Resumos
Hiperplasia adrenal congênita (HAC) é uma doença autossômica recessiva decorrente da alteração de enzimas que participam da síntese do cortisol. As manifestações podem ser causadas pela deficiência do cortisol e, em alguns casos, aldosterona e pelo acúmulo de precursores. O objetivo desta revisão é apresentar os mecanismos moleculares dos principais defeitos enzimáticos envolvidos na etiopatogênese da HAC. A deficiência da 21-hidroxilase (21OH) ocorre em 95% dos casos de HAC. Existem dois genes que codificam o P450c21: um ativo, CYP21, e um pseudogene CYP21P. Ambos são altamente homólogos (98%), o que favorece o emparelhamento desigual dos cromossomos homólogos durante a meiose, levando a duplicações e/ou deleções ou conversões desses genes. Adicionalmente, foram também descritas mutações de ponto, muitas delas presentes no pseudogene sugerindo microconversões. Mutações no gene CYP11B1 causam HAC por deficiência da 11beta-hidroxilase, forma esta que corresponde a 5% dos casos. Algumas mutações são recorrentes, situando-se principalmente entre os exons 6-8 que representaria uma área hot-spot no gene CYP11B1. A deficiência de 17-hidroxilase é causada por mutações no gene CYP17, que codificam uma proteína alterada, levando a deficiência total ou parcial de 17-hidroxilação e 17,20-liase ou deficiência isolada de 17,20-liase. Finalmente, deficiência de 3beta-HSD é causada por mutações no gene HSD3B2, que codifica a enzima 3beta-HSD tipo II e estas mutações têm sido associadas tanto com a forma clássica como com a forma não clássica da deficiência da 3beta-HSD.
Hiperplasia adrenal congênita; Deficiência de 21-hidroxilase; Deficiência de 11beta-hidroxilase; Deficiência de 17-hidroxilase; Deficiência de 3beta-HSD
Congenital adrenal hiperplasia (CAH) is a recessive autossomic disease caused by inherited defects in cortisol biosynthesis. The manifestations are caused both by the deficient synthesis of cortisol, and sometimes of aldosterone, and by accumulation of the precursor steroids. The objective of this review is to present the molecular mechanisms of the main enzymatic defects involved in the etiopathogenesis of CAH. Deficiency of 21-hydroxylase (21OH) accounts for more than 95% of all cases of CAH. The human genome contains two CYP genes: one active, CYP21, and a pseudogene, CYP21P. Both are highly homologous (98%), facilitating recombination events during meiosis, leading to duplication and/or deletion or conversion of these genes. Additionally, point mutations have also been described. Deficiency of 11beta-hydroxylase (11betaOH) is caused by mutations in the CYP11B1 gene, and accounts for 5% of all cases. Some mutations are recurrent, and mainly located on exons 6-8, which is considered a hot-spot area in CYP11B1 gene. Deficiency of 17alpha-hydroxylase (17OH) is caused by mutations in the CYP17 gene, producing a truncated or impaired protein. These mutations have been described in patients with combined deficiencies of 17OH and 17,20-lyase or with isolated 17,20-lyase. Finally, CAH caused by 3beta-HSD deficiency is the consequence of mutations in the gene HSD3B2 that encodes 3beta-HSD type II. In the classical form of the disease nonsense mutations, insertion and deletions have been described, while in non classical forms, mutations result in diminished enzyme affinity and loss of enzyme activity.
Congenital adrenal hyperplasia; 21-hydroxylase deficiency; 11beta-hydroxylase deficiency; 17-hydroxylase deficiency; 3beta-HSD deficiency
atualização
Bases Moleculares da Hiperplasia Adrenal Congênita
Maricilda Pallandi de Mello
Tânia A.S.S. Bachega
Marivânia da Costa-Santos
Lívia Mara Mermejo
Margaret de Castro
Centro de Biologia Molecular e
Engenharia Genética (CBMEG) -
UNICAMP (MPM), Campinas, SP;
Laboratório de Hormônios e Genética
Molecular, Disciplina de
Endocrinologia, Hospital das Clínicas
- FMUSP (TASSB); Universidade
Federal de São Paulo -UNIFESP/EPM
(MC-S), São Paulo, SP; e Divisão de
Endocrinologia, Departamento de
Clínica Médica - FMRP-USP (LMM,
MC), Ribeirão Preto, SP.
Recebido em 29/07/2002
Aceito em 02/08/2002
RESUMO
Hiperplasia adrenal congênita (HAC) é uma doença autossômica recessiva decorrente da alteração de enzimas que participam da síntese do cortisol. As manifestações podem ser causadas pela deficiência do cortisol e, em alguns casos, aldosterona e pelo acúmulo de precursores. O objetivo desta revisão é apresentar os mecanismos moleculares dos principais defeitos enzimáticos envolvidos na etiopatogênese da HAC. A deficiência da 21-hidroxilase (21OH) ocorre em 95% dos casos de HAC. Existem dois genes que codificam o P450c21: um ativo, CYP21, e um pseudogene CYP21P. Ambos são altamente homólogos (98%), o que favorece o emparelhamento desigual dos cromossomos homólogos durante a meiose, levando a duplicações e/ou deleções ou conversões desses genes. Adicionalmente, foram também descritas mutações de ponto, muitas delas presentes no pseudogene sugerindo microconversões. Mutações no gene CYP11B1 causam HAC por deficiência da 11b-hidroxilase, forma esta que corresponde a 5% dos casos. Algumas mutações são recorrentes, situando-se principalmente entre os exons 6-8 que representaria uma área hot-spot no gene CYP11B1. A deficiência de 17-hidroxilase é causada por mutações no gene CYP17, que codificam uma proteína alterada, levando a deficiência total ou parcial de 17-hidroxilação e 17,20-liase ou deficiência isolada de 17,20-liase. Finalmente, deficiência de 3b-HSD é causada por mutações no gene HSD3B2, que codifica a enzima 3b-HSD tipo II e estas mutações têm sido associadas tanto com a forma clássica como com a forma não clássica da deficiência da 3b-HSD. (Arq Bras Endocrinol Metab 2002;46/4:457-477)
Descritores: Hiperplasia adrenal congênita; Deficiência de 21-hidroxilase; Deficiência de 11b-hidroxilase; Deficiência de 17-hidroxilase; Deficiência de 3b-HSD
ABSTRACT
Molecular Bases of Congenital Adrenal Hyperplasia.
