential of adult neural stem cells. Science 288: 1660-1663.
- DUNNETT SB AND BJORKLUND A. 1999. Prospects for new restorative and neuroprotective treatments in Parkinson's disease. Nature 399: A32-A39.
- EINSTEIN O, GRIGORIADIS N, MIZRACHI-KOL R, REINHARTZ E, POLYZOIDOU E, LAVON I, MILONAS I, KARUSSIS D, ABRAMSKY O AND BEN-HUR T. 2006. Transplanted neural precursor cells reduce brain inflammation to attenuate chronic experimental autoimmune encephalomyelitis. Exp Neurol 198: 275-284.
- ERICKSON RI, PAUCAR AA, JACKSON RL, VISNVEI K AND KORNBLUM H. 2008. Roles of insulin and transferrin in neural progenitor survival and proliferation. J Neurosci Res 86: 1884-1894.
- ERIKSSON PS, PERFILIEVA E, BJORK-ERIKSSON T, ALBORN AM, NORDBORG C, PETERSON DA AND GAGE FH. 1998. Neurogenesis in the adult human hippocampus. Nat Med 4: 1313-1317.
- FORSBERG-NILSSON K, ERLANDSSON A, ZHANG XQ, UEDA H, SVENSSON K, NISTÉR M, TRAPP BD, PETERSON AC AND WESTERMARK B. 2003. Oligodendrocyte precursor hypercellularity and abnormal retina development in mice overexpressing PDGF-B in myelinating tracts. Glia 41: 276-289.
- GAGE FH. 1998. Cell therapy. Nature 392: 18-24.
- GAGE FH. 2000. Mammalian neural stem cells. Science 287: 1433-1438.
- GOLDMAN SA AND NOTTEBOHM F. 1983. Neuronal production, migration, and differentiation in a vocal control nucleus of the adult female canary brain. Proc Natl Acad Sci USA 80: 2390-2394.
- GOULD E, TANAPAT P, HASTINGS NB AND SHORS TJ. 1999. Neurogenesis in adulthood: a possible role in learning. Trends Cogn Sci 3: 186-192.
- ISHII Y, MATSUMOTO Y, WATANABE R, ELMI M, FUJIMORI T, NISSEN J, CAO Y, NABESHIMA Y, SASAHARA M AND FUNA K. 2008. Characterization of neuroprogenitor cells expressing the PDGF beta-receptor within the subventricular zone of postnatal mice. Mol Cell Neurosci 37: 507-518.
- KUHN HG AND SVENDSEN CN. 1999. Origins, functions, and potential of adult neural stem cells. Bioessays 21: 625-630.
- KUHN HG, DICKINSON-ANSON H AND GAGE FH. 1996. Neurogenesis in the dentate gyrus of the adult rat: agerelated decrease of neuronal progenitor proliferation. J Neurosci 16: 2027-2033.
- MCKAY R. 1997. Stem cells in the central nervous system. Science 276: 66-71.
- OSTENFELD T AND SVENDSEN CN. 2004. Requirement for neurogenesis to proceed through the division of neuronal progenitors following differentiation of epidermal growth factor and fibroblast growth factor-2-responsive human neural stem cells. Stem Cells 22: 798-811.
- REYNOLDS BA AND WEISS S. 1996. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol 175: 1-13.
- REYNOLDS BA, TETZLAFF W AND WEISS S. 1992. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci 12: 4565- 4574.
- SVENDSEN CN AND CALDWELL MA. 2000. Neural stem cells in the developing central nervous system: implications for cell therapy through transplantation. Prog Brain Res 127: 13-34.
- SVENDSEN CN AND SMITH AG. 1999. New prospects for human stem-cell therapy in the nervous system. Trends Neurosci 22: 357-364.
- SVENDSEN CN, TER BORG MG, ARMSTRONG RJ, ROSSER AE, CHANDRAN S, OSTENFELD T AND CALDWELL MA. 1998. A new method for the rapid and long term growth of human neural precursor cells. J Neurosci Methods 85: 141-152.
- SVENDSEN CN, CALDWELL MA AND OSTENFELD T. 1999. Human neural stem cells: isolation, expansion and transplantation. Brain Pathol 9: 499-513.
- THOMSON JA, ITSKOVITZ-ELDOR J, SHAPIRO SS WAKNITZ MA, SWIERGIEL JJ, MARSHALL VS AND JONES JM. 1998. Embryonic stem cell lines derived from human blastocysts. Science 282: 1145-1147.
- VAN PRAAG H, SCHINDER AF, CHRISTIE BR, TONI N, PALMER TD AND GAGE FH. 2002. Functional neurogenesis in the adult hippocampus. Nature 415: 1030-1034.
- WRIGHT LS, PROWSE KR, WALLACE K, LINSKENS MH AND SVENDSEN CN. 2006. Human progenitor cells isolated from the developing cortex undergo decreased neurogenesis and eventual senescence following expansion in vitro. Exp Cell Res 312: 2107-2120.
Publication Dates
-
Publication in this collection
19 Aug 2009 -
Date of issue
Sept 2009
History
-
Accepted
16 Dec 2008 -
Received
27 June 2008