Resumos
As ambigüidades genitais têm-se constituído em uma verdadeira emergência pediátrica e a adequada avaliação de cada caso pode evitar que o paciente seja criado num sexo inadequado, com interferência importante na sua saúde bio-psico-social. Os autores fazem uma abordagem da fisiopatologia da determinação gonadal, bem como dos mecanismos envolvidos na diferenciação sexual e fornecem elementos para o diagnóstico diferencial e conduta terapêutica.
Genitália ambígua; Intersexo; Gônadas; Diferenciação sexual
Genital ambiguity is considered a pediatric emergency and the patients successful bio-psycho-social follow-up depends on the adequate and timely intervention early in life. The authors review the pathophysiology of gonadal determination and sexual differentiation, allowing the possibility of differential diagnosis and proper conduct.
Genital ambiguity; Intersex; Gonads; Sexual differentiation
atualização
Genitália Ambígua: Diagnóstico Diferencial e Conduta
Durval Damiani
Nuvarte Setian
Hílton Kuperman
Thaís D. Manna
Vaê Dichtchekenian
Unidade de Endocrinologia Pediátrica
do Instituto da Criança,
Hospital das Clínicas da Faculdade de
Medicina da Universidade de
São Paulo, São Paulo, SP.
Recebido em 20/10/00
Aceito em 10/01/01
RESUMO
As ambigüidades genitais têm-se constituído em uma verdadeira emergência pediátrica e a adequada avaliação de cada caso pode evitar que o paciente seja criado num sexo inadequado, com interferência importante na sua saúde bio-psico-social. Os autores fazem uma abordagem da fisiopatologia da determinação gonadal, bem como dos mecanismos envolvidos na diferenciação sexual e fornecem elementos para o diagnóstico diferencial e conduta terapêutica.
Unitermos: Genitália ambígua; Intersexo; Gônadas; Diferenciação sexual.
ABSTRACT
Genital ambiguity is considered a pediatric emergency and the patients successful bio-psycho-social follow-up depends on the adequate and timely intervention early in life. The authors review the pathophysiology of gonadal determination and sexual differentiation, allowing the possibility of differential diagnosis and proper conduct.
Keywords: Genital ambiguity; Intersex; Gonads; Sexual differentiation.
O CAMINHO A SER PERCORRIDO para que se chegue a um desenvolvimento sexual normal, quer masculino ou feminino, é bastante longo e sujeito a erros em vários momentos. Não é somente necessário que os estímulos ocorram em determinadas estruturas embriológicas, mas também sua cronologia é absolutamente fundamental para que o processo se realize a contento. Podemos dividir didaticamente este processo de diferenciação em dois momentos distintos: a determinação gonadal, que é a transformação da gônada bipotencial, indiferenciada em testículo ou em ovário; e a diferenciação sexual, que ocorre a partir da gônada diferenciada e que leva o indivíduo ao seu fenótipo final, incluindo ductos internos e genitália externa. Se por um lado a determinação gonadal tem suscitado muitas dúvidas e podemos afirmar, seguramente, que ainda não conhecemos todos os mecanismos envolvidos nesse processo, por outro lado, o caminho percorrido a partir da gônada diferenciada tem se tornado mais claro. Sucintamente, vamos repassar estes dois processos para que se compreenda melhor a variedade de causas que podem levar a uma ambigüidade genital.
DETERMINAÇÃO GONADAL
Desde a década de 50, o papel do cromossomo Y tem sido reconhecido como vital para a determinação testicular, mas logo se reconheceu que não era o cromossomo Y como um todo, mas uma região situada no seu braço curto (região 1 A1, com 35Kb) que continha a seqüência sinalizadora para a gônada indiferenciada seguir a testículo. Esta pequena região passou a ser chamada de SRY (Sex-determining region on the Y chromosome) e se constitui num gene com apenas 1 exon, que codifica uma proteína com 223 aminoácidos (pertencente à família das proteínas com alta mobilidade ou High Mobility Group HMG), com uma porção altamente conservada de 80 aminoácidos (HMG box) (1-3). Após ligar-se a uma proteína nuclear chamada SIP-1 (SRY-interacting-protein 1), o SRY provoca um encurvamento de 82o no DNA, o que media seus efeitos. A identificação do TDF (o fator de determinação testicular) foi o auge de uma busca intensiva que se iniciou em 1959 quando Jacobs e Strong, bem como Ford e col., demonstraram que a presença do cromossomo Y era determinante do sexo masculino humano (4,5).
No entanto, a caracterização do SRY não esclareceu totalmente o problema da determinação gonadal porque logo ficou aparente que outros genes no cromossomo X (DAX-1, por exemplo), ou em autossomos, também deveriam estar implicados nesse processo que foi se revelando muito mais complexo do que imaginado inicialmente (figura 1). Anormalidades com o gene WT-1, no cromossomo 11 (Wilms tumour gene), são associadas ao desenvolvimento de tumor de Wilms (síndrome de Denys-Drash) e gonadoblastomas (síndrome de Frasier), bem como a associação tumor de Wilms, aniridia, anomalias genitais e retardo mental (WAGR). O gene WT-1 aparentemente está envolvido na formação da gônada bipotencial, sendo sua atuação anterior ao próprio SRY (upstream SRY). Também envolvido no desenvolvimento da gônada bipotencial e dos rins foi recentemente clonado o gene LIM-1. Mutações de LIM-1 no ser humano ainda não foram descritas.
Recentemente, têm havido novas implicações para o fator esteroidogênico 1 (SF1) na formação gonadal. Trata-se de um receptor nuclear órfão com uma estrutura que conta com dois "dedos de zinco" (zinc finger) e um domínio de ligação. Seu RNA mensageiro é detectado na crista urogenital, que vai formar gônadas e supra-renais e é também encontrado em regiões cerebrais em desenvolvimento. Os ratos que não apresentam SF1 não desenvolvem gônadas, adrenais e hipotálamo. Acredita-se que o SF1 também regule enzimas envolvidas na esteroidogênese, bem como a transcrição do hormônio anti-mülleriano (AMH).