Congenital adrenal hiperplasia (CAH) is a recessive autossomic disease caused by inherited defects in cortisol biosynthesis. The manifestations are caused both by the deficient synthesis of cortisol, and sometimes of aldosterone, and by accumulation of the precursor steroids. The objective of this review is to present the molecular mechanisms of the main enzymatic defects involved in the etiopathogenesis of CAH. Deficiency of 21-hydroxylase (21OH) accounts for more than 95% of all cases of CAH. The human genome contains two CYP genes: one active, CYP21, and a pseudogene, CYP21P. Both are highly homologous (98%), facilitating recombination events during meiosis, leading to duplication and/or deletion or conversion of these genes. Additionally, point mutations have also been described. Deficiency of 11b-hydroxylase (11bOH) is caused by mutations in the CYP11B1 gene, and accounts for 5% of all cases. Some mutations are recurrent, and mainly located on exons 6-8, which is considered a hot-spot area in CYP11B1 gene. Deficiency of 17a-hydroxylase (17OH) is caused by mutations in the CYP17 gene, producing a truncated or impaired protein. These mutations have been described in patients with combined deficiencies of 17OH and 17,20-lyase or with isolated 17,20-lyase. Finally, CAH caused by 3b-HSD deficiency is the consequence of mutations in the gene HSD3B2 that encodes 3b-HSD type II. In the classical form of the disease nonsense mutations, insertion and deletions have been described, while in non classical forms, mutations result in diminished enzyme affinity and loss of enzyme activity. (Arq Bras Endocrinol Metab 2002;46/4: 457-477)
Keywords: Congenital adrenal hyperplasia; 21-hydroxylase deficiency; 11b-hydroxylase deficiency; 17-hydroxylase deficiency; 3b-HSD deficiency
HIPERPLASIA ADRENAL CONGENITA POR DEFICIENCIA DA 21-HIDROXILASE
A HIPERPLASIA ADRENAL CONGÊNITA (HAC) por deficiência da 21-hidroxilase (21OH) é o mais freqüente dos defeitos enzimáticos adrenais, ocorrendo em 95% dos casos de HAC (1). A enzima 21OH participa da síntese dos glico e dos mineralocorticóides. Sua ação é converter a progesterona em desoxicorticosterona (DOC) e a 17OH-progesterona (17OHP) em 11-desoxicortisol (S), que por sua vez é convertido em cortisol sob ação da 11-hidroxilase (11OH). A redução da atividade da 21OH com decorrente diminuição da síntese de cortisol resulta em estimulação crônica do córtex adrenal pelo ACTH com hiperplasia adrenal e superprodução dos precursores do cortisol. Estes precursores são desviados para a biossíntese dos andrógenos, que não necessita da atividade da 21OH, causando os sinais de virilização característicos em indivíduos com esta deficiência enzimática.
Tradicionalmente é classificada em duas formas clínicas: clássica, que inclui os subgrupos perdedora de sal e virilizante simples; e não clássica, que inclui os subgrupos sintomática e assintomática (ou críptica). A forma virilizante simples caracteriza-se por graus variados de virilização pré-natal da genitália externa no sexo feminino e virilização pós-natal em ambos os sexos, com aumento do clitóris ou pênis, pubarca precoce e avanço da idade óssea com prejuízo na estatura final. A forma perdedora de sal, além da hiperprodução androgênica da forma anterior, apresenta deficiência mais severa na produção de aldosterona levando à desidratação com hiponatremia e hiperpotassemia nos primeiros 30 dias de vida que, se não tratada, resulta em choque e óbito. A forma de início tardio sintomática não apresenta virilização pré-natal e os sintomas iniciam-se em épocas variáveis, resultando em pubarca precoce, amenorréia primária ou secundária, hirsutismo, acne e infertilidade. A forma não clássica assintomática apresenta o mesmo perfil hormonal da forma sintomática, porém sem manifestações clínicas, sendo geralmente diagnosticada na investigação dos familiares de um paciente (2).
Os programas de rastreamento neonatais sugerem uma freqüência da forma clássica em 1:5.000 a 1:15.000 nascimentos na maioria das populações caucasianas, enquanto que a forma não clássica ocorre aproximadamente em 1 a 2% da população geral (3,4). Estas formas não representam diferentes doenças, mas um contínuo espectro na severidade da deficiência da 21OH.
Genética Molecular
A 21-hidroxilação é mediada por uma enzima mitocondrial específica denominada P450c21; seu gene codificador está localizado no locus do complexo principal de histocompatibilidade humana no braço curto do cromossomo 6, 6p21, especificamente dentro do locus do HLA classe III (5). Devido à íntima relação da 21OH com os genes do complexo HLA, observou-se que indivíduos afetados de uma mesma irmandade eram quase que invariavelmente HLA idênticos (6). Por esta razão, antes do advento da clonagem do gene da 21OH, a tipagem do HLA foi muito utilizada no diagnóstico pré-natal das gestações de risco.
Não existe um haplótipo que seja diagnóstico de uma forma clínica específica da deficiência da 21OH. Entretanto, algumas correlações podem ser feitas: o HLA-Bw47; DR7 está presente em 20% a 25% dos alelos da forma clássica e o HLA-B14; DR1 está presente em 70% dos alelos da forma não clássica, principalmente em judeus do leste europeu (7,8). Essas combinações recebem o nome de haplótipos. Posteriormente, com a clonagem dos genes da 21OH foram descobertas as mutações que esses haplótipos carregavam.
Existem dois genes para o P450c21, um pseudogene CYP21P e um gene ativo CYP21. Ambos contêm 10 exons, suas seqüências são 98% idênticas nos exons e 96% idênticas nos introns (9). O CYP21P é um pseudogene porque não codifica uma proteína devido à presença de várias mutações, sendo as mais comuns: deleção de 8 pares de bases no exon 3, inserção de T no exon 7 (as quais geram uma alteração na matriz de leitura), e uma substituição de C por T no exon 8 (que cria um "codon" prematuro de parada da leitura). O gene CYP21 codifica uma proteína com 494 aminoácidos.
