Abstract
Abstract: Cyclodextrin glycosyltransferases (CGTases) are important enzymes in the biotechnology field because they catalyze starch conversion into cyclodextrins and linear oligosaccharides, which are used in food, pharmaceutical and cosmetic industries. The CGTases are classified according to their product specificity in α-, β-, α/β- and γ-CGTases. As molecular markers are the preferred tool for bacterial identification, we employed six molecular markers (16S rRNA, dnaK, gyrB, recA, rpoB and tufA) to test the identification of a CGTase-producing bacterial strain (DF 9R) in a phylogenetic context. In addition, we assessed the phylogenetic relationship of CGTases along bacterial evolution. The results obtained here allowed us to identify the strain DF 9R as Paenibacillus barengoltzii, and to unveil a complex origin for CGTase types during archaeal and bacterial evolution. We postulate that the α-CGTase activity represents the ancestral type, and that the γ-activity may have derived from β-CGTases.
Key words
Cyclodextrin glycosyltransferases; housekeeping genes; Paenibacillus; 16S rRNA
INTRODUCTION
During a prospection of cyclodextrin glycosyltransferases (CGTase)-producing soil microorganisms, several bacteria were identified through biochemical tests (FerrarottiFERRAROTTI SA, ROSSO AM, MARÈCHAL MA, KRYMKIEWICZ N and MARÈCHAL L. 1996. Isolation of two strains (S-R type) of Bacillus circulans and purification of a cyclomaltodextrin-glucanotransferase. Cell Mol Biol 42: 653-657. S.A., unpublished data). Among them, a peculiar strain -DF 9R- attracted our attention because of its high cyclodextrin (CD) producing activity and its concomitant potential for industrial exploitation. Moreover, this strain produces a unique CGTase whose structure allowed clarifying the induced fit mechanism of these enzymes (CostaCOSTA H, DISTÉFANO AJ, MARINO-BUSLJE C, HIDALGO A, BERENGUER J, BISCOGLIO DE JIMÉNEZ BONINO M and FERRAROTTI SA. 2012. The residue 179 is involved in product specificity of the Bacillus circulans DF 9R cyclodextrin glycosyltransferase. Appl Microbiol Biotechnol 94: 123-130. et al. 2012). At the time, the strain DF 9R was identified as Bacillus circulans using morphological, physiological and biochemical characteristics (Ferrarotti et al. 1996). However, the current widespread use of molecular markers for bacterial species identification, and the numerous taxonomic re-classifications carried-out within the genus Bacillus (AshASH C, PRIEST FG and COLLINS MD. 1993. Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test. Proposal for the creation of a new genus Paenibacillus. Antonie Van Leeuwenhoek 64: 253-260. et al. 1993, KaulpiboonKAULPIBOON J, PRASONG W, RIMPHANITCHAYAKIT V, MURAKAMI S, AOKI K and PONGSAWASDI P. 2010. Expression and characterization of a fusion protein-containing cyclodextrin glycosyltransferase from Paenibacillus sp. A11. J Basic Microbiol 50: 427-435. et al. 2010, DeakDEAK T. 2011. A survey of current taxonomy of common foodborne bacteria. Acta Alimentaria 40: 95-116. 2011, ZhaoZHAO B, LU W, ZHANG S, LIU K, YAN Y and LI J. 2017. Reclassification of Bacillus saliphilus as Alkalicoccus saliphilus gen. nov., comb. nov., and description of Alkalicoccus halolimnae sp. nov., a moderately halophilic bacterium isolated from a salt lake. Int J Syst Evol Microbiol 67: 1557-1563. et al. 2017) led us to question the identification of strain DF 9R. As well, the classification of CGTase-producing bacteria has been under review (Kaulpiboon et al. 2010). All these revisions were stimulated by the availability of molecular tools (WeisburgWEISBURG WG, BARNS SM, PELLETIER DA and LANE DJ. 1991. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173: 697-703. et al. 1991, RajendhranRAJENDHRAN J and GUNASEKARAN P. 2011. Microbial phylogeny and diversity: Small subunit ribosomal RNA sequence analysis and beyond. Microbiol Res 166: 99-110. and Gunasekaran 2011). In particular, the small subunit of the ribosomal RNA gene (16S rRNA) has been widely used to detect, identify and classify microorganisms due to its ubiquity and ample nucleotidic variation range, based on the presence of conserved and variable regions (SantosSANTOS SR and OCHMAN H. 2004. Identification and phylogenetic sorting of bacterial lineages with universally conserved genes and proteins. Environ Microbiol 6: 754-759. and Ochman 2004, Rajendhran and Gunasekaran 2011, HwangHWANG M, KIM MS, PARK KU, SONG J and KIM EC. 2011. tuf gene sequence analysis has greater discriminatory power than 16S rRNA sequence analysis in identification of clinical isolates of coagulase-negative staphylococci. J Clin Microbiol 49: 4142-4149. et al. 2011, VosVOS M, QUINCE C, PIJL AS, DE HOLLANDER M and KOWALCHUK GA. 2012. A Comparison of rpoB and 16S rRNA as Markers in Pyrosequencing Studies of Bacterial Diversity. PLoS ONE 7: e30600. et al. 2012). Still, some drawbacks of rRNA genes have been raised in relation to their multigenic nature (i.e, the presence in multiple copies per bacterial genome) and potential contribution to intragenomic variation (LeeLEE ZM, BUSSEMA CA and SCHMIDT TM. 2009. Documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res 37: 489-493. et al. 2009, ZengZENG YH, KOBLÍŽEK M, LI YX, LIU YP, FENG FY, JI JD, JIAN JC and WU ZH. 2013. Long PCR-RFLP of 16S-ITS-23S rRNA genes: a high-resolution molecular tool for bacterial genotyping. J Appl Microbiol 114: 433-447. et al. 2013). Nowadays, housekeeping genes have become the preferred complementary tools for modern bacterial taxonomy (TanabeTANABE Y, KASAI F and WATANABE MM. 2007. Multilocus sequence typing (MLST) reveals high genetic diversity and clonal population structure of the toxic cyanobacterium Microcystis aeruginosa. Microbiology 153: 3695-3703. et al. 2007, PorwalPORWAL S, LAL S, CHEEMA S and KALIA VC. 2009. Phylogeny in Aid of the Present and Novel Microbial Lineages: Diversity in Bacillus. PLoS One 4: e4438. et al. 2009, Hwang et al. 2011, Vos et al. 2012, GomesGOMES ACSM, SANTOS SRD, RIBEIRO MC, CRAVO PVL, VIEIRA JDG, SOUZA KMC and AMARAL AC. 2018. Is there still room to explore cyclodextrin glycosyltransferase producers in Brazilian biodiversity? An Acad Bras Cienc 90: 1473-1480. et al. 2018); they are single copy markers that evolve faster than the 16S rRNA and show scarce indel events (Santos and Ochman 2004).