A progressão posterior da gônada indiferenciada a testículo passa a ser mediada tanto por SRY como por genes autossômicos. Há evidências de que o SRY se liga ao promotor do gene do AMH e controla a expressão de enzimas esteroidogênicas (interaçãoWT-1/SF1) (6). O gene SOX 9 (SRY-box-related) está relacionado à condrogênese e à diferenciação gonadal, sendo localizado no cromossomo 17. Este gene é transcrito nas estruturas gonadais masculinas seguindo a expressão do SRY. Adicionalmente, é um ativador do gene do colágeno tipo II, que, por sua vez, é essencial para a formação da matriz extracelular da cartilagem. Defeitos no SOX 9 levam à reversão sexual em indivíduos 46,XY, bem como a alterações esqueléticas conhecidas como displasia campomélica.
Deleções no braço curto do cromossomo 9 (envolvendo os genes DMRT1 e DMRT2) têm sido associadas a reversão sexual em indivíduos 46,XY, bem como anomalias faciais (fechamento precoce da sutura frontal), hidronefrose e retardo de desenvolvimento. Deleções no braço longo do cromossomo 10 (10q) também têm sido envolvidas em reversão sexual e retardo mental.
No braço curto do cromossomo X estão genes cuja "dose" é importante para a determinação testicular (DSS dose-sensitive sex reversal) e, juntamente com genes envolvidos na diferenciação da glândula supra-renal, constituem o DAX-1 (DSS/Adrenal hypoplasia congenita/no cromossomo X, região 1). Em situações de duplicação do DAX-1, ocorre reversão sexual em indivíduos 46,XY. Em contraste, mutações em DAX-1 diminuindo sua atividade levam à falta de formação da glândula supra-renal e hipogonadismo hipogonadotrófico (7).
Dessa forma, vemos que uma verdadeira cascata de determinação gonadal tem sido construída e talvez ainda estejamos longe de ter este mapa totalmente concluído. No entanto, a partir do momento em que temos uma gônada diferenciada, fica mais fácil entendermos o que ocorre daí em diante para resultar no fenótipo final do indivíduo (figura 1).
DIFERENCIAÇÃO SEXUAL
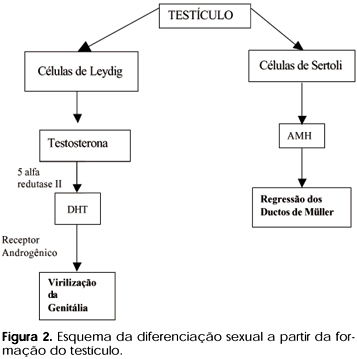
Existe uma tendência "intrínseca" das estruturas, tanto gonadais quanto dos ductos internos e da genitália externa, a seguir um caminho para sexo feminino. Assim, a diferenciação para o sexo masculino exige a atuação ativa, em momentos decisivos, de fatores envolvidos no processo de diferenciação sexual. A partir da diferenciação testicular, as células de Sertoli secretam o hormônio anti-mülleriano (AMH) cujo papel, neste momento (8 a 12 semanas de vida intra-uterina), é promover apoptose das células dos ductos de Müller e evitar que se diferenciem a fímbrias, trompas de Fallópio, útero e terço proximal de vagina.
Já as células de Leydig iniciam a produção de Testosterona (T), responsável, através de ação parácrina, pelo desenvolvimento dos ductos de Wolff, que darão origem aos ductos deferentes, epidídimo, vesículas seminais e ductos ejaculatórios. A conversão de T a dihidrotestosterona (DHT) pela enzima 5-alfa redutase II (que ocorre nas células da genitália externa) leva à masculinização da genitália externa.
Quando ocorre o desenvolvimento ovariano, a falta de AMH permite que os ductos de Müller se desenvolvam (fímbrias, trompas, útero e terço proximal de vagina). Por outro lado, não havendo T, os ductos de Wolff não se desenvolvem e sofrem atrofia. Como não há produção de T, não haverá conversão a DHT e a genitália externa seguirá seu caminho "natural" ao sexo feminino (figura 2).

AMBIGÜIDADES GENITAIS - CRITÉRIOS DIAGNÓSTICOS
Dentre as várias situações que podem configurar uma emergência pediátrica no recém-nascido, as ambigüidades genitais surgem com uma importância enorme tanto do ponto de vista imediato, já que algumas etiologias (hiperplasia adrenal congênita, síndromes malformativas) colocam a vida da criança em risco, como a longo prazo, em que uma situação de definição de sexo mal resolvida acarretará prejuízos irreparáveis ao bem-estar psico-social do paciente.
A questão se complica com a verificação de que não se dá, via de regra, a atenção devida ao exame da genitália por parte do médico que atende esta criança recém-nascida e não é incomum que a "suspeita de que alguma coisa está errada com os genitais da criança" seja levantada por um membro da família.
Num estudo realizado por Kaefer e col. com 79 pacientes em que coexistiam criptorquidismo e hipospádia, a incidência de intersexualidade chegou a 27%. Se a gônada não era palpável ao exame clínico, o risco de uma condição intersexual era três vezes maior do que quando se palpava a gônada. De forma análoga, quanto mais posteriormente posicionado o meato uretral (portanto, quanto maior o grau de hipospádia), maior era a probabilidade de se detectar uma condição intersexual (8).
