Resumos
A tríade clássica da Síndrome de McCune Albright (SMA) - displasia fibrosa poliostótica (DFP), máculas cutâneas "café au lait" e hiper-função autônoma de uma ou mais glândulas endócrinas - envolve mutações ativadoras do gene GNAS1, codificador da proteína Galfas e conseqüente aumento da produção do sinalizador intracelular AMPc em diferentes tecidos. Embora a pseudo-puberdade precoce (PPP) seja a manifestação clínica mais freqüente, são as alterações ósseas que conferem a maior morbidade à síndrome. Apresentamos o seguimento clínico endocrinológico e a evolução da doença óssea em 4 pacientes portadores da SMA, das quais 3 delas estão sendo tratadas com bisfosfonato (Pamidronato, 2mg/kg). Três das 4 pacientes apresentaram pseudo-puberdade precoce e duas delas desenvolveram hipertiroidismo. A variação do nível de fosfatase alcalina (FA) foi o indicador bioquímico da doença óssea, cujas complicações foram dor e fratura além de obstrução do canal lacrimal em uma e amaurose unilateral em outra como conseqüência das deformidades crânio-faciais. A infusão de pamidronato endovenoso produziu melhora das dores ósseas e reduziu (37% - 54%) os níveis de FA em todas as pacientes. Entretanto, a melhora da qualidade do osso com o uso do bisfosfonato ainda é incerta e para a qual necessitamos de estudos randomizados e com longo prazo de observação.
Displasia fibrosa óssea; McCune Albright; Puberdade precoce; Pamidronato
McCune Albright syndrome is a rare disorder characterized by polyostotic fibrous dysplasia, café-au-lait spots and some endocrine hyperfunction, mostly precocious puberty. It is caused by activating mutations of the GNAS1 gene that codifies for the Galphas protein, leading to overproduction of cAMP messenger at different tissues. Although pseudo-precocious puberty is the most frequent disfunction, the skeletal lesions are responsible for the highest morbidities such as bone pain, fractures and deformations. Clinical follow up focusing the bone disease is reported in four patients, three of them been treated with intravenous bisphosphonate (Pamidronate, 2mg/kg). Precocious puberty occurred in three and hyperthyroidism developed in two of the patients. Alkaline phosphatase (AP) levels were used as biochemical markers of the osteolytic lesions, clinically presented by local pain, fracture and bone deformities. In addition, lachrymal obstruction in one patient and unilateral blindness in another appear to be due to deformed skull and facial bones. Intravenous infusion of Pamidronate has been improving bone pain and decreased (37% - 54%) basal AP levels. However, a long-term randomized follow-up study determining improvement in bone quality still needs to be completed.
Bone fibrous dysplasia; McCune Albright syndrome; Precocious puberty; Pamidronate
artigo original
Evolução da Displasia Fibrosa Óssea na Síndrome de McCune Albright
Helena L. Farhat
Magnus Régios D. da Silva
Marise Lazaretti-Castro
Ieda T. N. Verreschi
Disciplina de Endocrinologia,
Universidade Federal de São Paulo -
Escola Paulista de Medicina,
São Paulo, SP.