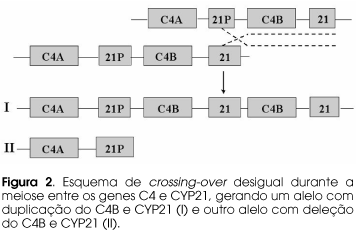
Os genes CYP21 e CYP21P se estendem sobre uma região de aproximadamente 30Kb, adjacentes e alternando com os genes C4A e C4B que codificam o quarto componente do complemento sérico (5,10). Recentemente foram descobertos outros pares de genes XA, XB, YA, YB, ZA e ZB (figura 1) (11). Os genes XA e XB sobrepõem-se ao último exon do CYP21P e CYP21 respectivamente. Estes genes são transcritos em sentido oposto aos genes da 21OH e XB codifica uma importante matriz protéica extracelular. Os genes YA e YB superpõem-se aos genes CYP21P e CYP21 e o YA usa o promotor do CYP21P, sugerindo que o pseudogene possa ter alguma função no córtex adrenal. Os genes ZA e ZB apresentam transcritos adrenais, mas como os dos genes YA e YB, suas funções ainda não são conhecidas. Esta região de genes duplicados C4A-ZA-CYP21P-XA-YA; C4B-ZB-CYP21-XB-YB apresenta um extremo grau de identidade de nucleotídeos e favorece o emparelhamento desigual dos cromossomos homólogos durante a meiose, o que pode levar ao aparecimento de duplicações e/ou deleções desses genes.
A técnica de Southern blotting seguida de hibridação com sonda específica permite a verificação do número de cópias de um gene. Através desta metodologia podemos pesquisar a deleção do gene CYP21, que foi a primeira mutação descrita responsável pela deficiência da 21OH. Em decorrência de um emparelhamento desigual dos cromossomos durante a meiose, ocorre a formação de um alelo com duplicação da unidade C4B-CYP21 e outro com perda desta unidade, que é o alelo responsável pela HAC-21OH (10,12) (figura 2). Em 98,2% dos casos a deleção compreende 30Kb estendendo-se de algum ponto entre os exons 3 e 8 do CYP21P, através do C4B até o ponto correspondente no CYP21. Assim, o alelo resultante é um híbrido onde a porção 5' apresenta a seqüência do CYP21P e seu extremo 3' a seqüência do CYP21. Estima-se que as deleções do gene CYP21 ocorram em 25% dos alelos nas formas perdedoras de sal em diversas populações caucasianas e do oeste europeu. Baixa freqüência de deleções têm sido restritas a populações específicas tais como: nas populações mexicana e japonesa com freqüência <2%, na população argentina com freqüência de 7% (13-15). No Brasil, onde a maioria da população tem ascendência portuguesa e casamentos interraciais com negros e índios são freqüentes, também observamos uma freqüência baixa de deleções, da ordem de 4% e de 7% em dois estudos populacionais (16,17).
A segunda mutação descrita na HAC-21OH foi a conversão gênica, que também ocorre por um emparelhamento desigual dos genes durante a meiose. Neste caso ocorre a troca de somente uma fita de DNA entre os genes CYP21 e CYP21P, transferindo seqüências deletérias do pseudogene para o gene ativo (18) (figura 3). A freqüência das conversões varia de 10 a 15% entre as diversas populações, estando presente principalmente na forma clássica (tabela 1). Na população brasileira sua freqüência foi de 17% em estudo que incluiu pacientes com a forma clássica (16) e de 7% em estudo que incluiu as três formas clínicas da HAC-21OH (17). A freqüência menor de mutações do tipo grandes rearranjos na nossa população sugere freqüência maior das mutações de ponto.
São descritas também deleções e duplicações do pseudogene envolvendo principalmente o gene C4B, os quais aparecem também na população normal, não sendo portanto causa da doença. Estes rearranjos são considerados como pré-mutação, por facilitar o emparelhamento desigual dos genes durante a meiose. Estudos populacionais demonstraram que em 65 a 90% dos alelos estudados em pacientes com HAC-21OH não foram evidenciadas as grandes mutações do tipo deleção do CYP21 e conversão gênica, indicando que deveria existir outro tipo de alteração responsável pela patologia.
Através do seqüenciamento destes genes e posteriores estudos de atividade enzimática in vitro evidenciou-se a presença de mutações de ponto, que quando em homozigose ou heterozigose composta poderiam ser responsáveis pela deficiência da 21OH (19). Atualmente estas mutações podem ser facilmente identificadas através de técnicas de rastreamento como hibridação oligo-específica (20) ou reação de polimerização em cadeia (PCR) alelo-específico (21).
Foram descritas mais de 40 mutações de ponto responsáveis pela deficiência da 21OH (22). Destas, 9 são as mais freqüentes nas diversas populações estudadas (tabela 2) e estão presentes normalmente no pseudogene, sugerindo que foram transferidas para o gene ativo através de eventos de microconversões, devidas ao emparelhamento desigual destes genes durante a meiose (23,24).
A substituição de uma adenina por guanina no nucleotídeo 668 do intron 2 constitui a mutação mais freqüente causadora da deficiência da 21OH. Esta mutação cria um novo sinal de término do intron e 19 nucleotídeos que deveriam ser retirados são retidos no RNAm alterando toda a rede de leitura, impedindo a síntese de uma proteína ativa. Ocorre entre 26 a 42% dos alelos de indivíduos afetados por HAC. A maioria dos pacientes homo ou hemizigotos para esta mutação tem a forma perdedora de sal. Ocasionalmente, a apresentação da perda de sal pode ser retardada para alguns meses de vida nos portadores desta mutação. Em um paciente de nossa amostra, homozigoto para esta mutação, a primeira crise ocorreu com seis meses de vida.
A deleção de 8 pares de bases no exon 3 leva também a uma alteração na rede de leitura e codifica uma enzima completamente ineficiente, sendo encontrada em 3 a 10% dos alelos das formas perdedoras de sal.
Um grupo de 3 mutações ocorre ao mesmo tempo (cluster) no exon 6 nos codons 236, 237 e 239 e causa a substituição de 3 aminoácidos: Ile, Val, Met ® Asn, Glu, Lis, respectivamente. Confere grave deficiência enzimática estando portanto mais associada à forma perdedora de sal. Ocorre com uma freqüência de 3 a 17% entre todos os alelos.
A mutação Q318X é devida a uma substituição de citosina por timina, cria um codon prematuro de parada de leitura e gera uma proteína truncada que é inefetiva. Ocorre em 4 a 7% dos alelos da forma perdedora de sal.
A mutação R356W decorrente de uma substituição de citosina por guanina altera a ligação da enzima com o substrato e ocorre em 14% dos alelos.
A mutação I172N resulta de uma substituição de timina por adenina, originando a troca de um aminoácido polar por um não polar e interrompendo a ligação da enzima com o retículo endoplasmático. A atividade da enzima ao redor de 3% permite que seja produzida uma pequena quantidade de aldosterona, suficiente para impedir a crise de perda de sal, o que faz com que esta mutação esteja associada à forma virilizante simples. Apresenta-se numa freqüência de 5 a 10% dos alelos afetados.