Na Unidade de Endocrinologia Pediátrica do Instituto da Criança do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo temos adotado os critérios propostos por Danish, em 1982 (9), que definem uma ambigüidade genital se qualquer das manifestações listadas a seguir estiver presente:
Numa genitália de aspecto masculino:
1. Gônadas não palpáveis;
2. Tamanho peniano esticado abaixo de -2,5 DP da média de tamanho peniano normal para a idade (tabela 1);
3. Gônadas pequenas, ou seja, maior diâmetro inferior a 8mm;
4. Presença de massa inguinal que poderá corresponder a útero e trompas rudimentares;
5. Hipospádia.
Numa genitália de aspecto feminino:
1. Diâmetro clitoriano superior a 6mm;
2. Gônada palpável em bolsa labioescrotal;
3. Fusão labial posterior;
4. Massa inguinal que possa corresponder a testículos.
Problemas relacionados ao determinismo gonadal abrangem:
Hermafroditismo verdadeiro (HV) presença, no mesmo indivíduo, de tecido testicular e tecido ovariano em qualquer combinação: ovário + testículo (forma lateral); ovário + ovotéstis ou testículo + ovotéstis (forma unilateral); ovotéstis bilaterais (forma bilateral). A associação mais freqüente é, na casuística revista por van Niekerk & Retief, ovário de um lado, testículo do outro (11), enquanto outros autores têm encontrado a forma unilateral com ovário de um lado, ovotéstis do outro (12,13). Novamente enfatizamos que muitos hermafroditas verdadeiros podem não apresentar ambigüidade da genitália externa.
Um dado que não pode ser esquecido é que a caracterização de tecido testicular e de tecido ovariano deve ser clara do ponto de vista histológico. Assim, os critérios mínimos para HV são: folículos ovarianos ou presença de corpora albicantia para definir estrutura ovariana; túbulos seminíferos ou espermatozóides definem a existência de tecido testicular. Somente a presença de células de Leydig ou células hilares não é suficiente para definir tecido testicular, da mesma forma que estroma fibroso não é suficiente para definir estrutura ovariana.
Disgenesia gonadal mista também chamada disgenesia gonadal assimétrica, descrita por Sohval e col. em 1963, constitui um grupo heterogêneo de pacientes com ambigüidade da genitália externa, apresentando um testículo com graus variáveis de disgenesia de um lado e uma gônada em fita (streak) do outro. O cariótipo mais freqüente é o mosaicismo 45,X/ 46,XY. Derivados müllerianos não só do lado do streak mas também do lado do testículo podem estar presentes. Dependendo do grau de função das células de Leydig (produtoras de testosterona) e de Sertoli (produtoras de AMH) há graus maiores ou menores de desenvolvimento dos ductos de Wolff e de Müller.
Homem XX neste caso, em um indivíduo com sexo genético 46,XX desenvolvem-se testículos com capacidade de produção de testosterona e de virilização da genitália externa. Na verdade, cerca de 1/5 desses pacientes apresentam ambigüidade genital, o que permite que o diagnóstico seja feito na faixa etária pediátrica. De outra forma, como todos são inférteis, seus diagnósticos acabam sendo realizados quando da procura de clínicas de infertilidade. Em cerca de 80% desses pacientes detecta-se a presença do SRY. Como os genes responsáveis pela espermatogênese estão no braço longo do Y, que esses pacientes não têm, todos são inférteis. Este detalhe, ou seja, a incapacidade de os testículos produzirem espermatozóides, justifica a colocação desses pacientes no grupo das disgenesias gonadais ou dos "distúrbios de diferenciação gonadal". De outra forma, ao menos nos casos em que a genitália externa é ambígua, poderiam ser classificados como uma forma de pseudohermafroditismo masculino.
Quando a gônada está definida e a genitália externa é ambígua, temos duas categorias sindrômicas de pacientes:
Pseudohermafroditismo masculino (PHM) aqui o cariótipo é 46,XY, desenvolveram-se testículos bilaterais mas algum, ou alguns, dos passos necessários para completar a diferenciação da genitália externa não ocorreu de forma adequada e chegamos a uma ambigüidade. Alguns autores definem o quadro pela presença de testículos + genitália externa ambígua; outros, presença de cariótipo 46,XY com genitália externa ambígua. Como o homem XX seria a exceção (46,XX + testículos) mas já o colocamos no grupo dos distúrbios da determinação gonadal, poderemos adotar uma definição eclética, em que PHM é um indivíduo com ambigüidade de sua genitália externa, em presença de testículos e cariótipo 46,XY.
Pseudohermafroditismo feminino (PHF) neste caso, ocorre virilização de um feto programado para evoluir para o sexo feminino: a genitália externa é ambígua, em presença de ovários e de um cariótipo 46,XX. A grande etiologia dos PHF são as hiperplasias congênitas de supra-renais e que, nas formas perdedoras de sal, constituem-se em uma situação de risco de vida.
DIAGNÓSTICO DIFERENCIAL
Na Unidade de Endocrinologia Pediátrica do Instituto da Criança, temos utilizado uma classificação que agrupa as etiologias de anomalias da diferenciação sexual em quatro grandes grupos:
(1) Distúrbios da determinação gonadal; (2) Distúrbios da função testicular; (3) Distúrbios dos tecidos-alvo dependentes de andrógenos; e (4) Distúrbios da diferenciação do sexo feminino devidos à virilização anormal.
Temos partido do cariótipo para a "montagem" de um verdadeiro quebra-cabeças, que vai permitindo encaixar as várias peças para que se chegue a uma etiologia e, por via de conseqüência, para que se possa fazer a opção de sexo de criação, medicação hormonal quando pertinente e correção cirúrgica para o sexo escolhido.