Recebido em 20/05/99
Aceito em 13/08/99
RESUMO
A tríade clássica da Síndrome de McCune Albright (SMA) - displasia fibrosa poliostótica (DFP), máculas cutâneas "café au lait" e hiper-função autônoma de uma ou mais glândulas endócrinas - envolve mutações ativadoras do gene GNAS1, codificador da proteína Gas e conseqüente aumento da produção do sinalizador intracelular AMPc em diferentes tecidos. Embora a pseudo-puberdade precoce (PPP) seja a manifestação clínica mais freqüente, são as alterações ósseas que conferem a maior morbidade à síndrome. Apresentamos o seguimento clínico endocrinológico e a evolução da doença óssea em 4 pacientes portadores da SMA, das quais 3 delas estão sendo tratadas com bisfosfonato (Pamidronato, 2mg/kg). Três das 4 pacientes apresentaram pseudo-puberdade precoce e duas delas desenvolveram hipertiroidismo. A variação do nível de fosfatase alcalina (FA) foi o indicador bioquímico da doença óssea, cujas complicações foram dor e fratura além de obstrução do canal lacrimal em uma e amaurose unilateral em outra como conseqüência das deformidades crânio-faciais. A infusão de pamidronato endovenoso produziu melhora das dores ósseas e reduziu (37% - 54%) os níveis de FA em todas as pacientes. Entretanto, a melhora da qualidade do osso com o uso do bisfosfonato ainda é incerta e para a qual necessitamos de estudos randomizados e com longo prazo de observação. (Arq Bras Endocrinol Metab 1999;43/5: 351-359)
Unitermos: Displasia fibrosa óssea; McCune Albright; Puberdade precoce; Pamidronato
ABSTRACT
McCune Albright syndrome is a rare disorder characterized by polyostotic fibrous dysplasia, café-au-lait spots and some endocrine hyperfunction, mostly precocious puberty. It is caused by activating mutations of the GNAS1 gene that codifies for the Gas protein, leading to overproduction of cAMP messenger at different tissues. Although pseudo-precocious puberty is the most frequent disfunction, the skeletal lesions are responsible for the highest morbidities such as bone pain, fractures and deformations. Clinical follow up focusing the bone disease is reported in four patients, three of them been treated with intravenous bisphosphonate (Pamidronate, 2mg/kg). Precocious puberty occurred in three and hyperthyroidism developed in two of the patients. Alkaline phosphatase (AP) levels were used as biochemical markers of the osteolytic lesions, clinically presented by local pain, fracture and bone deformities. In addition, lachrymal obstruction in one patient and unilateral blindness in another appear to be due to deformed skull and facial bones. Intravenous infusion of Pamidronate has been improving bone pain and decreased (37% - 54%) basal AP levels. However, a long-term randomized follow-up study determining improvement in bone quality still needs to be completed. (Arq Bras Endocrinol Metab 1999;43/5: 351-359)
Keywords: Bone fibrous dysplasia; McCune Albright syndrome; Precocious puberty; Pamidronate
A SÍNDROME DE McCUNE ALBRIGHT (SMA) foi descrita em 1937 por McCune, Bruch (1) e Albright (2), caracterizando-se pela tríade clássica de máculas cutâneas café com leite não elevadas, puberdade precoce e displasia fibrosa óssea poliostótica (DFP). Outras manifestações de hiperfunção endócrina podem também estar presentes como: hipersomatotropismo (3,4), hiperprolactinemia (5,6), hipertiroidismo (3,7,8), hiperparatiroidismo (8) e hipercortisolismo (9,10). No sistema esquelético, além da DFP que caracteriza a síndrome, foram descritos casos associados de raquitismo hipofosfatêmico (11,12). As lesões displásicas podem, ainda que raramente, evoluir para osteossarcoma (13). Em casos mais graves poder-se-á encontrar participação de múltiplos órgãos, tornando o controle difícil e podendo evoluir ao óbito (14).
Relataremos o quadro clínico evolutivo de quatro pacientes com SMA e DFP acompanhadas no serviço de endocrinologia da UNIFESP, com especial ênfase para a doença óssea e acompanhado da revisão de literatura pertinente.
CASUÍSTICA E METODOLOGIA
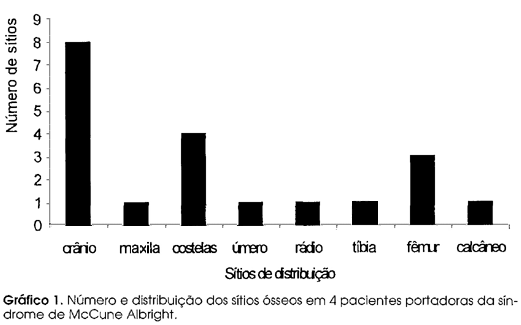
As quatro pacientes desse estudo foram seguidas no ambulatório de endocrinologia da Escola Paulista de Medicina -UNIFESP desde o diagnóstico. O tempo de seguimento variou de 4 a 26 anos, média de 15 anos. A idade atual das pacientes variou de 6 a 28 anos, mediana de 11,5 anos. Todas elas apresentavam DFO forma poliostótica. O quadro clínico inicial assim como a distribuição das lesões ósseas estão detalhadas nas tabelas 1 e 2, e no gráfico 1.
O pamidronato (Aredia®, Novartis) foi administrado em três pacientes, via endovenosa, na dose de 2mg/kg nas crianças e 60mg/dia na paciente adulta, infundido continuamente em 250mL de soro glicosado 5%, ao longo de 4 horas. A periodicidade de infusão foi orientada pela elevação dos níveis plasmáticos de fosfatase alcalina (FA), na tentativa de mantê-los mais próximos do valor superior da normalidade, embora não haja critérios bem definidos na literatura.