Bacterial CGTases are key metabolic enzymes produced by a wide variety of microorganisms such as Bacillus, Brevibacillus, Geobacillus, Gracilibacillus, Paenibacillus, Solibacillus, Klebsiella, Anaerobranca, Pyrococcus, Thermoanaerobacter, Thermoanaerobacterium and Thermococcus (CostaCOSTA H, GASTÓN JR, LARA J, MARTINEZ CO, MORIWAKI C, MATIOLI G and FERRAROTTI SA. 2015. Cyclodextrin glycosyltransferase production by free cells of Bacillus circulans DF 9R in batch fermentation and by immobilized cells in a semi-continuous process. Bioprocess Biosyst Eng 38: 1055-1063. et al. 2015, Gomes et al. 2018). These enzymes are members of the Glycoside Hydrolase family 13, also known as the α-amylase family (LombardLOMBARD V, GOLACONDA RAMULU H, DRULA E, COUTINHO PM and HENRISSAT B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42: 490-495. et al. 2014), and catalyze the bioconversion of starch. This polysaccharide is the main and most ubiquitous plant reserve substance, and a significant source of energy for many animals and microorganisms. The CGTases are classified as α, β, α/β and γ according to their product specificity (Costa et al. 2012). The product of CGTases´ activity consists of a mixture of cyclic, linear and limit dextrins, among which the CDs are the most important. These are non-reducing maltooligosaccharides with a hydrophilic external surface and a hydrophobic central cavity. Because CDs molecules can form inclusion complexes with several compounds, they are extensively used in food, pharmaceutical and cosmetic industries (KurkovKURKOV SV and LOFTSSON T. 2013. Cyclodextrins. Int J Pharm 30: 167-180. and Loftsson 2013, Li et al. 2014LI Z, CHEN S, GU Z, CHEN J and WU J. 2014. Alpha-cyclodextrin: Enzymatic production and food applications. Trends Food Sci Technol 35: 151-160.). The most common are the α-, β-, and γ-CDs, which comprise six, seven or eight glucose residues, respectively, linked by α-1,4 bonds (Costa et al. 2015). The relative composition of CDs mixtures mainly depends on the type of the CGTase involved in the catalytic reactions. In the present study we firstly confronted the identification as B. circulans of the rotten potato-isolated bacterial strain DF 9R by employing six molecular markers and a phylogenetic analytical context. We also explored, for this particular strain, the occurrence of intragenomic variation within the 16S rRNA gene. Afterwards, we investigated on the phylogenetic relationships of CGTase-types across an ample taxonomic sampling of bacteria with experimentally proved product specificity.
MATERIALS AND METHODS
STRAIN AND CULTURE CONDITIONS
The strain DF 9R was isolated from rotten potatoes (Ferrarotti et al. 1996) and deposited in the “Colección de Cultivos Microbianos, FFyB, UBA”, catalog number CCM-A-29:1290 from the World Federation for Culture Collections. The strain was cultured in a minimum saline medium with starch, consisting of 1.5% cassava starch, 0.4% (NH4)2SO4, 100 mM phosphate buffer pH 7.6, 0.002% MgSO4 and 0.002% FeSO4 and incubated at 37°C and 120 rpm for 48 h (RossoROSSO AM, FERRAROTTI SA, KRYMKIEWICZ N and NUDEL BC. 2002. Optimization of batch culture conditions for cyclodextrin glucanotransferase production from Bacillus circulans DF 9R. Microb Cell Factories 1: 1-10. et al. 2002).