Duas mutações de menor gravidade, P30L e V281L, estão mais associadas à forma de início tardio. A mutação P30L no exon 1 ocorre devido à troca de uma citosina por uma timina e está presente em 17% dos alelos nas formas de início tardio. A mutação V281L é devida a uma substituição de guanina por timina causando uma troca conservativa de aminoácidos, o que origina uma mudança na conformação da proteína, reduzindo sua atividade enzimática. Sua freqüência nas formas de início tardio associada ao HLA-B14 varia de 78 a 84% dos alelos. A mutação P30L em geral tem expressão clínica mais grave do que a mutação V281L, todas as nossas pacientes portadoras desta mutação apresentaram clitoromegalia.
A tabela 3 apresenta dados referentes às mutações de ponto em diversas populações. A freqüência das mutações de ponto em dois estudos populacionais brasileiros foi semelhante (25,26). As mutações mais freqüentes foram I2 splice, I172N e V281L e estiveram associadas significativamente com as formas perdedora de sal, virilizante simples e não clássica respectivamente (p<0,01) (25). Em geral as freqüências descritas em outros estudos não diferem das nossas, poucos casos onde diferenças foram observadas podem refletir variações individuais da amostra ou diferenças populacionais (26).
O gene CYP21 pode ser convertido em grandes segmentos do pseudogene contendo duas ou mais mutações de ponto, ou ainda, um mesmo alelo pode receber eventos diferentes de microconversão ao longo das gerações. Em nossa amostra, 6% dos alelos apresentaram duas ou três mutações de ponto. Por este motivo, recomenda-se, para adequado diagnóstico molecular, pesquisar no mínimo as nove mutações de ponto derivadas do pseudogene em todos os pacientes. Também é importante a segregação das mutações no DNA dos pais. Além de caracterizar a distribuição das mutações por alelo, permite diferenciar homozigose de hemizigose (indivíduos que têm uma mutação de ponto em um alelo e deleção no outro alelo).
A pesquisa dos grandes rearranjos e das 9 mutações de ponto mais freqüentes são responsáveis por 80 a 85% dos alelos na população brasileira (25,26). Os demais alelos devem apresentar mutações novas, as quais não foram identificadas por terem sido empregadas técnicas de estudo dirigidas para mutações específicas. Estudos de seqüenciamento do gene CYP21 têm identificado mutações novas nas diferentes populações (22). De forma geral as mutações novas não estão presentes no pseudogene e representam eventos mutagênicos casuais, sendo descritas em casos isolados.
Na população brasileira foram identificadas as mutações G424S e H28+C (27,28). A mutação G424S foi identificada em 7 pacientes portadores da forma virilizante simples, em desequilíbrio de ligação com o HLA-DR17 e deleção dos genes CYP21P e C4A. A presença do mesmo haplótipo em todos os portadores da mutação sugere uma origem comum ou seja, efeito de gene fundador.
A mutação H28+C foi identificada em homozigose em uma menina portadora da forma perdedora de sal. Ocorreu a inserção de um nucletídeo C no codon 28, levando a uma alteração de toda a rede de leitura e a formação de uma parada prematura de leitura no codon 78, gerando uma proteína truncada. Ambas mutações não foram encontradas no estudo do pseudogene, sugerindo que não são provenientes de eventos de microconversão. Outras mutações novas em pacientes brasileiros estão em estudo quanto à possibilidade de gene fundador. Como a nossa população apresenta grau alto de miscigenação, é provável que estas sejam identificadas também em outras populações.
Estudos de seqüenciamento complementando as técnicas anteriores identificaram mutações em 100% dos alelos da forma clássica, enquanto que a forma não clássica, em nossa amostra, permanece ainda com freqüência alta (23%) de alelos sem mutações identificadas (29). Nenhum paciente portador de forma não clássica e com valor da 17OHP pós-ACTH <17ng/mL teve seu genótipo definido. Em outros estudos populacionais, onde significante número de pacientes com a forma não clássica foi estudado, esta forma clínica também apresenta porcentagem alta de alelos sem mutações identificadas. Estes dados sugerem que o atual critério diagnóstico desta forma clínica, 17OHP >10ng/mL, possa estar superestimando o seu diagnóstico (30).
Para investigar esta possibilidade, foi realizado o estudo de 59 heterozigotos obrigatórios quanto à presença de manifestações hiperandrogênicas, valores da 17OHP pós-ACTH e pesquisa de mutações no gene CYP21. Manifestações hiperandrogênicas foram encontradas em 10% dos heterozigotos, em freqüência semelhante à da população normal. Identificamos dois indivíduos sem manifestações hiperandrogênicas e com valores da 17OHP pós-ACTH de 11 e de 15ng/mL, os quais são valores diagnósticos da forma não clássica da 21OH. Para afastar a possibilidade de serem afetados pela forma não clássica assintomática, foi realizado o seqüenciamento do gene CYP21 destes indivíduos, e confirmou-se a presença de apenas uma mutação em heterozigose, Q318X e G424S, respectivamente. Observa-se que existe uma superposição dos valores da 17OHP pós-ACTH entre os indivíduos heterozigotos para mutações com severo comprometimento da atividade enzimática e os portadores da forma não clássica (31). Assim, como heterozigose para a 21OH não causa manifestações hiperandrogênicas, sugerimos que estes pacientes não devem ser tratados com glicocorticóide. O estudo molecular do gene CYP21 está indicado para se diferenciar heterozigose de forma não clássica da 21OH nos indivíduos com manifestações hiperandrogênicas e com valores da 17OHP pós-ACTH entre 10 e 17ng/mL.
Correlação do Genótipo com Fenótipo
Os estudos de mutagênese in vitro permitiram quantificar a redução da atividade enzimática conferida por cada mutação (19,32) e sua correlação com as diferentes formas clínicas da doença. Speiser e cols. (20) dividiram as mutações em três grupos de acordo com a gravidade do comprometimento da atividade enzimática e correlacionaram com as diferentes formas clínicas. O grupo A (severo comprometimento) incluiu mutações com atividade enzimática ausente ou mínima. O grupo B (moderado comprometimento) incluiu a mutação I172N que confere entre 3 a 7% de atividade enzimática. O grupo C (leve comprometimento) incluiu as mutações P30L e V281L, as quais conferem atividade enzimática > 18%.