O seguimento era feito em intervalos variáveis de 2 a 6 meses. Medidas de FA total sérica foram obtidas de todas as pacientes previamente e posteriormente à terapêutica (gráfico 2). As funções paratiroidiana, gonadal, hipofisária e tireoidiana foram avaliadas em todas pacientes periodicamente através do exame clínico, radiológico, ultrassonográfico e das dosagens hormonais.
Como valores normais para FA variaram segundo os métodos laboratoriais utilizados, os resultados serão expressos como "Acima do Limite Referencial Máximo" (ALRM) para o método (gráfico 2). O controle radiológico foi feito pelo mesmo observador segundo comparação com os exames iniciais. As reavaliações de imagem foram solicitadas esporadicamente segundo indicação clínica e após pelo menos 6 meses da infusão do pamidronato.
DESCRIÇÃO DOS CASOS
Caso 1
EZ, 27 anos, sexo feminino, iniciou quadro de pseudo-puberdade precoce aos 2 anos com aparecimento de telarca e menarca, tratada com antiandrogênico dos 2,5 aos 8 anos e posteriormente com testolactona até os 21 anos. Neste momento, optou-se pelo uso de agonista de LHRH durante 6 meses, na tentativa de se controlar melhor o aparecimento de cistos ovarianos.
Aos 7 anos foi constatada a presença de assimetria facial, evoluindo com lesões ósseas em crânio, úmero e posteriormente em costelas, bacia, clavícula, fêmur e finalmente vertebras, ao longo dos últimos 20 anos. Pelo aparecimento de dores ósseas em costelas e quadril e na tentativa de se evitar a progressão das deformidades, aos 25 anos de idade, foi instituído pamidronato endovenoso, na dose de 60mg por aplicação. Apresentava atividade de FA inicial de 4,6 ALRM. Após a primeira infusão relatou melhora das dores óssea, com redução significativa da atividade de FA, cujo valor após 1 ano de tratamento era de 2,3 ALRM.
Caso 2
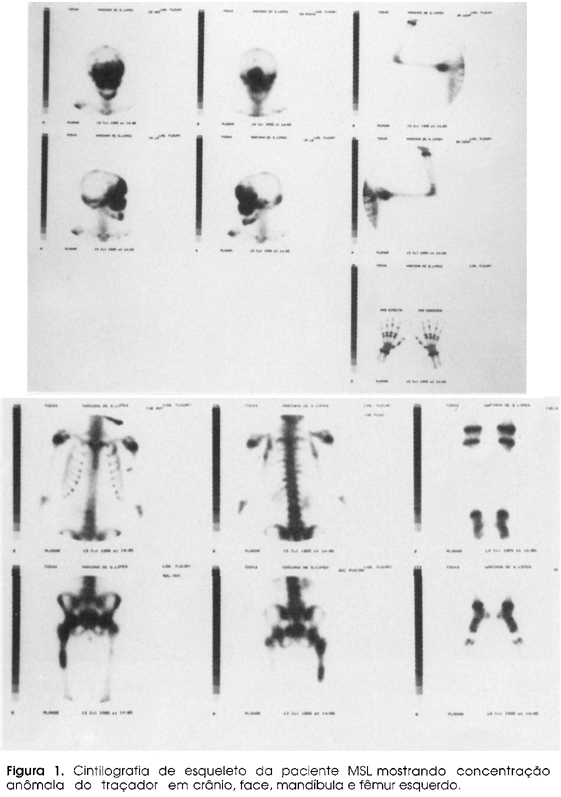
MSL, 9 anos, sexo feminino, nasceu com manchas café com leite em face, pescoço e região dorsal do tronco. O diagnóstico inicial foi realizado a partir de uma deformidade em crânio, notada aos 6 anos. Nesta ocasião identificou-se também lesão displásica em diáfise de fêmur D (Figura 1), e após alguns meses iniciou claudicação progressiva, ao mesmo tempo em que apresentava telarca. Devido ao agravamento da dor e risco de fratura com deformidade de fêmur, foi indicada infusão de pamidronato endovenoso na dose de 2mg/kg peso. A FA inicial era de 3,5 x ALRM, reduzindo para 1,8 x ALRM um mês após, aproximando-se do limite superior da normalidade após 2 meses da infusão (1,3 x ALRM), com diminuição da claudicação e melhora radiológica, inclusive com espessamento da cortical no local da lesão 6 meses após o início do tratamento (Figuras 2 e 3). Aos 7 anos e 6 meses, apresentou um único episódio de sangramento vaginal, com a presença de cisto ovariano demonstrado pela ultrassonografia e que desapareceu espontaneamente. Um nódulo tiroidiano foi observado na mesma ocasião e permaneceu com nível de TSH suprimido, porém com hormônios tiroidianos normais durante vários meses.