AMPLIFICATION AND SEQUENCING OF MOLECULAR MARKERS
We employed five housekeeping genes, dnaK (coding a heat-shock protein of 70 kDa), gyrB (subunit β of DNA gyrase), recA (recombinase A protein), rpoB (β subunit of RNA polymerase) and tufA (elongation factor Tu), and the 16S rRNA. Sense and antisense primers were designed for dnaK, recA, rpoB and tufA using Primer3Plus software (UntergasserUNTERGASSER A, NIJVEEN H, RAO X, BISSELING T, GEURTS R and LEUNISSEN JAM. 2007. Primer3Plus, an enhanced web interface to Primer3. Nucl Acids Res 35: W71-W74. et al. 2007). The rpoB gene was amplified in two non-overlapping parts (rpoBi and rpoBf). For 16S rRNA and gyrB we used primers taken from the literature (Weisburg et al. 1991, YamamotoYAMAMOTO S and HARAYAMA S. 1995. PCR amplification and direct sequencing of gyrB genes with universal primers and their application to the detection and taxonomic analysis of Pseudomonas putida strains. Appl Environ Microbiol 61: 1104-1109. and Harayama 1995, GürtlerGÜRTLER V and STANISICH VA. 1996. New approaches to typing and identification of bacteria using the 16S-23S rDNA spacer region. Microbiology 142: 3-16. and Stanisich 1996).
Genomic DNA was extracted as described previously (Costa et al. 2012). PCR amplifications were carried out in a 25 μL final reaction mixture, containing 0.2 mM each dNTPs, 1.0 μM primers, 2.5 U Pfx DNA polymerase (Thermo Fisher Scientific), 1X Pfx amplification buffer, 1 mM MgSO4 and 20 ng genomic DNA. Amplification conditions were: 3 min at 94°C, followed by 35 cycles of 40 s at 94°C, 30 s at 52°C (for dnaK, tufA and recA), at 55°C (for rpoBi and rpoBf ), at 58.5°C (for the gyrB) or at 60°C (for 16S rRNA), and extension for 1 min (for recA and tufA) or 2 min (for the dnaK, gyrB, rpoBi, rpoBf and 16S rRNA) at 68°C. PCR products were separated through 1% agarose gels electrophoreses and visualized under UV light after staining with ethidium bromide; selected bands were subsequently purified with the Wizard® SV Gel and PCR Clean-Up System (Promega) according to manufacturer’s instructions. Nucleotide sequences were obtained via bidirectional automated sequencing using ABI PRISM® BigDye Terminator Cycle Sequencing Ready Reaction Kit reagent (Applied Biosystems, Foster City, CA, USA) in an Applied Biosystems 3130xl Genetic Analyzer. The primers used for sequencing were the same used for amplification (Table I). The nucleotide sequences of dnaK, gyrB, recA, rpoB, tufA and 16S rRNA obtained herein from strain DF 9R, were deposited in GenBank (Table SI).
To investigate the presence of 16S rRNA intragenomic variants, an aliquot of the purified PCR product was cloned into a pCR2.1-TOPO Cloning vector with the TOPO TA cloning kit (Thermo Fisher Scientific) according to the manufacturer´s protocols. Recombinant clones were selected on Luria Broth agar plates supplemented with 100 μg/mL ampicilin and 50 μg/mL X-Gal, and subsequently isolated using the Wizard® Plus SV Minipreps DNA Purification System (Promega). To confirm the presence of the insert, plasmids were digested with 1 U of EcoRI (Thermo Fisher Scientific) for 60 min at 37 ºC, and then visualized on agarose gel electrophoresis, as detailed above. Twenty recombinant clones were sequenced, as above, using M13 primers.
SEQUENCE AND PHYLOGENETIC ANALYSES
Sequences were manually edited for ambiguity in Bioedit version 7.2.5 (HallHALL TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp 41: 95-98. 1999). The identity of each amplified region was verified through Blast searches against the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi) using a stringent threshold e-value ≤ 10-130.
A matrix was generated consisting solely of the 20 cloned 16S rRNA sequences from strain DF 9R. After manual alignment, it was used to estimate sequence variation as the number of observed differences over the total length (i.e., uncorrected p-distance) in MEGA 6.0 (TamuraTAMURA K, STECHER G, PETERSON D, FILIPSKI A and KUMAR S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30: 2725-2729. et al. 2013).
To generate data matrices (one per marker region) we selected curated nucleotide sequences from the leBIBI (FlandroisFLANDROIS J, PERRIÈRE G and GOUY M. 2015. leBIBIQBPP: A set of databases and a web tool for automatic phylogenetic analysis of prokaryotic sequences. BMC Bioinformatics 16: 251. et al. 2015) and Uniprot (BatemanBATEMAN A et al. 2014. The UniProt Consortium. UniProt: a hub for protein information. Nucl Acids Res 43: 204-212. et al. 2014) databases that included several sequences from type strains. Sequences data from all the bacteria included are detailed in Supplementary Material (Table SI, SII) SUPPLEMENTARY MATERIAL .
Alignments were carried out using MUSCLE program (EdgarEDGAR RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32: 1792-1797. 2004) with default settings, as implemented in MEGA 6.0 (Tamura et al. 2013).