A HAC-21OH é uma doença com herança autossômica recessiva causada por várias mutações que estão presentes com freqüência alta na população. Por este motivo, os indivíduos afetados apresentam geralmente mutações diferentes em cada um dos alelos (heterozigotos compostos) e uma minoria são homozigotos para uma mutação. A forma clínica no heterozigoto composto é conferida pelo alelo com maior atividade enzimática. Assim, indivíduos com o genótipo severo/severo apresentam, principalmente, a forma perdedora de sal, com os genótipos severo/moderado e moderado/moderado apresentam, principalmente, a forma virilizante simples e os com os genótipos severo/leve, moderado/leve e leve/leve apresentam, principalmente, a forma não clássica.
Existe boa correlação do genótipo com o fenótipo. Em amostra de 93 pacientes com deficiência da 21-hidroxilase foi observado 62% de correlação do genótipo severo/severo com a forma perdedora de sal. Isto porque 60% dos pacientes deste grupo possuíam a mutação I2 splice em pelo menos um alelo. Estudos in vitro da mutação do I2 splice demonstram splicing alternativos resultando em níveis variados de 21-hidroxilase normal, o que explicaria sua associação também com a forma virilizante simples (19). Houve 96% de correlação dos genótipos severo/moderado e moderado/moderado com a forma virilizante simples e 88% de correlação dos genótipos severo/leve, moderado/leve e leve/leve com a forma não clássica. Estas correlações também ocorreram de forma estatisticamente significativa com os valores hormonais basais da 17OH-progesterona e da testosterona, os quais se traduziram no grau de virilização da genitália externa (25).
Estudos de correlação do genótipo com o fenótipo (20,21,33) verificaram que nem sempre ocorre a associação esperada. Os fatores que podem ser responsáveis por esta disparidade são a presença de mutações raras no mesmo alelo, bem como atividade de 21-hidroxilação extra-adrenal mediada por outras enzimas além da P450c21. Outros fatores que podem ser responsáveis por esta ausência de correlação e que devem ser estudados são anormalidades em regiões promotoras e regulatórias do gene CYP21, atividade de fatores de transcrição e de proteínas transportadoras.
A deficiência da 21-hidroxilase é uma doença que apresenta um complexo mecanismo molecular, por apresentar um gene ativo e um pseudogene altamente homólogos, localizados em uma região de genes duplicados que favorece eventos mutagênicos. Entretanto, apresenta bem elucidado seu mecanismo fisiológico e os conhecimentos de biologia molecular num futuro próximo poderão ser direcionados para a terapia gênica. Já foi realizado estudo com terapia gênica em camundongos com completa ausência da atividade da 21-hidroxilase e esta possibilitou restauração da capacidade de secreção de cortisol durante 40 dias (34). Mas, até o momento, na prática clínica são consolidadas as seguintes aplicações do estudo molecular do gene CYP21: 1) aconselhamento genético; 2) diagnóstico pré-natal das gestações de risco para a forma clássica; 3) na diferenciação entre heterozigose de forma não clássica da 21-hidroxilase; 4) em parceiro de paciente com as diferentes formas clínicas para se determinar a probabilidade de terem filhos afetados com a forma clássica.
Diagnóstico Prenatal
A principal complicação da HAC-21OH é a virilização intra-útero da genitália externa de fetos femininos afetados. O diagnóstico pré-natal vem sendo utilizado há duas décadas em gestações de risco, isto é, de mães que já possuem uma criança com a forma clássica da deficiência da 21-hidroxilase, com o objetivo de instituir a terapêutica a fim de suprimir a adrenal fetal e evitar a virilização da genitália externa em fetos femininos afetados. Inicialmente o diagnóstico pré-natal foi realizado através de determinações da 17OH-progesterona ou da tipagem do HLA a partir da 15ª semana de gestação (35,36). Entretanto estas metodologias ocasionalmente forneciam resultados falso negativos, nas formas clínicas de menor gravidade quando se utilizava a dosagem hormonal (37) ou mesmo na tipagem do HLA por eventos de recombinação intra-HLA. O inconveniente destes testes que utilizam a amniocentese é que são realizados tardiamente, somente a partir da 15ª semana de gestação, quando já pode ter ocorrido a virilização da genitália externa.
O aprimoramento das técnicas de identificação das mutações responsáveis pela deficiência da 21-hidroxilase associado à metodologia de biópsia de vilo coriônico permitiu sua aplicação clínica em testes de diagnóstico pré-natal, melhorando a sensibilidade e precocidade diagnóstica. A extração de DNA obtido de tecido de vilo coriônico possibilitou o diagnóstico genético a partir da 10ª semana de gestação, tornando possível a intervenção terapêutica. Como a diferenciação da genitália pode iniciar-se antes da 10ª semana de gestação, o tratamento deve ser iniciado assim que se confirmar a gestação (4ª a 5ª semana).
O protocolo de tratamento pré-natal seguido mundialmente consiste em orientar as mães de risco para procurar o médico com uma a duas semanas de atraso menstrual. Após a confirmação da gestação, pelo bhCG positivo, deve ser iniciada a terapia com dexametasona, na dose de 20µg/Kg/dia, dividida em três vezes. A partir da 10ª a 12ª semana de gestação realiza-se a biópsia de vilo coriônico para análise da determinação do sexo e estudo dos genes CYP21. Em caso de sexo masculino não há risco de problemas na genitália e a terapia deve ser suspensa. Em caso de sexo feminino prossegue-se o tratamento e o estudo para determinação no feto das mutações presentes no caso índex. Se o feto apresentar mutações em ambos alelos deve-se continuar a terapêutica até o nascimento, caso contrário o tratamento é interrompido. Durante a gestação a supressão adrenal fetal deve ser avaliada periodicamente com dosagens de estriol urinário. Em caso de feto masculino sugere-se também a investigação das mutações, o que possibilitaria o início da terapêutica de reposição, nos casos afetados, logo após o nascimento, podendo-se evitar a crise de perda de sal. A figura 4 apresenta o diagrama da conduta diagnóstica e terapêutica em gestações de risco para a HAC -21OH.
A vantagem do diagnóstico através do estudo de vilosidade coriônica é a sua precocidade, o que permite interromper precocemente a exposição da mãe à dexametasona em caso do feto não afetado. Speiser e cols. (38), estudando 24 gestações de risco com técnicas gene-específicas como Southern blotting, PCR e hibridação alelo-específico para as 9 mutações de ponto mais freqüentes, demonstraram serem estes métodos bastante eficientes, pois foram detectadas mutações em 95% dos cromossomos estudados com uma eficácia de 96% confirmada em exame pós-natal. Erro com o diagnóstico molecular ocorreu em 5%. Este erro pode ser decorrente de cross-contaminação das amostras se não for realizado rigoroso controle na execução da reação. Também pode ocorrer falha de amplificação de um alelo (allelic dropout) normal ou mutante durante a PCR. Para minimizar este problema, o estudo de microssatélites altamente polimórficos que flanqueiam o gene da 21-hidroxilase tem sido utilizado no diagnóstico pré-natal em adjunto ao estudo molecular do gene CYP21 (39).