Seguiram-se novas infusões assim que a FA tornava a se elevar. Apesar disto, após 3 anos de tratamento, fraturou o femur após uma queda sobre o mesmo, tendo sofrido correção cirúrgica. Devido a deformidade dos ossos do crânio, desenvolveu obstrução do canal lacrimal, com formação de abcesso local, sendo necessária a drenagem cirúrgica. Durante este episódio, apresentou tirotoxicose, sendo iniciado metimazol e programada cirurgia para retirada da glândula.
Caso 3
TFP, 5 anos, sexo feminino. Ao nascimento apresentava manchas cutâneas café com leite em região glútea e coxa D. Aos 2 anos iniciou telarca com aceleração do desenvolvimento estatural. A avaliação radiológica revelou aos 3,2 anos, DFP acometendo diáfise da fíbula D, submetafisário do fêmur D e terço proximal do rádio D. Recebeu testolactona na dose de 20mg/kg/dia. A atividade de FA inicial era de 2,1 x ALRM quando foi introduzido alendronato 5mg/dia, elevando-se após um mês para 2,7 x ALRM. Nessa ocasião recebeu 2mg/Kg Pamidronato EV, melhorando os níveis de FA para 1,3 x ALRM ao cabo de 6 meses. Foi mantido o esquema de infusão conforme controle da FA. Devido ao bom controle da PPP manteve-se a testoiactona. No último seguimento ambulatorial foi diagnosticado bócio multinodular, hipoacusia leve e abaulamento craniano frontal e parietal D. Exames hormonais revelaram hipertiroidismo com anticorpos tiroidianos negativos, sendo orientado tiroidectomia total, atualmente em terapia de reposição com L-Tiroxina.
Caso 4
CMP, 14 anos, sexo feminino. Apresentava manchas café com leite em maxila D e coxa E. Aos 7 anos de idade passou a apresentar cefaléia frontal esporádica e em peso, acompanhado de abaulamento fronto-parietal D com exoftalmia D e tumoração em extremidade superior da tíbia D, indolores, a última com 2cm de tamanho. A telarca ocorreu aos 10 anos e a menarca aos 11 anos. O abaulamento estendeu-se até região supra-orbitária, era de consistência endurecida e media 10cm em seu maior diâmetro, causando diminuição da acuidade visual D. A RNM revelou alteração óssea compatível com DFP em ossos frontal, etmoidal, esfenoidal, temporal e parietal, determinando proptose do olho D. A cintilografia mostrou incremento no ritmo de remodelação em calota e base craniana assim como em zigomático e mandíbula D. Submetida à neurocirurgia para curetagem de material ósseo fibrodisplásico em região fronto-parietal e teto da órbita, reconstituído com enxerto ósseo. O estudo anátomo-patológico revelou displasia fibrosa óssea. A atividade de FA inicial era de 1,6 x ARLM e se manteve sem oscilações importantes. Evoluiu estável do quadro ósseo e sem outras hiperfunções endócrinas até última avaliação ambulatorial, embora com seqüelas oculares após a cirurgia, como proptose e redução da acuidade visual D definitiva.
DISCUSSÃO
A base molecular da SMA reside na mutação ativadora do gene GNAS1 que codifica a sub-unidade a da proteína G (15,16). Dessa forma há uma ativação constitutiva da proteína Gas, que estimula, portanto, de forma autônoma o sistema da adenil-ciclase, levando à hiperfunção dos tecidos afetados (17). O espectro variável da síndrome (18,19) e a ausência de hereditariedade sugerem que a mutação seja somática ou pós zigótica, postulando-se que esta alteração genética tenha ocorrido antes do desenvolvimento do disco trilaminar pois estão afetados tecidos tanto de origem ectodérmica, endodérmica como mesodérmica (20). Estas mutações são resultantes da substituição dos resíduos de aminoácido arginina (codon 201) e glicina (codon 227), nos exons 8 e 9 respectivamente (15).
Todos os tecidos afetados contêm essas mutações, entretanto, ainda não foram identificadas no tecido ósseo displásico e nas manchas cutâneas, possivelmente devido ao mosaicismo genético e a distribuição randômica de células mutantes (16,18).