For coding regions, we firstly translated the nucleotides to protein sequences in MEGA and then we performed the alignments; afterwards aligned sequences were converted to nucleotidic bases for further analyses. Thus, the data matrix for dnaK consisted of 1857 bp, gyrB of 1155 bp, recA of 816 bp, rpoB of 2520 bp, tufA of 957 bp and that for 16S rRNA of 1446 bp. Subsequently, a combined matrix was constructed by concatenating the pre-aligned individual marker matrices in Bioedit; it spanned 8838 bp. For the phylogenetic analyses, we firstly selected the best-fitting substitution model for each matrix by using the information criteria implemented in MEGA. Analyses were accomplished under the Maximum Likelihood criterion, with Subtree Pruning and Regrafting (SPR level 3) heuristic search method and a strong swapping filtering option. All positions with less than 50% site coverage were eliminated from the analyses. For rooting purposes, we included sequences from outgroup taxa, these are indicated in the corresponding figure caption. Bootstrap support values were estimated with 500 pseudoreplicates in MEGA.
PHYLOGENETIC ANALYSIS OF CGTASES
To evaluate the relationship of the different CGTase-types, only bacterial sequences with demonstrated enzymatic activity (i.e., published) and with clearly established product specificity were used to construct a data matrix. These sequences were retrieved from the Carbohydrate Active Enzymes (CAZy) database (Lombard et al. 2014). Sequence data used for this analysis are detailed in Table SII. CGTase nucleotide sequences were translated into protein sequences and then aligned with MUSCLE, using default settings; this yielded a matrix of 680-aligned residues. After model selection, the CGTase matrix was analyzed under Maximum Likelihood, as previously described. We included sequences from Pyrococcus furiosus, Thermococcus kodakaraensis, Thermococcus sp., Klebsiella pneumoniae and Haloferax mediterranei, for rooting purposes.
In addition, the ancestral CGTase activity-type was obtained at each node (i.e., optimized) over the Maximum Likelihood phylogram. For this, only the branching pattern for the species (i.e., the topology) was imported into TNT version 1.1 (GoloboffGOLOBOFF P, FARRIS J and NIXON K. 2008. TNT: tree analysis using new technology. Cladistics 24: 1-13. et al. 2008) in parenthetical notation. Optimization was accomplished using FarrisFARRIS J. 1970. Methods for computing Wagner trees. Syst Zool 19: 83-92. (1970) optimization under the Maximum Parsimony criterion as implemented in TNT, considering the CGTase activity-types as a single multistate character (states: α-, β-, γ-, α/β-), and with equally weighted transformations among the four states. The effect of uncertainty was evaluated by considering the different resolutions and by enumerating all possible most parsimonious reconstructions in the case of ambiguity.
The GenBank accession number of the dnaK, gyrB, recA, rpoB, tufA and 16S rRNA sequences from strain DF 9R reported in this paper are KM357898, KM357899, KM357900, KM357902, KM357901 and KM357896, respectively.
RESULTS
Two variants of the 16S rRNA gene were detected in the bacterial genome of strain DF 9R, differing in six positions along the gene (one C-T and four A-G transitions, and one A-T transversion; p-distance = 0.424%).
Phylogenetic analyses performed on single-marker matrices generated topologies which were congruent in the unequivocal location of strain DF 9R within Paenibacillus clades (97.6-100% of bootstrap support, BS) (Supplementary Material, Figures S1-S6). Moreover, strain DF 9R showed a sister relationship with P. barengoltzii when the inferences were derived from dnaK, gyrB, recA and tufA (58-94.8% BS) (Supplementary Material, Figures S2-S4 and S6, respectively). The rpoB topology did not contradict this (Supplementary Material, Figure S5).
The analysis of the 35 taxa concatenated matrix verified that the strain DF 9R locates within a Paenibacillus clade (100% BS; Fig. 1), and its relationship with P. barengoltzii was retrieved with full support (100% BS). Also, a strong sister-group relationship was recovered between Paenibacillus and the clade formed by Brevibacillus and Aneurinibacillus (100% BS). Furthermore, three lineages were detected for the members of Bacillus, one related to bacteria from Geobacillus, Anoxybacillus and Planococcus, and two lineages formed solely by Bacillus representatives.

Maximum likelihood phylogram derived from the multi-locus concatenated matrix. The phylogram (lnL= -138304.54) was obtained by applying the general time reversible (NeiNEI M and KUMAR S. 2000. Molecular Evolution and Phylogenetics. Oxford University Press. and Kumar 2000) substitution model together with a discrete Gamma distribution (G= 0.48, with 5 categories) to model evolutionary rate differences among sites, and 30.71% of invariant sites (I); 8838 positions were considered. Branch lengths are in number of substitutions per site. Lactobacillus acidophilus was used for rooting. Bootstrap support values > 50% are shown on the branches. The arrow indicates strain DF 9R.