Aproximadamente 70% das meninas tratadas pré-natalmente nascem com genitália externa normal ou ligeiramente virilizada, não necessitando de cirurgia plástica. São descritas falhas da terapia, através de meninas que nasceram mais virilizadas necessitando de correção cirúrgica. As causas que poderiam contribuir para a falha da terapêutica pré-natal são: 1) início tardio da terapia; 2) dose inadequada de dexametasona; 3) interrupção da terapia no meio da gestação; 4) não aderência; 5) passagem transplacentária do glicocorticóide, que pode ser influenciada pela sua afinidade pela transcortina; e 6) a função esteroidogênica adrenal fetal pode não ser regulada somente pelo ACTH em fases precoces da gestação.
Não é referida na literatura a presença de complicações fetais decorrentes do tratamento com dexametasona. A incidência de abortos espontâneos foi igual à da população geral. Da mesma forma não foram encontradas alterações nos parâmetros antropométricos (altura, peso e perímetro cefálico ao nascimento); porém a maioria destas crianças ainda não alcançou a estatura final ou a idade puberal para conclusões finais serem estabelecidas. É observada uma freqüência maior de abortos após a biópsia de vilo corial, para evitar isto a biópsia deve ser realizada apenas por profissionais com experiência, entre a 10ª e 12ª semana de gestação (22).
Com relação às mães, em geral a terapia é bem tolerada sendo citadas complicações em 4 a 9% dos casos. Ocorrem queixas de ganho excessivo de peso apesar do uso de dieta hipocalórica e hipossódica. Também são referidas estrias violáceas, crescimento de pêlos em rosto, hiperglicemia, hipertensão e fácies cushingóide. A maioria dos efeitos colaterais desaparecem com a interrupção da terapia. O tratamento pré-natal deve ser considerado apenas para as mães sadias e estas devem ser cuidadosamente monitoradas quanto ao ganho de peso, pressão arterial e à tolerância à glicose (40).
No serviço de Endocrinologia da FMUSP foram acompanhadas 22 gestações de risco para a deficiência da 21-hidroxilase, sendo que dentre elas ocorreu um aborto no primeiro trimestre não relacionado à biópsia de vilo, 21 gestações chegaram a termo. No momento do diagnóstico da gestação as mães iniciaram a terapêutica com dexametasona 20µg/kg/dia. Duas delas apresentaram excessivo ganho de peso nos primeiros 3 meses da gestação. Foi predito que em 11 gestações o feto era do sexo feminino, dentre elas três afetadas pela forma clássica e foi mantida a terapia pré-natal com dexametasona. Em todas o diagnóstico foi confirmado após o nascimento e as meninas nasceram com genitália externa normal, mostrando a eficácia do tratamento (41). Em 10 gestações foi predito que o feto era do sexo masculino, sendo um afetado pela forma perdedora de sal. A terapia pré-natal com dexametasona foi descontinuada e logo após o nascimento o diagnóstico foi confirmado, possibilitando a instituição da terapia com glico e mineralocorticóide antes da crise de perda de sal.
A terapêutica pré-natal ainda é controversa. Por ser uma metodologia recente ainda não existem dados com relação à puberdade, estatura final e perfil psicológico das crianças submetidas ao tratamento. Somente um entre 8 fetos será uma menina afetada pela deficiência da 21-hidroxilase, sendo assim, sete gestações serão submetidas desnecessariamente ao tratamento com dexametasona, antes que seja determinado o sexo e o genótipo. Por outro lado, a terapia é importante porque permite a ocorrência de uma genitália externa normal evitando ou diminuindo o trauma cirúrgico, evitando erros de atribuição do sexo ao nascimento e ainda por evitar a ação excessiva de andrógenos no cérebro feminino fetal, que poderia ser responsável por um dimorfismo sexual cerebral fetal. Dados da literatura referem que meninas com deficiência da 21-hidroxilase se casam menos, têm menos filhos, têm menor interesse sexual e maior incidência de lesbianismo. Estas complicações podem ser resultado do imprinting de andrógenos no cérebro pré e/ou pós-natal, como também do trauma cirúrgico da genitoplastia.
O consenso adotado hoje em dia é que os benefícios da terapia pré-natal suplantam os riscos. Até que estas crianças submetidas ao tratamento com dexametasona pré-natal, afetadas ou não, atinjam a idade adulta e possam ser tomadas conclusões definitivas, este tratamento deve ser empregado apenas em hospitais-escola. As mães devem ser informadas sobre os potenciais riscos da terapia e também quanto à possibilidade da falta de benefícios.
Hiperplasia Congênita de Adrenal por Deficiência de 11b-Hidroxilase
A deficiência da 11b-hidroxilase (11b-OH) foi primeiramente relatada por Eberlein e Bongiovani em 1955. Na maioria dos países corresponde a no máximo 5% dos casos de hiperplasia congênita da adrenal, com freqüência em torno de 1:100.000 nascimentos (42). No entanto, em populações com alto grau de endogamia uma freqüência maior é registrada: 1:30-40.000 em Israel, 1:5-7.000 em judeus provenientes do Marrocos (43) e pode chegar a mais de 25% dos casos de HCA na Arábia Saudita (44).
Na deficiência de 11b-OH, além dos sinais de hiperandrogenismo, um sinal clínico importante é a hipertensão arterial com alcalose hipercalêmica devida ao excesso de produção de deoxicorticosterona (DOC), o que a distingue da deficiência da 21-OH. Entretanto a hipertensão arterial pode ou não estar presente e é observada em 30-60% dos casos (42,43). A hipertensão, muitas vezes, só se manifesta nas fases mais tardias da infância ou adolescência e é atribuída ao excesso de DOC. A perda de sal na deficiência de 11b-OH pode ocorrer na infância, quando os níveis de DOC ainda são relativamente baixos e há maior vulnerabilidade à perda de sal. A perda de sal também pode ocorrer após o tratamento com glicocorticóides, que suprimem a DOC. O diagnóstico da forma clássica da deficiência da 11b-OH é feito pela constatação de níveis basais elevados de três hormônios principais: 11-deoxicortisol sérico, DOC e da substância S-tetra-hidro-urinária (THS). Em alguns casos ocorre a deficiência da 11b-OH denominada não clássica ou parcial. Esta é uma etiologia relativamente incomum. Entretanto, a sutileza com que esta variante se manifesta e as dificuldades associadas com o seu diagnóstico podem retardar sua identificação e resultar em uma redução significante na estatura adulta se não tratada precocemente com reposição hormonal. O diagnóstico da forma não clássica da deficiência da 11b-OH é baseado em elevação do 11-desoxicortisol em pelo menos 3 vezes os valores máximos obtidos pelo percentil 95% da população após estímulo com ACTH exógeno.