CLASSIFICAÇÃO DAS ANOMALIAS DA DIFERENCIAÇÃO SEXUAL
CARIÓTIPO 46,XY
I. Distúrbios da determinação gonadal
Hermafroditismo verdadeiro
Disgenesia gonadal mista
Disgenesia gonadal pura XY
Disgenesia testicular
Síndrome da regressão testicular
- Precoce
Agonadismo (regressão entre 8a e 12a semanas)
Testículos rudimentares (regressão entre 14a e 20a semanas)
- Tardia
Anorquia (regressão após 20a semana)
Agenesia ou Hipogenesia de células de Leydig
II. Distúrbios da função testicular
Deficiência ou anormalidade de LH ou de seu receptor
Síndrome da persistência dos ductos de Müller
Defeitos de síntese de Testosterona
Deficiência enzimática
20,22 desmolase (CYP11A)
3b-hidroxiesteróide-desidrogenase tipo 2 (3bHSD2)
17-hidroxilase (CYP17)
17,20 desmolase (CYP17)
17b-hidroxiesteróide-desidrogenase tipo 3 (17bHSD3)
Interferência transplacentária da biossíntese de testosterona por ingestão hormonal materna
III. Distúrbios dos tecidos-alvo dependentes de andrógenos
Deficiência de 5a redutase tipo 2 (SRD5A2)
Síndrome da insensibilidade androgênica (AIS)
Forma completa (CAIS)
Forma parcial (PAIS)
IV. Idiopática
CARIÓTIPO 46,XX
I. Distúrbios da determinação gonadal
Hermafroditismo verdadeiro
Disgenesia gonadal pura tipo XX
Homem XX
II. Distúrbios da diferenciação sexual feminina
Hiperplasia congênita de supra-renais
21 hidroxilase (CYP21)
11 hidroxilase (CYP11B1)
3b hidroxiesteróide desidrogenase (3BHSD2)
20,22 desmolase (CYP11A)
17 hidroxilase (CYP17)
Deficiência de aromatase
Andrógenos maternos ingeridos e/ou produzidos
Idiopática
MOSAICISMOS
Hermafroditismo verdadeiro
Disgenesia gonadal mista
ANEUPLOIDIAS
Síndrome de Klinefelter e suas variantes
Síndrome de Turner e suas variantes
DEFEITOS EMBRIOGENÉTICOS NÃO ATRIBUÍVEIS A GÔNADAS OU HORMÔNIOS OU A ALTERAÇÕES CARIOTÍPICAS
Epispádia
Transposição peno-escrotal
Pênis bífido associado a extrofia vesical
Agenesia de pênis associada a ânus imperfurado
Ausência congênita de vagina
Tumor de Willms com cariótipo 46,XY (mutação no gene WT-1)
Denys-Drash
WAGR
Frasier
Agenesia renal com cariótipo 46,XX
Quadros sindrômicos
Dados de anamnese, exame físico, avaliação hormonal, exames de imagens e uma discussão multidisciplinar vão permitir que se tome a melhor conduta com relação as sexo de criação de uma criança com ambigüidade genital e torna-se desnecessário enfatizar a urgência de todo esse processo. A seguir, vamos ressaltar alguns aspectos para que se chegue ao diagnóstico da etiologia do processo, permitindo-se uma tomada de conduta mais consistente.
ANAMNESE Uma boa anamnese é sempre um bom começo para se chegar à etiologia de uma ambigüidade genital. Os pontos a seguir devem constar obrigatoriamente de toda história de uma criança com genitália ambígua:
Ingestão materna de drogas potencialmente virilizantes (andrógenos, progesterona) ou feminizantes (ciproterona, progestágenos) em período crítico da embriogênese, ou seja, entre 8 e 12 semanas de gestação.
Verificar se há casos semelhantes na família ou se houve mortes inexplicadas por desidratação, o que pode sugerir a presença de casos com hiperplasia congênita de supra-renal. Muitas das etiologias das ambigüidades genitais apresentam transmissão genética autossômica recessiva, ligada ao cromossomo X, autossômica dominante limitada ao sexo masculino, de modo que a presença de familiares afetados pode se constituir em pista diagnóstica útil. Apenas para exemplificar, os defeitos de receptor androgênico são transmitidos por gene recessivo no cromossomo X, de modo que em caso de suspeita diagnóstica (paciente 46,XY, níveis elevados de testosterona e eventualmente, LH, boa conversão de testosterona a dihidrotestosterona) a presença de caso semelhante que seja compatível com herança ligada ao X (portanto de ocorrência na família da mãe) permite o diagnóstico, mesmo que não se tenha avaliado a função do receptor ou mesmo seu seqüenciamento. Lembrar que pais consangüíneos têm maior probabilidade de gerar um filho com hiperplasia adrenal congênita, por exemplo, já que a herança é autossômica recessiva.
Verificar a presença de doença virilizante materna que teria o mesmo efeito na criança que uma ingestão materna de hormônios virilizantes. Há relato de crianças que nasceram totalmente virilizadas devido à produção materna de andrógenos. Também é importante sabermos se houve virilização materna durante a gestação, pois defeitos de aromatase podem se expressar em nível placentário e virilizar a mãe. A criança apresentará um pseudohermafroditismo feminino, já que as meninas serão virilizadas pela dificuldade de transformar testosterona em estradiol (papel da enzima aromatase, que transforma esteróides de 19 carbonos como androstenediona e testosterona em esteróides de 18 carbonos como estrona, estradiol e estriol). Esta é uma entidade descrita pela primeira vez em 1991 numa paciente japonesa por Shozu e col (15).