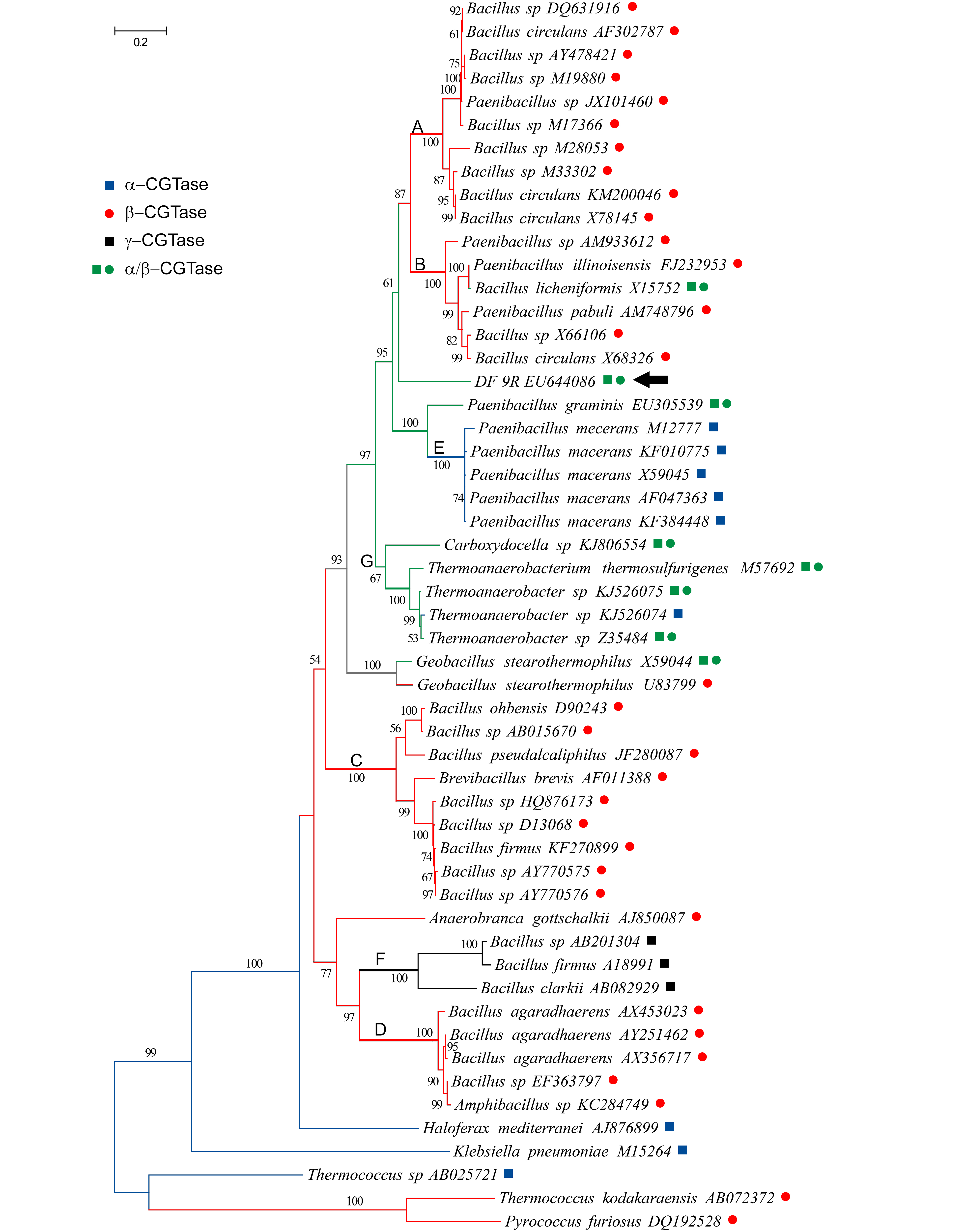
The phylogenetic analysis of 53 CGTase amino acid sequences showed that the most abundantly sampled CGTases, the β-type, were scattered over the phylogram, forming at least four highly supported clades (A-D in Fig. 2). The CGTase protein sequence from strain DF 9R appeared as the sister group of the clades A and B, though with moderate support (61% BS). The α-CGTases formed a clade composed by accessions of Paenibacillus macerans (clade E, 100% BS), whereas the γ-CGTases formed another clade (F, 100% BS). Finally, the clade G (67% BS) included four α/β-CGTases and one sequence of an α-CGTase producing bacteria.

Maximum likelihood phylogram derived from the analysis of CGTase protein sequences. The phylogram (lnL= -21347.60) shown was obtained by applying the LeLE SQ and GASCUEL O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol 25: 1307-1320. and Gascuel (2008) model plus a discrete Gamma distribution (G= 2.12, with 5 categories) were used to model evolutionary rate differences among sites. A total of 680 residues were considered. Branch lengths are measured in number of substitutions per site. Bootstrap support values > 50% are shown on the branches. For rooting we used archeal taxa indicated in Materials and Methods section. The inference of ancestral CGTase activities (optimization, see Materials and Methods section) is indicated with symbols and colors: a blue square indicates α-CGTase activity; a red dot, β-CGTase; a black square, γ-CGTase; green square and dot, α/β-CGTase; branches in grey indicate ambiguous optimization. Bold capital letters (A-G) mark the clades described in the text. The arrow indicates strain DF 9R.
When the CGTase activity types were optimized over the topology obtained, the ancestral activity appeared to be the α-type (in blue, Fig. 2). Then, this enzymatic activity re-appeared twice, in parallel. The β-activity (in red) also appeared in parallel, at least four times; twice from an α-CGTases ancestor and twice from α/β-CGTase precursor (in green). The γ-CGTases (in black) appeared once from β-forms. The α/β-CGTases may have originated two or three times depending on the most parsimonious reconstruction of the ambiguous node (marked in grey, Fig. 2), but in either case, it was from β-activities.