Nossas pacientes apresentaram-se invariavelmente com doença óssea poliostótica, cuja evolução tem-se mostrado progressiva, com o aparecimento de novas lesões ao longo da vida. Segundo o observado em nossas pacientes, o comprometimento dos tecidos endócrinos é variável, podendo estar presente ou não, ser único ou múltiplo, e ainda surgir em momentos diferentes ao longo do segmento. Portanto, durante seu acompanhamento, a avaliação das funções endócrinas deve ser realizada periodicamente.
Os pacientes portadores da síndrome apresentam geralmente problemas psicológicos de aceitação social devido às deformidades ósseas, sobretudo quando ocorre assimetria facial, além do comprometimento neurológico ou incapacitação que as lesões ósseas apendiculares podem induzir. Portanto, uma alternativa terapêutica para controlar a evolução da doença óssea vem sendo esperada ansiosamente pelos pacientes bem como pelos médicos que os acompanham.
As alternativas terapêuticas das quais dispomos ainda se limitam à abordagem sintomática. A progressão da doença óssea tem sido tratada através da cirurgia ortopédica com curetagem, enxertos ósseos, fixações internas de ossos longos e redução de fraturas (21). Drogas como a calcitonina, mitramicina e etidronato foram empregadas porém sem sucesso (22,23). O relato recente de bons resultados com o uso do pamidronato (24) para controle das lesões nos entusiasmou a utilizá-lo em nossas pacientes. O resultado obtido nas nossas três pacientes foi satisfatório. Houve redução nos níveis da FA, o principal parâmetro bioquímico de atividade da doença, em todas as pacientes que realizaram a infusão, e ainda melhora da dor óssea. A dose, periodicidade e forma de seguimento clínico-laboratorial sob terapia com Pamidronato ainda não está completamente ratificada na literatura científica. Muitos desses parâmetros são reflexos do tratamento com essa droga na Doença de Paget.
Na paciente n° 2 observamos espessamento cortical no local da lesão após tratamento com pamidronato. Entretanto, isto não a impediu de fraturar o fêmur após queda sobre o membro afetado. A paciente n° 3 apresentou um discreto alargamento epifisário 6 meses após a infusão do bisfosfonato, e por cautela interpretamos como sendo um defeito de mineralização induzido pela droga. Optamos por associar o uso de calcitriol sem interromper o pamidronato, devido ao elevado grau de comprometimento esquelético. Após 1 ano, a evolução radiológica mantém-se adequada.
Apesar do curto tempo de uso sistemático dessas drogas anti-reabsortivas em crianças e adolescentes, poucos efeitos colaterais foram detectados, tais como: hipertermia 24 a 48 h após a infusão; hipocalcernia sintomática e alargamento da placa epifisária de crescimento. Estes efeitos adversos são controláveis com o aumento do intervalo de administração, diminuição da dose, associação com calcitriol ou suspensão temporária da droga (24-26).
O pamidronato é um bisfosfonato de segunda geração que atua diminuindo o turnover ósseo, usado com sucesso na doença de Paget, hipercalcemia associada à malignidade, metástases ósseas, mieloma múltiplo e osteoporose (24). A administração endovenosa é preferida devido à absorção intestinal ser reduzida, além dos efeitos colaterais gastrointestinais quando administrada por via oral.
Embora reconhecendo a necessidade de estudos controlados e randomizados, o presente relato, utilizando um esquema terapêutico racional com drogas anti-reabsortivas em dose e intervalos conforme controle clínico, radiológico e bioquímico, torna-se valioso em função da raridade da síndrome.
Em recente revisão feita por Matthew e col. (20) de 158 pacientes com SMA relatados na literatura até 1995, constataram que 98% deles apresentavam lesões ósseas displásicas únicas ou múltiplas, sendo a DFP a característica mais freqüente da síndrome. Estas lesões apareciam também tipicamente na 1a década de vida, constituindo o principal fator de morbidade, ocasionado pelas deformidades progressivas, fraturas e compressão nervosa.
Os pacientes podem ser clinicamente assintomáticos ou sintomáticos, sofrendo de dores ósseas constantes ou claudicantes, surdez neurossensorial ou condutiva, secundária à alteração óssea (27,28). As lesões ósseas são mais freqüentes em femur, pelve e crânio. O aspecto radiográfico é de hiperostose em base do crânio, lesões líticas e expansivas tipo "vidro fosco" em ossos longos, bordas irregulares devido à erosão endosteal e afinamento da cortical (20,27,29).