EXAME FÍSICO Ao exame clínico, verificar a presença de malformações, particularmente ano-retais e de coluna terminal. Nesses casos, a ambigüidade genital pode ser apenas mais uma malformação sem base hormonal. O estado de hidratação, a pilificação corpórea e a pressão arterial são outros elementos auxiliares na caracterização do processo. Um cuidado especial deve ser dado ao exame dos genitais. Apesar de as características clínicas da genitália externa não permitirem um diagnóstico etiológico, são muito úteis para dirigir a priorização de exames e de testes funcionais que deverão ser realizados naquele paciente. Os seguintes elementos deverão ser caracterizados ao exame físico:
Gônadas localização, tamanho e consistência. Gônadas palpáveis em bolsa labioescrotal ou são testículos ou ovotéstis, constituindo-se no elemento mais elucidativo do exame físico. A ausência de gônadas palpáveis deixa PHF ou HV como hipóteses diagnósticas sindrômicas mais prováveis; a presença de gônadas indica PHM como causa sindrômica mais provável. Quanto à consistência, a palpação de um pólo mais macio e um mais rígido pode levantar a suspeita de tecido ovariano e testicular presentes e, portanto, HV.
Falo caracterização do tamanho em relação às medidas consideradas normais (Tabela 1)
Posicionamento do meato uretral aliado ao tamanho do falo, este dado é de grande importância na conduta a ser tomada quanto ao sexo de criação, evidentemente desde que o sexo social não esteja ainda estabelecido quando então, assume a maior importância na decisão final quanto à atribuição do sexo.
A caracterização da genitália externa pode seguir os critérios de Prader, numa tentativa de comparação entre as diversas casuísticas, em diferentes serviços. Apesar de a classificação de Prader ter sido elaborada em 1954 para hiperplasia congênita de supra-renal (HCSR), ela pode ser utilizada nos dias de hoje e pode ser ampliada para outros casos de ambigüidade genital. Prader classificou as genitálias externas dos pacientes com HCSR de acordo com o grau de virilização que essas meninas sofriam: do mais leve (Prader I) até o mais virilizado (Prader V) (16).
Prader I - aumento isolado do clitóris, indicando que a virilização tenha ocorrido após 20 semanas de vida intra-uterina (VIU);
Prader II - aumento do clitóris associado a um intróito vaginal em forma de funil, podendo visualizar-se aberturas uretral e vaginal distintas, indicando virilização iniciada com 19 semanas de VIU;
Prader III - aumento de clitóris associado a um intróito profundo, em forma de funil, com a uretra esvaziando-se na vagina, como um pseudo seio urogenital. Há vários graus de fusão lábio-escrotal indicando uma virilização ocorrida com 14-15 semanas de VIU;
Prader IV - clitóris fálico com abertura urogenital em forma de fenda na base do falo, indicando virilização ocorrida cp, 12-13 semanas de VIU;
Prader V - fusão lábio-escrotal completa e uretra peniana, indicando virilização ocorrida com 11 semanas de VIU.
Em 1997, Sinnecker e col propuseram uma classificação para os casos de PHM por insensibilidade androgênica, que sugere 5 tipos, cada um com dois sub-tipos ("a"e "b"), o que se traduz em 10 fenótipos. Como Prader, esta classificação parte dos pacientes com menor grau de defeito (tipo 1) para os de maior grau de defeito (tipo 5). Ocorre que, como os pacientes com PHM com defeito no receptor androgênico vão sofrendo "progressiva" falta de virilização, o tipo 5 de Sinnecker apresenta os pacientes com fenótipo feminino, enquanto o tipo 1 apresenta os pacientes com fenótipo masculino, bem ao contrário do que ocorre na classificação de Prader (o tipo V é totalmente virilizado e o tipo I é francamente feminino), o que gera uma grande confusão. Além disso, a terminologia utilizada pelo grupo de Sinnecker confunde a própria definição de ambigüidade genital : tipo 1 fenótipo masculino; tipo 2 fenótipo "predominantemente masculino" ; tipo 3- fenótipo ambíguo; tipo 4- fenótipo "predominantemente feminino" ; tipo 5- fenótipo feminino. Fica difícil entender como um fenótipo "predominantemente" masculino ou feminino não seja ambíguo (18). Acreditamos que esta classificação, além de não contribuir para e uniformização das classificações clínicas das ambigüidades genitais, traz mais confusão do que propõe resolver, de modo que somos da opinião de que devemos continuar utilizando a classificação de Prader, lembrando sempre que o aspecto clínico da genitália externa não é suficiente para permitir um diagnóstico etiológico.
Quando avaliamos uma genitália externa, a direção inicial para os exames de laboratório pode ser dada pela presença ou ausência de gônadas palpáveis. Assim, podemos selecionar três situações :
1. Não há gônadas palpáveis o diagnóstico mais provável é de pseudo-hermafroditismo feminino devido à hiperplasia congênita de supra-renal.
Hermafroditismo verdadeiro ou Homem XX nesse caso, a caracterização histológica das gônadas, através de laparoscopia ou mesmo laparotomia exploradora, é essencial ao diagnóstico. A estimulação prévia com gonadotrofina coriônica humana (hCG) com dosagem de testosterona antes e após o estímulo pode revelar elevação dos níveis, podendo-se, a partir daí, inferir a presença de tecido testicular, reforçando ainda mais o diagnóstico. Mais específica do que a própria dosagem de testosterona, a dosagem de hormônio anti-mülleriano pode demonstrar a presença de células de Sertoli, melhor marcador de tecido testicular do que a presença de células de Leydig. A presença de um cariótipo 46,XX, presente em 60% dos pacientes com HV, associada a marcadores da presença de tecido testicular (testosterona ou hormônio anti-mülleriano) praticamente deixam HV e homem XX como as únicas opções diagnósticas.
Disgenesia gonadal mista nesses casos, a apresentação mais comum é de um testículo de um lado e de um streak (gônada fibrosa semelhante à da síndrome de Turner) do outro e o cariótipo mais comum é o mosaicismo 46,XY/45,X. Também nesses casos a laparotomia exploradora se faz necessária para definição diagnóstica.