DISCUSSION
Results obtained herein allow us to postulate that the cyclodextrin glycosyltransferase-producing bacterial strain DF 9R is a member of Paenibacillus, being P. barengoltzii the favored species identification. Several hydrolases produced by species of Paenibacillus have been described. Particularly, a pullulanase produced by P. barengoltzii has been reported (LiuLIU J, LIU Y, YAN F, JIANG Z, YANG S and YAN Q. 2016. Gene cloning, functional expression and characterisation of a novel type I pullulanase from Paenibacillus barengoltzii and its application in resistant starch production. Protein Expression and Purification 121: 22-30. et al. 2016). Pullulanases, like CGTases, belong to the family 13 of glycoside hydrolases (Lombard et al. 2014). As no CGTase activity has been documented for P. barengoltzii so far, the present report would be the first record. Moreover, our results suggested that the genus Bacillus seems to be an artificial arrange of species (i.e., not forming a monophyletic group) and thus, additional surveys would be needed to generate a classification based on natural groups.
The molecular markers used in this study proved useful for bacterial taxonomic identification when assayed in a phylogenetic context. MartensMARTENS M, DAWYNDT P, COOPMAN R, GILLIS M, DE VOS P and WILLEMS A. 2008. Advantages of multilocus sequence analysis for taxonomic studies: a case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int J Syst Evol Microbiol 58: 200-214. et al. (2008) recommended using at least two housekeeping genes for bacterial identification, in order to minimize the effect of lateral gene transfer instances. We herein analyzed five housekeeping markers, and the congruent phylogenies obtained reflected a common evolutionary history for these genes, indicating negligible lateral genetic transfer.
Even though results derived from the simultaneous analysis of the six markers were concordant with single-marker analyses, we demonstrated that multi-locus simultaneous analysis yields more robust identification and phylogenetic hypotheses. It is almost certain that in a near future whole genome sequencing will be the ideal tool for bacterial identification. However, as those methodologies are still unaffordable, at least in some countries, multi-locus analyses will continue to be a valuable source of data.
The detection of 16S rRNA intragenomic variants for strain DF 9R is in agreement with the polycistronic nature of ribosomal genes, and also with results of Zeng et al. (2013). These authors encountered up to 14 copies per bacterial genome within Phylum Firmicutes, to which the genus Paenibacillus belongs.
The phylogenetic analysis performed on carefully selected bacterial CGTase sequences with proven product specificity allowed us to postulate that the α-CGTase (or an α-CGTase-like enzyme) could be the ancestral activity from which the other types have diversified (Fig. 2). In the same way, the γ-CGTase activity which appeared as derived might have been acquired during the evolution of β-CGTases. Then, the mixed α/β-CGTases could have originated α- or β-CGTases in several occasions by mutations that could rise or diminish one of the complementary activities. Our protein-based analysis also revealed that the β-CGTase produced by Brevibacillus brevis is more closely related to enzymes from Bacillus (clade C) than to any other β-producer, in accordance to the results of KellyKELLY RM, DIJKHUIZEN L and LEEMHUIS H. 2009. The evolution of cyclodextrin glucanotransferase product specificity. Appl Microbiol Biotechnol 84: 119-133. et al. (2009). We encountered that α-CGTases from different strains and accessions of Paenibacillus macerans are likely to have a common origin (clade E), and that in this group the enzymatic activity may had derived from a protein showing α/β-CGTase activity; the close relationship between P. macerans and P. graminis (α/β-CGTase producer) coincides with results of VolluVOLLU RE, DA MOTA FF, GOMES EA and SELDIN L. 2008. Cyclodextrin production and genetic characterization of cyclodextrin glucanotransferase of Paenibacillus graminis. Biotechnol Lett 30: 929-35. et al. (2008). The two accessions of Geobacillus stearothermophillus used herein were reported to exhibit different activities, namely α/β-CGTase (FujiwaraFUJIWARA S, KAKIHARA H, WOO KB, LEJEUNE A, KANEMOTO M, SAKAGUCHI K and IMANAKA T. 1992. Cyclization characteristics of cyclodextrin glucanotransferase are conferred by the NH2-terminal region of the enzyme. Appl Environ Microbiol 58: 4016-4025. et al. 1992) and β-CGTase (ChungCHUNG HJ et al. 1998. Characterization of a thermostable cyclodextrin glucanotransferase isolated from Bacillus stearothermophilus ET1. J Agricult Food Chem 46: 952-959. et al. 1998). If the data provided by Chung et al. (1998) indicating that G. stearothermophillus are capable of producing α- and β-CDs in comparable amounts is considered, then the ambiguous optimization encountered here (grey branches in Fig. 2) could be solved in favor of an α/β-CGTase ancestral activity for the species involved across clades A, B, E and G, plus the aforementioned P. graminis and P. barengoltzii. Chemical and genetic modifications have been made to understand the CGTases´ structural characteristics determining product specificity (Kelly et al. 2009) and to alter the type of CD generated (LeemhuisLEEMHUIS H, KELLY RM and DIJKHUIZEN L. 2010. Engineering of cyclodextrin glucanotransferases and the impact for biotechnological applications. Appl Microbiol Biotechnol 85: 823-835. et al. 2010), suggesting that the diversity of CDs could be a consequence of specific amino acid substitutions, insertions, and/or deletions in precise active sites (Kelly et al. 2009, Li et al. 2009LI Z, ZHANG J, WANG M, GU Z, DU G, LI J, WU J and CHEN J. 2009. Mutations at subsite -3 in cyclodextrin glycosyltransferase from Paenibacillus macerans enhancing α-cyclodextrin specificity. Appl Microbiol Biotechnol 83: 483-490., XieXIE T, YUE Y, SONG B, CHAO Y and QIAN S. 2013. Increasing of product specificity of gamma-cyclodextrin by mutating the active domain of alpha-cyclodextrin glucanotransferase from Paenibacillus macerans sp. 602-1. Sheng Wu Gong Cheng Xue Bao 29: 1234-1244. et al. 2013, WangWANG L, DUAN X and WU J. 2016. Enhancing the α-Cyclodextrin Specificity of Cyclodextrin Glycosyltransferase from Paenibacillus macerans by Mutagenesis Masking the Subsite -7. Appl Environ Microbiol 82: 2247-2255. et al. 2016).