O estudo com radioisótopos mostra maior captação do marcador nas áreas afetadas e apresenta sensibilidade suficiente para utilizar este exame como mapeador das lesões no esqueleto (30). A tomografia computadorizada e a ressonância nuclear magnética podem ser úteis no diagnóstico diferencial com malignidade (31,32).
O estudo histológico revela um tecido ósseo impróprio e com excesso de matriz fibrótica desorganizada, além de extensas áreas de fibrose medular (33). Os índices histomorfométricos mostram um aumento acentuado da superfície osteóide (matriz óssea pouco calcificada) todavia com diminuição da superfície de osteoblastos maduros (33,34). O principal defeito, portanto, parece ser uma maturação incompleta dos osteoblastos a partir das células estromais da medula óssea (34).
A Pseudo Puberdade Precoce (PPP) é a mais freqüente das hiperfunções endócrinas na SMA, principalmente em meninas. O perfil hormonal é geralmente caracterizado por níveis elevados de estradiol com gonadotrofinas hipofisárias suprimidas ou normais, resultante da autonomia ovariana (35). A resposta de LH e FSH ao teste do GnRH é do tipo pré-puberal ou suprimida, orientando o diagnóstico para pseudo-puberdade precoce (não dependente de GnRH). A presença dos cistos ovarianos autônomos pode ser observada pela ultrassonografia pélvica.
O tratamento da PPP com testolactona, um inibidor da aromatase, tem sido a única alternativa eficaz no controle da evolução dos caracteres sexuais secundários (36). Os análogos do GnRH não são indicados em virtude da independência do eixo gonadotrófico. Três das quatro meninas desenvolveram PPP, sendo que duas delas utilizaram testolactona VO por tempo prolongado.
O excesso do hormônio de crescimento (GH) e da prolactina (PRL) são as mais freqüentes das hiperfunções pituitárias na síndrome, não apresentada até o momento por nenhuma das nossas pacientes. Apenas 65% dos pacientes apresentam tumor visível radiologicamente, variando entre adenoma e hiperplasia lactosomatotrófica (37), o que dificulta a abordagem cirúrgica, sobretudo quando somado à esclerose óssea da base do crânio. Também tem sido proposto, além da abordagem cirúrgica transesfenoidal, complementação radioterapêutica e ou análogos da somatostatina e bromocriptina visando redução tumoral (3,6,37,38).
O hipercortisolismo ocorre por autonomia nodular ou hiperplasia da glândula adrenal (9). A Síndrome de Cushing, embora muito rara, já foi descrita em recém-nascidos, adolescentes e adultos (7,9,10). Geralmente apresenta nível elevado de cortisol, não supressível pela dexametasona e ACTH baixo ou indetectável. Nenhuma das pacientes desenvolveu disfunção hipofisária ou adrenal até último seguimento, mesmo assim mantemos o controle periódico dos níveis de GH, PRL e cortisol, anualmente.
A disfunção tiroidiana foi estimada em 40% segundo os estudos de Feuillan et al. (8). Os achados ecográficos revelaram bócio com áreas heterogêneas e lesões císticas (3,8). Em todos os casos deve-se ter anticorpo estimulador anti-receptor do TSH negativos (TRAB), resposta do TSH ao TRH suprimida, captação e hormônios tiroidianos elevados com TSH baixo. O hipertiroidismo é insidioso e embora pouco agressivo não se deve aguardar remissão com drogas anti-tiroidianas ou terapia com iodo radioativo, visto que a história natural de autonomia glandular sempre persiste. A opção cirúrgica de tiroidectomia é a mais indicada. As pacientes 2 e 3 desenvolveram hipertiroidismo ainda na infância e receberam drogas anti-tiroidianas provisoriamente. Uma delas foi submetida à tiroidectomia total e a outra paciente aguarda cirurgia.
O raquitismo ou osteomalácea hipofosfatêmicos se desenvolvem nos pacientes acometidos pela DFP com ou sem o fenótipo de SMA. Estes apresentam alteração tubular renal que leva à perda de fosfato pela urina (hiperfosfatúria com hipofosfatemia), com conseqüente defeito de mineralização óssea. A base fisiopatológica permanece desconhecida, contudo duas hipóteses são descritas. A primeira relata a possível existência de um fator fosfatúrico circulante produzido pelo tecido osteóide fibrótico e a segunda sugere a existência de um defeito intrínseco de reabsorção tubular renal de fosfato (11,12,38,39).