Virilização por hormônios ingeridos ou produzidos pela mãe, incluindo defeito de aromatase nos casos de ingestão, o diagnóstico é sempre de exclusão, nunca sendo possível afirmar que tal medicação ingerida em época crítica da embriogênese foi a real causadora do problema em questão. Lembremos que as formas idiopáticas não podem ser descartadas nesses casos.
Agenesia renal situação que responde por algumas ambigüidades genitais, razão por que sempre a urografia excretora ou um exame ultra-sonográfico renal devam fazer parte da exploração desses casos.
2. Ambas as gônadas são palpáveis o diagnóstico mais provável é pseudo hermafroditismo masculino, não se podendo excluir, no entanto, hermafroditismo verdadeiro ou disgenesia gonadal mista.
Cariótipo 46,XY a avaliação inicial neste caso será a da integridade da via sintética de testosterona. Para tal, utiliza-se gonadotrofina coriônica humana na dose de 1000 U/dia, via intramuscular, por cinco dias, dosando-se os hormônios da via sintética de testosterona, tanto antes quanto depois do estímulo. Considera-se uma resposta adequada uma elevação de 150ng/dL de T acima do valor basal ou um nível mínimo de 160ng/dl após estímulo. Se a resposta for boa, estarão excluídos todos os defeitos de síntese de T, hipogenesia/agenesia de células de Leydig ou anorquia. Relação T/DHT normal (12+/-3) exclui defeito de conversão periférica, enquanto uma relação superior a 35-40 firma o diagnóstico de deficiência de 5-alfa-redutase. Caso não ocorra elevação de T após hCG, há duas possibilidades :
Defeito de síntese de T a elevação do precursor imediato ao bloqueio localiza o defeito enzimático. As relações entre os compostos imediatamente pré e pós-defeito apresentam um valor ainda maior na caracterização do defeito enzimático. Dentre os defeitos de síntese de T, três enzimas são comuns à via sintética do cortisol (P450scc, 3 beta HSD e 17hidroxilase), enquanto duas enzimas são exclusivas da via sintética de T ( 17,20 desmolase e 17HSD).
Falta de produção de T por disgenesia testicular, anorquia ou hipoplasia de células de Leydig - situações em que não ocorre elevação de precursores. Em casos de disgenesia testicular, os níveis de AMH são mais baixos que o normal para a idade do paciente.
Por exclusão, podemos ter as insensibilidades parciais a andrógenos, ou seja, ocorre adequada produção de T, conversão de T a DHT, mas a atuação periférica de DHT está comprometida pela falta ou pela incapacidade funcional dos receptores citosólicos ou intranucleares. Com cultura de fibroblastos de pele genital ou pelo seqüenciamento do gene do receptor androgênico é possível caracterizar-se o defeito de receptor androgênico. Nesses casos, o AMH tende a ser elevado (19). Como as insensibilidades androgênicas são transmitidas por gene recessivo ligado ao cromossomo X, a presença de parentes da mãe com o mesmo problema é forte evidência diagnóstica, mesmo sem os estudos do receptor androgênico. Restarão as formas idiopáticas, que respondem por um número apreciável de casos de PHM.
Cariótipo 46,XX o diagnóstico provável é de hermafroditismo verdadeiro ou homem XX, necessitando-se de biópsia gonadal para o diagnóstico de certeza. A estimulação gonadal com gonadotrofina menopáusica humana (hMG) pode dar informação sobre a presença de tecido ovariano (20), de modo que a combinação de resposta de T ou presença de AMH + resposta de estradiol ao FSH pode dar o diagnóstico de HV antes mesmo da biópsia gonadal (diagnóstico de certeza).
Mosaicismos - nesses casos, sempre necessitaremos de biópsia gonadal para elucidação diagnóstica.
3. Apenas uma gônada palpável pode tratar-se de disgenesia gonadal mista, hermafroditismo verdadeiro ou PHM, necessitando-se de laparoscopia/ laparotomia e biópsia gonadal para diganóstico definitivo. O esquema abaixo resume o roteiro diagnóstico baseado na presença ou ausência de gônadas palpáveis.
CONDUTA TERAPÊUTICA NAS ANOMALIAS DA DIFERENCIAÇÃO SEXUAL
A questão de maior importância no tratamento de crianças com diferenciação sexual anômala é a escolha do sexo de criação e, a partir daí, todas as outras condutas terapêuticas, no que concerne ao tratamento clínico e ao tratamento cirúrgico, são decorrência da opção tomada. Além do sexo social, o diagnóstico etiológico e as condições anatômicas presentes, permitindo uma abordagem cirúrgica eficiente, desempenham parte importante nessa decisão.
O sexo social deve ser avaliado com muito cuidado e por profissionais experimentados em tal abordagem já que, uma vez estabelecido, passa a ter papel preponderante no sexo definido a ser escolhido para a criança. Acreditamos que, mais do que a idade do paciente, a avaliação da identidade sexual pode dar a informação desejada. Classicamente, a partir de 3 anos de idade, o sexo social já está plenamente estabelecido pois ele acompanharia, do ponto de vista de desenvolvimento, a aquisição da fala. No entanto, nós já tivemos um paciente com 9 anos de idade sem identidade sexual estabelecida, o que permitiu a modificação do seu sexo de criação numa idade em que, teoricamente, tal conduta seria impossível.
O hermafroditismo verdadeiro, apesar de ter uma apresentação fenotípica mais para o lado masculino na maior parte dos casos, deverá ter opção preferencial para o sexo feminino, preservando-se as estruturas femininas eventualmente presentes como útero, trompas, cavidade vaginal e ovários. Em situações em que o ovotéstis esteja presente, a tentativa de separar cirurgicamente a porção ovariana da testicular poderá preservar a capacidade funcional desses tecidos ovarianos, com possibilidades de fertilidade. Esta conduta baseia-se no fato de o tecido ovariano no HV ser funcional e anatomicamente íntegro, ao passo que o tecido testicular é usualmente disgenético. Em nossos 19 pacientes com HV, esta conduta foi tomada em 6, três das quais encontram-se em puberdade e menstruando, com possibilidades de fertilidade. Uma dessas adolescentes apresenta curva térmica sugestiva de ovulação. As outras três pacientes ainda não estão em idade puberal.