Forthcoming phylogenetic approaches, like those performed herein and based on our curated protein database, will certainly aid in predicting enzymatic activities when additional sequences from putative or novel CGTases be tested and even could assist in delineating downstream procedures.
ACKNOWLEGMENTS
We thank to Agencia Nacional de Promoción Científica y Tecnológica and Universidad Nacional de Luján. AMG, HC and OAT are career members of the Argentine Council of Scientific and Technical Research (CONICET). JDLMC is a PhD fellow of the Commission of Scientific Research of the Province of Buenos Aires (CIC) at Universidad Nacional de Luján. SCL is PhD student of Universidad Nacional de Luján. This study was funded by Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), PICT 2013-0880 and PICT 2016-0240.
REFERENCES
- ASH C, PRIEST FG and COLLINS MD. 1993. Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test. Proposal for the creation of a new genus Paenibacillus. Antonie Van Leeuwenhoek 64: 253-260.
- BATEMAN A et al. 2014. The UniProt Consortium. UniProt: a hub for protein information. Nucl Acids Res 43: 204-212.
- CHUNG HJ et al. 1998. Characterization of a thermostable cyclodextrin glucanotransferase isolated from Bacillus stearothermophilus ET1. J Agricult Food Chem 46: 952-959.
- COSTA H, DISTÉFANO AJ, MARINO-BUSLJE C, HIDALGO A, BERENGUER J, BISCOGLIO DE JIMÉNEZ BONINO M and FERRAROTTI SA. 2012. The residue 179 is involved in product specificity of the Bacillus circulans DF 9R cyclodextrin glycosyltransferase. Appl Microbiol Biotechnol 94: 123-130.
- COSTA H, GASTÓN JR, LARA J, MARTINEZ CO, MORIWAKI C, MATIOLI G and FERRAROTTI SA. 2015. Cyclodextrin glycosyltransferase production by free cells of Bacillus circulans DF 9R in batch fermentation and by immobilized cells in a semi-continuous process. Bioprocess Biosyst Eng 38: 1055-1063.
- DEAK T. 2011. A survey of current taxonomy of common foodborne bacteria. Acta Alimentaria 40: 95-116.
- EDGAR RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32: 1792-1797.
- FARRIS J. 1970. Methods for computing Wagner trees. Syst Zool 19: 83-92.
- FERRAROTTI SA, ROSSO AM, MARÈCHAL MA, KRYMKIEWICZ N and MARÈCHAL L. 1996. Isolation of two strains (S-R type) of Bacillus circulans and purification of a cyclomaltodextrin-glucanotransferase. Cell Mol Biol 42: 653-657.
- FLANDROIS J, PERRIÈRE G and GOUY M. 2015. leBIBIQBPP: A set of databases and a web tool for automatic phylogenetic analysis of prokaryotic sequences. BMC Bioinformatics 16: 251.
- FUJIWARA S, KAKIHARA H, WOO KB, LEJEUNE A, KANEMOTO M, SAKAGUCHI K and IMANAKA T. 1992. Cyclization characteristics of cyclodextrin glucanotransferase are conferred by the NH2-terminal region of the enzyme. Appl Environ Microbiol 58: 4016-4025.
- GOLOBOFF P, FARRIS J and NIXON K. 2008. TNT: tree analysis using new technology. Cladistics 24: 1-13.
- GOMES ACSM, SANTOS SRD, RIBEIRO MC, CRAVO PVL, VIEIRA JDG, SOUZA KMC and AMARAL AC. 2018. Is there still room to explore cyclodextrin glycosyltransferase producers in Brazilian biodiversity? An Acad Bras Cienc 90: 1473-1480.
- GÜRTLER V and STANISICH VA. 1996. New approaches to typing and identification of bacteria using the 16S-23S rDNA spacer region. Microbiology 142: 3-16.
- HALL TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp 41: 95-98.
- HWANG M, KIM MS, PARK KU, SONG J and KIM EC. 2011. tuf gene sequence analysis has greater discriminatory power than 16S rRNA sequence analysis in identification of clinical isolates of coagulase-negative staphylococci. J Clin Microbiol 49: 4142-4149.
- KAULPIBOON J, PRASONG W, RIMPHANITCHAYAKIT V, MURAKAMI S, AOKI K and PONGSAWASDI P. 2010. Expression and characterization of a fusion protein-containing cyclodextrin glycosyltransferase from Paenibacillus sp. A11. J Basic Microbiol 50: 427-435.
- KELLY RM, DIJKHUIZEN L and LEEMHUIS H. 2009. The evolution of cyclodextrin glucanotransferase product specificity. Appl Microbiol Biotechnol 84: 119-133.
- KURKOV SV and LOFTSSON T. 2013. Cyclodextrins. Int J Pharm 30: 167-180.
- LE SQ and GASCUEL O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol 25: 1307-1320.