Genética Molecular
A enzima 11b-OH é uma das enzimas da família do citocromo P-450 chamada CYP11. Em humanos há 2 isoenzimas, uma responsável pela síntese de cortisol e outra pela aldosterona. A CYP11B1 catalisa a conversão de 11-desoxicortisol a cortisol na camada fasciculada, demonstrada em estudos de cultura de células e por hibridização in situ (45), a CYP11B2, por sua vez, age na conversão de desoxicorticosterona (DOC) a corticosterona, 18-hidroxicorticosterona e aldosterona na camada glomerulosa (46-49).
Mutações no gene CYP11B1, que codifica a enzima 11b-hidroxilase (11b-OH), causam HCA, ao passo que as mutações no gene CYP11B2, que codifica a enzima CYP11B2, são as causas da deficiência de corticosterona metil oxidase ou deficiência da aldosterona sintase. Por outro lado, a síndrome conhecida como hiperaldosteronismo supressível por glicocorticóides é resultado de uma recombinação intergênica, justapondo-se o promotor do CYP11B1 com seqüências codificadoras do CYP11B2 formando um gene híbrido (50,51). Neste processo o gameta resultante apresenta o gene híbrido CYP11B1/CYP11B2 intercalado aos genes CYP11B2 e CYP11B1. Nesses casos uma questão que se levanta é porquê um alelo que possui três cópias gênicas, duas delas com estrutura molecular inalterada, estaria produzindo um fenótipo alterado devido a uma cópia gênica quimérica. O gameta com deleção no locus de CYP11B, gerado como produto recíproco no processo de cruzamento desigual, produzindo o híbrido CYP11B2/CYP11B1 deve produzir a deficiência de 11b-OH (52,53). Apenas recentemente foram descritos dois pacientes com deficiência de 11b-OH apresentando deleções com a formação de genes quiméricos deste tipo (54,55). Mutações deletérias no gene CYP11B1, entretanto, são encontradas nas análises de DNA de pacientes com deficiência de 11b-OH e são correlacionadas com alterações em resíduos importantes para a atividade enzimática ou com interrupções da transcrição normal do gene gerando mRNA alterados e, conseqüentemente, proteínas truncadas (52,56-62).
Algumas mutações são recorrentes, principalmente, em certos grupos étnicos com alto grau de endogamia (judeus iranianos e marroquinos), o que sugere um forte efeito fundador na origem e irradiação dos alelos mutantes (63). No Brasil a mutação Q356X foi a primeira a ser descrita em um paciente da raça negra (64). Posteriormente, a mesma mutação foi encontrada em outros cinco pacientes brasileiros negros não relacionados indicando um possível efeito fundador nesse grupo étnico na população brasileira (65). A mutação Q356X havia sido anteriormente descrita em pacientes americanos de origem africana (52,66). A tabela 4 resume as principais mutações descritas e suas prováveis ações na transcrição e ação catalítica da enzima.
Curiosamente, 15/36 das mutações deletérias para a hidroxilação em c11 da 11b-OH estão situadas entre os exons 6-8 (52), formando um cluster de mutações no gene CYP11B1. Duas possibilidades têm sido levantadas para explicar essa concentração: mutações nessas regiões aconteceriam com a mesma freqüência que no resto dos exons, porém teriam maior probabilidade de serem deletérias na atividade enzimática, ou a região do cluster representaria uma área hot-spot no gene de CYP11B1, estando por alguma razão mais susceptível a mutações. Na verdade, quatro das oito mutações pontuais mais freqüentes (T318M, R374Q, R384Q e R448H) são transições CpGÞTpG, sendo este o grupo de transições mais comuns nos eventos de mutações pontuais do genoma humano (67). Sugestivamente, 49% (23/49) dos dinucleotídios CpG das regiões codificadoras de CYP11B1 estão dentro da região que engloba os exon 6 ao 8. Se comparado ao genoma, em geral, que apresenta 1,5% de CpG (67), a região do cluster de mutações de CYP11B1 apresenta uma porcentagem duas vezes maior (~3%) dessa composição nucleotídica, sugerindo que a alta taxa mutacional nessa região possa ser devida a uma área hot-spot (52). No entanto, o fato da região entre os exons 6-8 codificar domínios importantes para a ação enzimática, como o de ligação ao substrato à adrenodoxina, entre os codons 362-367, e o de ligação ao grupo heme, entre os codons 443-463, reforça a primeira hipótese.
Outra característica interessante que se observa nesse quadro de mutações é a ocorrência de várias mutações em um mesmo codon. Os codons 384 e 448, por exemplo, apresentam duas mutações diferentes, respectivamente. Outros dois codons que parecem ser alvos de múltiplas mutações são os codons 267 e 318 que, coincidentemente, fazem parte da seqüência consenso doadora de splicing, dos exons 4 e 5, respectivamente. Portanto, a expressão biológica de mutações nestes codons se deve mais a um processo de splicing anômalo do que a uma alteração na atividade enzimática causada por uma troca de aminoácido. Esta afirmação encontra respaldo em um trabalho publicado recentemente, no qual foi demonstrado que uma mutação conservativa no codon 318 (T(ACG) a T(ACC)) provocou a eliminação dos exons 3-7 no mRNA isolado da glândula adrenal da paciente (68). Num estudo realizado no Brasil foi identificada uma terceira mutação no codon 267, que troca uma glicina por serina (69). O códon 267 é formado pelo último nucleotídeo do éxon 4 e pelos dois primeiros nucleotídeos do éxon 5. Portanto, duas mutações, G267S e G267R, resultam, respectivamente, da troca de G®A e G®C no último nucleotídeo do éxon 4. Por outro lado, a mutação G267D ocorre no primeiro nucleotídeo do éxon 5. Desta forma, além de provocarem a troca do aminoácido, ocorrem na seqüência consenso do sítio doador e aceptor de splice do íntron 4 e podem afetar o processo para a formação do RNAm (70). Uma nova mutação foi encontrada em uma paciente de origem árabe no Brasil. Trata-se da inserção de uma citosina no éxon 8; isto faz com que o quadro de leitura para a síntese protéica sofra um deslocamento, a arginina do códon 404 é substituída por uma prolina e na posição 421 aparece um códon de terminação (71). Considera-se que a proteína produzida a partir do gene mutado seja anômala e tenha, portanto, sua atividade enzimática comprometida.