A disgenesia gonadal mista também tem opção preferencial para o sexo feminino, com retirada do "streak" e do tecido testicular. O risco de degeneração maligna das gônadas nessa condição é muito alto e, mesmo em situações em que se optou por sexo masculino devido a outros fatores (sexo social, por exemplo), a remoção das gônadas com colocação de prótese testicular é a opção preferencial. Nem sempre o exame clínico do testículo permite a detecção precoce de uma neoplasia que nela se esteja desenvolvendo, de modo que, muitas vezes, a metástase já é o primeiro sinal de que houve uma neoplasia testicular, particularmente em casos de disgerminoma ou seminoma.
Nos casos de malformações onde não temos uma base hormonal para a alteração genital, a opção se sexo dependerá da avaliação das estruturas presentes e da possibilidade de correção cirúrgica para este ou aquele sexo. Num exemplo extremo, como o de agenesia de pênis, a opção evidentemente será para o sexo feminino, já que nenhuma técnica cirúrgica atualmente disponível permitiria a criação de um pênis normalmente funcionante nessas crianças.
O grupo de anomalias da diferenciação sexual com cariótipo 46,XY é o que apresenta as maiores dificuldades quanto à opção de sexo de criação, excetua das desse grupo as situações já discutidas isoladamente nos parágrafos precedentes. Nos defeitos de síntese de T, nos distúrbios dos tecidos-alvo dependentes de andrógenos e mesmo nas formas idiopáticas que respondem por uma proporção apreciável de casos nesse grupo (30% em algumas casuísticas), serão as características da genitália externa que dirigirão primariamente a opção de sexo de criação. Voltamos a frisar novamente que estamos supondo que o sexo social ainda não esteja plenamente desenvolvido, ou seja, que o diagnóstico se fez em idade precoce como é desejável em todos os casos de ambigüidade genital. Há controvérsia na literatura a respeito da estimulação dos genitais com T exógena para prever a capacidade desses tecidos de responderem ao estímulo puberal e atingirem tamanho e função normais. O tamanho do falo e o posicionamento do meato uretral deverão ser os parâmetros para a opção a sexo masculino: um falo com tamanho acima de -2 desvios-padrão da média com uretra não perineal, pode ter uma função adequada no sexo masculino e ser passível de uma adequada correção cirúrgica. No entanto, todo caso em que haja dúvidas quanto à correta função no sexo masculino, deverá ter a opção para o sexo feminino, pois em princípio, é mais fácil criar-se uma vagina funcionante do que um pênis funcionante. Em alguns de nossos casos de PHM com opção para o sexo masculino, a necessidade de várias cirurgias reparadoras com resultado estético e funcional discutíveis, alerta para os problemas que uma opção para o sexo masculino sem base anatômica adequada pode acarretar ao paciente, à família e à própria equipe médica.
Nos defeitos de síntese de T a resposta periférica a T exógena é muito boa e deve ser usada para melhorar as condições anatômicas, no sentido de permitir uma correção eventual para o sexo masculino. Já o mesmo não ocorre nas insensibilidades parciais a andrógenos, onde há certo grau de resistência dos tecidos-alvo à T exógena, tornando muitas vezes aconselhável a opção feminina. Nas deficiências de 5 alfa redutase, o aspecto externo é mais feminino e a opção feminina deverá ser preferida. Evidentemente, apenas ressaltamos em linhas gerais as opções preferenciais, mas cada caso deverá ser avaliado individualmente e com muito critério para que a opção tomada seja definitiva.
Uma vez escolhido o sexo de criação, a correção cirúrgica virá a seguir, removendo-se todas as estruturas que não digam respeito ao sexo escolhido. Para exemplificarmos, na insensibilidade parcial a andrógenos com opção para o sexo feminino, os testículos deverão ser removidos. A cirurgia estética da genitália externa muitas vezes é feita num primeiro tempo logo após o diagnóstico, deixando-se para época posterior ao estirão pubertário, a correção definitiva.
Quanto à terapia hormonal, a única etiologia de ambigüidade genital que põe em risco a vida do paciente em idade precoce é a hiperplasia congênita de supra-renais e seu tratamento deve ser iniciado a partir do diagnóstico, com terapêutica substitutiva de glicocorticóides e, nos casos de perda de sal, de mineralocorticóides (vide artigo "Aspectos Terapêuticos da Hiperplasia Adrenal Congênita", neste mesmo número dos ABE&M).