- LEE ZM, BUSSEMA CA and SCHMIDT TM. 2009. Documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res 37: 489-493.
- LEEMHUIS H, KELLY RM and DIJKHUIZEN L. 2010. Engineering of cyclodextrin glucanotransferases and the impact for biotechnological applications. Appl Microbiol Biotechnol 85: 823-835.
- LI Z, CHEN S, GU Z, CHEN J and WU J. 2014. Alpha-cyclodextrin: Enzymatic production and food applications. Trends Food Sci Technol 35: 151-160.
- LI Z, ZHANG J, WANG M, GU Z, DU G, LI J, WU J and CHEN J. 2009. Mutations at subsite -3 in cyclodextrin glycosyltransferase from Paenibacillus macerans enhancing α-cyclodextrin specificity. Appl Microbiol Biotechnol 83: 483-490.
- LIU J, LIU Y, YAN F, JIANG Z, YANG S and YAN Q. 2016. Gene cloning, functional expression and characterisation of a novel type I pullulanase from Paenibacillus barengoltzii and its application in resistant starch production. Protein Expression and Purification 121: 22-30.
- LOMBARD V, GOLACONDA RAMULU H, DRULA E, COUTINHO PM and HENRISSAT B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42: 490-495.
- MARTENS M, DAWYNDT P, COOPMAN R, GILLIS M, DE VOS P and WILLEMS A. 2008. Advantages of multilocus sequence analysis for taxonomic studies: a case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int J Syst Evol Microbiol 58: 200-214.
- NEI M and KUMAR S. 2000. Molecular Evolution and Phylogenetics. Oxford University Press.
- PORWAL S, LAL S, CHEEMA S and KALIA VC. 2009. Phylogeny in Aid of the Present and Novel Microbial Lineages: Diversity in Bacillus. PLoS One 4: e4438.
- RAJENDHRAN J and GUNASEKARAN P. 2011. Microbial phylogeny and diversity: Small subunit ribosomal RNA sequence analysis and beyond. Microbiol Res 166: 99-110.
- ROSSO AM, FERRAROTTI SA, KRYMKIEWICZ N and NUDEL BC. 2002. Optimization of batch culture conditions for cyclodextrin glucanotransferase production from Bacillus circulans DF 9R. Microb Cell Factories 1: 1-10.
- SANTOS SR and OCHMAN H. 2004. Identification and phylogenetic sorting of bacterial lineages with universally conserved genes and proteins. Environ Microbiol 6: 754-759.
- TAMURA K, STECHER G, PETERSON D, FILIPSKI A and KUMAR S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30: 2725-2729.
- TANABE Y, KASAI F and WATANABE MM. 2007. Multilocus sequence typing (MLST) reveals high genetic diversity and clonal population structure of the toxic cyanobacterium Microcystis aeruginosa. Microbiology 153: 3695-3703.
- UNTERGASSER A, NIJVEEN H, RAO X, BISSELING T, GEURTS R and LEUNISSEN JAM. 2007. Primer3Plus, an enhanced web interface to Primer3. Nucl Acids Res 35: W71-W74.
- VOLLU RE, DA MOTA FF, GOMES EA and SELDIN L. 2008. Cyclodextrin production and genetic characterization of cyclodextrin glucanotransferase of Paenibacillus graminis. Biotechnol Lett 30: 929-35.
- VOS M, QUINCE C, PIJL AS, DE HOLLANDER M and KOWALCHUK GA. 2012. A Comparison of rpoB and 16S rRNA as Markers in Pyrosequencing Studies of Bacterial Diversity. PLoS ONE 7: e30600.
- WANG L, DUAN X and WU J. 2016. Enhancing the α-Cyclodextrin Specificity of Cyclodextrin Glycosyltransferase from Paenibacillus macerans by Mutagenesis Masking the Subsite -7. Appl Environ Microbiol 82: 2247-2255.
- WEISBURG WG, BARNS SM, PELLETIER DA and LANE DJ. 1991. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173: 697-703.
- XIE T, YUE Y, SONG B, CHAO Y and QIAN S. 2013. Increasing of product specificity of gamma-cyclodextrin by mutating the active domain of alpha-cyclodextrin glucanotransferase from Paenibacillus macerans sp. 602-1. Sheng Wu Gong Cheng Xue Bao 29: 1234-1244.
- YAMAMOTO S and HARAYAMA S. 1995. PCR amplification and direct sequencing of gyrB genes with universal primers and their application to the detection and taxonomic analysis of Pseudomonas putida strains. Appl Environ Microbiol 61: 1104-1109.
- ZHAO B, LU W, ZHANG S, LIU K, YAN Y and LI J. 2017. Reclassification of Bacillus saliphilus as Alkalicoccus saliphilus gen. nov., comb. nov., and description of Alkalicoccus halolimnae sp. nov., a moderately halophilic bacterium isolated from a salt lake. Int J Syst Evol Microbiol 67: 1557-1563.
- ZENG YH, KOBLÍŽEK M, LI YX, LIU YP, FENG FY, JI JD, JIAN JC and WU ZH. 2013. Long PCR-RFLP of 16S-ITS-23S rRNA genes: a high-resolution molecular tool for bacterial genotyping. J Appl Microbiol 114: 433-447.
Publication Dates
-
Publication in this collection
07 Oct 2019 -
Date of issue
2019
History
-
Received
5 June 2018 -
Accepted
10 Jan 2019