Outra temida complicação, principalmente em adultos jovens, é a esclerose ou displasia fibrosa do osso temporal levando à surdez de condução neural e é difícil a abordagem cirúrgica para descompressão das fibras nervosas.
Devido a multiplicidade de manifestações e implicações da doença, acreditamos que os pacientes devam ser seguidos por uma equipe multiprofíssional, incluindo o pediatra, o endocrinologista, o ortopedista, o psicólogo e o constante apoio da família.
Endereço para correspondência:
Helena L. Farhat
Rua Pedro de Toledo, 781, 12° andar
04023-000 São Paulo, SP.
- 1. McCune DJ, Bruch H. Osteodystrophia fibrosa. Am J Dis Child 1937;54:806-48.
- 2. Albright F, Butler AM, Hampton AO, Smith P. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation, and endocrine dysfunction, with precocious puberty in females. N Engl J Med 1937;216:727-41.
- 3. Abs R, Beckers A, Van de Vyver FL, De Schepper A, Stevenaert A, Hennen G. Acromegaly, multinodular goiter, and silent polyostotic dysplasia. A variant of the McCune Albright Syndrome. J Endocrinol Invest 1990; 13:671-5.
- 4. Albin J, Wu R. Abnormal hypothalamic pituitary function in polyostotic fibrous dysplasia. Clin Endocrinol 1981;14:435-43.
- 5. Chung KF, Alaghb-Zadeh J, Guz A. Acromegaly and hyperprolactinemia in McCune Albright Syndrome. Am J Dis Child 1983;137:134-6.
- 6. Cremonini N, Graziano E, Chiarini V, Sforza A, Zampa GA. Atypical McCune Albright Syndrome associated with growth hormone-prolactin pituitary adenoma: Natural history, long-term follow-up and SMS 201-995 bromocriptine combined treatment results. J Clin Endocrinol Metab 1991;75:1166-9.
- 7. Cavanah SF, Dons RF. McCune Albright Syndrome: How many endocrinopathies can one patient have? South Med J 1993;86:364-7.
- 8. Feuillan PP, Shawker T, Rose SR, Jones J, Jeevanram RK, Nisula BC. Thyroid abnormalities in the McCune Albright Syndrome: Ultrasonography and hormonal studies. J Clin Endocrinol Metab 1990;71:1596-601.
- 9. Boston BA, Mandel S, LaFranchi S, Bliziotes M. Activating mutation in the stimulatory guanine nucleotide-binding protein in an infant with Cushing's Syndrome and nodular adrenal hyperplasia. J Clin Endocrinol Metab 1994;79:890-3.
- 10. Yoshimoto M, Nakayanma M, Baba T, Uehara Y, Niikawa N, Ito M. A case of neonatal McCune Albright Syndrome with Cushing's Syndrome and hyperthyroidism. Acta Pediatr Scand 1991;80:984-7.
- 11. Hahn SB, Lee SB, Kim DH. Albright Syndrome with hypophosphatemia rickets and hyperthyroidism- a case report. Yonsei Med J 1991;32:179-83.
- 12. Lever EG, Pttingale KW. Albright Syndrome associated with soft-tissue myxoma and hypophosphatemic osteomalacia. J Bone Joint Surg 1983;65B:621-6.
- 13. Ruggieri P, Sim FH, Bond JR, Unni KK. Malignancies in fibrous dysplasia. Cancer 1994;73:1411-24.
- 14. Shenker A, Weinstein LS, Moran A, Pescovitz OH, Charest NJ, Boney CM, et al. Severe Endrocrine and nonendocrine manifestations of the McCune Albright Syndrome associated with activating mutations of the stimulatory G protein Gs. J Pediatr 1993;123:509-18.
- 15. Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatiory G protein in the McCune Albright Syndrome. N Engl J Med 1991;325:1688-95.
- 16. Schwindinger WF, Francomano CA, Levine MA. Identification of a mutation in the gene encoding the a subunit of the stimulatory G protein of adenylyl cyclase in McCune Albright Syndrome. Proc Natl Acad Sci USA 1992;89:5152-6.
- 17. Spiegel MA, Genetic Basis of Endocrine Disease: Mutations in G Protein-Coupled Receptors in Endocrine Disease. J Clin Endocrinol Metab 1996;81(7):2434-42.
- 18. Happle R. The McCune Albright Syndrome: A lethal gene surviving by mosaicism. Clin Genet 1986;29:321-4.