Em época de puberdade, os casos que não apresentem gônadas funcionantes deverão ter uma puberdade induzida por hormônios exógenos. Assim, numa idade óssea de 11 a 12 anos, inicia-se no menino, o uso de T exógena na forma de depósito, em injeções mensais na dose de 100 a 250 mg por via intra-muscular. Alternativas de implantação de pellets de testosterona ou de testosterona transdérmica podem ser utilizadas, dependendo da experiência de cada autor. A monitorização da idade óssea e a avaliação clínica das características sexuais secundárias e o crescimento físico irão determinar os ajustes das doses empregadas. Alguns autores acreditam que se consegue um melhor resultado de estatura final e de evolução puberal com doses menores tal como 25mg intra-muscular a cada 15 dias no primeiro ano de tratamento, passando-se a 100mg/mês no segundo ano. Nas meninas, a indução da puberdade deverá contemplar a possibilidade ou não para estas adolescentes menstruarem. Quando houve remoção do útero ou quando não houve desenvolvimento uterino, evidentemente não necessitaremos de terapia hormonal cíclica, e o uso de estrógenos conjugados eqüinos em esquema contínuo permitirá um bom desenvolvimento das características sexuais secundárias. Em nossa experiência, esta terapêutica mostra-se superior ao uso do próprio etinil-estradiol. Temos usado de 0,15 a 0,625mg/dia ajustando a dose de acordo com a progressão da idade óssea, crescimento físico e dos caracteres sexuais secundários (desenvolvimento mamário, particularmente). Quando a possibilidade de menstruação existe, a terapêutica com estrógenos e progesterona deve ser instituída. O esquema que temos utilizado é estrógeno do dia 1 ao 23, na dose de 0,3 a 0,625mg e medroxiprogesterona na dose de 2,5 a 5mg dos dias 17 a 23. Nenhuma medicação é administrada entre os dias 24 e o primeiro dia do ciclo seguinte. Um esquema alternativo é o uso de três semanas de estrógeno (0,3-0,625mg/dia) seguido de uma semana de progesterona (2,5-5mg/dia), iniciando-se a seguir um novo ciclo de três semanas de estrógeno. Atualmente, temos a opção do uso de estrógenos transdérmicos, associados a progesterona quando queremos instituir ciclos menstruais.
Um aspecto importante do tratamento é o seguimento psicológico que essas crianças e os familiares deverão ter. As dúvidas que surgem, tanto para os familiares quanto para a própria criança, devem ser respondidas por uma equipe treinada no tratamento de tais casos. As consultas deverão dispor de tempo adequado para que todos possam expressar claramente suas questões, seus medos, suas inseguranças e o papel da equipe multiprofissional é de apoio e compreensão com o objetivo de contribuir para a criação de um indivíduo adulto adaptado ao contexto social em que vive e à sua própria condição.
Endereço para correspondência:
Durval Damiani
Rua Bela Cintra 2117, apto. 9
01.415-002 São Paulo, SP
e.mail: durvald@csf.com.br
- 1. Gubbay J, Collignon J, Koopman P, Capel B, Economou A, Münsterberg A, et al. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryologically expressed genes. Nature 1990;346:245-50.
- 2. Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MH, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990;346:240-4.
- 3. Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R. Male development of chromosomally female mice transgenic for Sry. Nature 1991;351:117-21.
- 4. Jacobs P, Strong J. A case of human intersexuality having a possible XXY sex determining mechanism. Nature 1959;183:302-3.
- 5. Ford C, Polani P, Briggs J, Bishops P. A presumptive human XXY/XX mosaic. Nature 1959;183:1030-1.
- 6. Nachtigal MW, Hirokawa Y, Enyeart-VanHouten DL, Flanagan JN, Hammer GD, Ingraham HA. Wilmstumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1 in sex-specific gene expression. Cell 1998;93:445-54.
- 7. Hiort O, Holterhus PM. The molecular basis of male sexual differentiation. Eur J Endocrinol 2000;142:101-10.
- 8. Kaefer M, Diamond D, Hendren WH, Vemulapalli S, Bauer SB, Peters CA, et al. The incidence of intersexuality in children with cryptorchidism and hypospadias : stratification based on gonadal palpability and meatal position. J Urol 1999;162:1003-6.
- 9. Danish RK. Intersex problems in the neonate. Indian J Perdiatr 1982;49:555-75.
- 10. Lee PA, Mazur T, Danish R, Amrhein J, Blizzard RM, Money J, et al. Micropenis. I- Criteria, etiologies, and classification. Johns Hopk Med J 1980;146:156-63.
- 11. Van Niekerk WA, Refief AE. The gonads of human true hermaphrodites. Hum Genet 1981;58:117-22.
- 12. Krob G, Braun A, Kuhnle U. True hermaphroditism: geographical distribution, clinical findings, chromosomes and gonadal histology. Eur J Pediat 1994;153:2-10.
- 13. Damiani D, Fellous M, McElreavey K, Barbaux S, Barreto ESA, Dichtchekenian V, et al. True hermaphroditism: clinical aspects and molecular studies in 16 cases. Eur J Endocrinol 1997;136:201-4.
- 14. Sohval AR. Mixed gonadal dysgenesis: a variety of hermaphroditism. Am J Hum Genet 1963;15:155.
- 15. Shozu M, Akasoju K, Harada N. A new cause of female pseudohermaphroditism: placental aromatase deficiency. J Clin Endocrinol Metab 1991;72:560-6.
- 16. Prader A. Der genitalbefund beim pseudo-hermaphroditismus femininus des kongenitalen adrenogenitalen syndrome. Helv Paediat Acta 1954;9:231.
- 17. Damiani D. Estados intersexuais. Pediatria Moderna 1995;31:945-80.
- 18. Sinnecker GHG, Hiort O, Nitsche EM, Holterhus PM, Kruse K. Functional assessment and clinical classification of androgen sensitivity in patients with mutations of the androgen receptor genr. Eur J Pediatr 1997;156:7-14.
- 19. Rey RA, Belville C, Hihoul-Fékété C. Evaluation of gonadal function in 107 intersex patients by means of serum antimüllerian hormone measurement. J Clin Endocrinol Metab 1999;84:627-31.
- 20. Mendez JP, Schiavon R, Diaz-Cueto L, Ruiz AI, Canto P, Söderlund D, et al. A reliable endocrine test with human menopausal gonadotropins for diagnosis of true hermaphroditism in early infancy. J Clin Endocrinol Metab 1998;83:3523-6.
Datas de Publicação
-
Publicação nesta coleção
29 Jun 2001 -
Data do Fascículo
Fev 2001
Histórico
-
Recebido
Out 2000 -
Aceito
10 Jan 2001