- 19. Endo M, Yamada Y, Matsuura N, Niikawa N. Monozigot-ic Twins Discordant for the Major Signs of McCune Albright Syndrome. A J Med Genetics 1991;41:216-20.
- 20. Ringel MD, Schwindinger WF, Levine MA. Clinical Implications of Genetic Defects in G Protein: The Molecular Basis of McCune Albright Syndrome and Albright Hereditary Osteodystrophy In: Williams & Wilkins, editors. Williams Textbook of Endocrinology. Philadelphia: Saunders, 1996:171-84.
- 21. Stephenson RB, London MD, Hankin FM, Kaufert H. Fibrous dysplasia: an analysis of options for treatment. J Bone Joint Surg 1987;69A:400-9.
- 22. Bell NH, Avery S, Johnston CC Jr. Effects of calcitonin in Paget's disease and polyostotic fibrous dysplasia. J Clin Endocrinol Metab 1970;31:283-90.
- 23. Long A, Longhlin T, Towers RP, Mc Kenna TJ. Polyostotic fibrous dysplasia with contrasting responses to calcitonin and mythramycin: aetiological and therapeutic implications. Int J Med Sci 1988;157(7):229-34.
- 24. Chapurlat R, Liens D, Delmas PD, Meunier PJ. Long-Term Effects of Intravenous Pamidronate in Fibrous Dysplasia of Bone. J Bone Min Res 1997;120(10):1746-52.
- 25. Liens D, Delmas PD, Meunier PJ. Long-Term Effects of Intravenous Pamidronate in Fibrous Dysplasia of Bone. Lancet 1994;343:953-4.
- 26. Adamson BB, Gallacher SJ, Byars J, Ralston SH, Boyle IT, Boyce BF. Mineralisation defects with pamidronate therapy for Paget's disease. Lancet 1993;342:1459-60.
- 27. Harris WH, Dudley HR, Barry MD. The natural history of fibrous dysplasia. J Bone Joint Surg 1962;44A:207-33.
- 28. Firat D, Stutzman L. fibrous dysplasia of the bone: Review of twenty-four cases. Am J Med 1968;44:421-9.
- 29. Lee PA, Van Dop C, Migeon CJ. McCune Albright Syndrome: Long term follow-up. JAMA 1986;256:2980-4.
- 30. Malloy PC, Scott WW, Hruban RH. Case report 769. Skel Rad 1993;22:66-9.
- 31. Daffner RH, Kirks DR, Gehweiler JA, Heaston DK. Computed tomography of fibrous dysplasia. Am J Roentgen 1982;8:939-43.
- 32. Utz JA, Kransdorf MJ, Jelinek JS, Moser RP, Berrey BH. MR appearance of fibrous dysplasia. J Comput Assist Tomogr 1989;13:845-51.
- 33. Grabias SL, Campbell CJ. Fibrous dysplasia. Orthop Clin North Am 1977;8:771-83.
- 34. Marie PJ, Pollak C, Chanson P, Lomri A. Increased Proliferation of Osteoblastic Cells Expressing the Activating Gsa Mutation in Momostotic and Polyostotic Fibrous Dysplasia 1997;150(3): 1059-68.
- 35. Oliveira KS, Verreschi ITN, Rodrigues de Virna G. A funçăo ovariana na Síndrome de Albright: uma autonomia questionada. Arq Bras Endocrinol Metab 1994;38:82-96.
- 36. Feuillan PP et al. Treatment of Precocious Puberty in McCune Albright Syndrome with the Aromatase Inhibitor Testalactone. N Engl J Med 1986;315(18):1115-9.
- 37. Chanson P. et al. McCune Albright Syndrome and Acromegaly: Clinical studies and responses to treatment in five cases. Eur J Endocrinol 1994;131:229-34.
- 38. Econs MJ et al. Tumor-induced osteomalacia-unveiling a new hormone. N Engl J Med 1994;330:1679-981.
- 39. Zung A et al. Urinary Cyclic Adenosine 3'5'-Monophosphate Response in McCune-Albright Syndrome: Clinical Evidence for Altered Renal Adenylate Cyclase Activity. J Clin Endocrinol Metab 1995;80(12):3576-81.
Datas de Publicação
-
Publicação nesta coleção
04 Ago 2006 -
Data do Fascículo
Out 1999
Histórico
-
Aceito
13 Ago 1999 -
Recebido
20 Maio 1999