Hiperplasia Adrenal Congênita por Deficiência de 17-Hidroxilase/17,20 liase
A deficiência de 17-hidroxilase (D17OH) foi descrita pela primeira vez em 1966 (73). A paciente com 27 anos, cariótipo 46XX, apresentava ausência de desenvolvimento puberal e hipertensão arterial, com níveis diminuídos de cortisol, aldosterona e potássio. Considerada uma doença muito rara, a D17OH é comumente reportada na literatura através de casos isolados. Sua incidência estimada é de 1:50.000 (74), provavelmente, 1% de todos os casos de hiperplasia adrenal congênita (HAC). O bloqueio enzimático da 17-hidroxilase impede a produção de cortisol pela zona fasciculada adrenal e o conseqüente aumento do ACTH estimula a hipersecreção dos precursores imediatos ao bloqueio enzimático, como progesterona, desoxicorticosterona (DOC) e corticosterona (B) e, também, de esteróides com secreção geralmente limitada como 18OH-B, 18OH-DOC e 19-nor-DOC) (75). A ação mineralocorticóide exercida pela DOC é responsável pelos achados clínicos de hipertensão e hipocalemia, na presença de supressão do sistema renina-angiotensina-aldosterona. A deficiência dos esteróides gonadais, por sua vez, determina a ausência de desenvolvimento dos caracteres sexuais secundários no paciente 46XX ou presença de pseudohemafroditismo masculino. Alguns aspectos relacionados à fisiopatologia da HAC por D17OH ainda não estão totalmente elucidados, como: 1) níveis elevados de aldosterona em alguns pacientes japoneses (76); 2) os possíveis casos de deficiência parcial da enzima, com genitália ambígua, menarca, níveis pressóricos ou de potássio dentro da normalidade; 3) definição clínica e laboratorial da deficiência isolada de 17,20 liase (77); e 4) a associação de achados fortuitos como restos de tecido adrenal em testículo (78), mielolipoma (79), adenoma adrenal (80), gônadas disgenéticas e desenvolvimento incompleto de ductos müllerianos (81).
Genética Molecular
Vinte anos após a descrição do primeiro paciente com D17OH, o gene CYP17, constituído por 13kb, 8 exons e 7 introns, foi mapeado no cromossomo 10q24-q25 (82) e, desde então, o entendimento das bases moleculares desta doença, de herança autossômica recessiva, tem se intensificado. Trinta e nove mutações que afetam o gene estrutural do citocromo P450c17 foram descritas na literatura (tabela 5). A maioria dessas mutações (56%) constitui-se por substituições simples de bases, que promovem a troca de um aminoácido ou introduzem um códon de parada de transcrição do RNAm. Doze deleções que afetam o gene CYP17 (31% das mutações) promovem frameshift, com conseqüente expressão de uma proteína muito truncada. A maior mutação descrita no gene CYP17 é uma extensa deleção que acomete três membros de uma mesma família italiana (83). A deleção de 518 pb associada à inserção de 469 pb de DNA não humano acomete o exon II, todo o intron II e parte do exon III. Dois outros pacientes italianos apresentam homozigose para deleção de 24 pb, excluindo os aminoácidos 70-77 (84). As demais mutações consistem em pequenas duplicações e ainda, duas mutações do tipo splice, que substituem G ®T no intron 2 (85) e G ®A no intron 7 (86), afetando o sítio doador para o splice do RNAm.
O gene CYP17 humano é responsável pela codificação de uma única enzima - o citocromo P450c17, que exerce atividades de 17-hidroxilação e 17,20 liase nos tecidos adrenais e gônadas. Os seus fatores de transcrição ainda não foram totalmente elucidados. Baseando-se em estudos com roedores, possíveis sítios de interação com o SF-1 e com alguns receptores nucleares (COUP-TF, NGF-IB e fatores esteroidogênicos 1 e 2) (87), foram identificados na região promotora do gene CYP17. Experimentos com células adrenais Y1 de camundongo (88) demonstraram que as regiões -235/-184 e -184/-104 desempenham papel essencial na transcrição primária do gene e na sua expressão induzida por hormônio, respectivamente. Estudos recentes com células humanas adrenais NCI-H295A comprovaram que os elementos Sp1 e Sp3 interagem com a região promotora do gene CYP17, especificamente nos sítios -227/-184, assim como as proteínas NF-1C, atuam em sítios mais proximais (-178/-152 e -107/-85), ambos exercendo papel essencial na transcrição do gene, na espécie humana (87). Entretanto, até o momento, não há relato de mutação que afete a região promotora do gene CYP17 e apresente, por conseguinte, expressão clínica de D17OH.
O citocromo P450c17, codificado pelo gene CYP17, se localiza no retículo endoplasmático liso. O mesmo RNAm de 2.1kb se expressa tanto em adrenais como em gônadas (89) e codifica uma proteína constituída por 508 aminoácidos, peso molecular de 57kd. Os citocromos P-450 necessitam de ancoramento adequado à membrana microssomal, ligação de substrato, oxigênio e grupo heme, assim como transferência de elétrons, para o exercício adequado de suas funções. A ação do citocromo P450c17 é modulada por vários fatores específicos: 1) o tipo de substrato disponível, interfere diretamente na eficiência de catálise da enzima; 2) o tecido alvo e a espécie animal; 3) a disponibilidade do citocromo b5 e adrenodoxina redutase (90); e 4) a transferência de elétrons na presença de fosforilação (84).
A 17-hidroxilação de pregnenolona e progesterona é realizada com a mesma eficiência metabólica pelo citocromo P450c17. Entretanto, a quebra do carbono na posição 17,20 é 50 vezes mais eficiente para a formação de DHEA do que androstenediona, e depende diretamente da ação do citocromo b5, como foi demonstrado em estudos realizados em diferentes tipos de células e organismos (91,92). O citocromo b5 atua como um cofator alostérico (91), ao contrário do citocromo adrenodoxina redutase que atua por transferência de elétrons. A presença do citocromo b5 tem a capacidade de aumentar em até 10 vezes a função de 17,20 liase, mas ainda mantêm a preferência para metabolização da via ç5 (93) que, por sua vez, tem importância vital na espécie humana, principalmente a partir dos 8 anos de idade com a elevação de DHEA e início da adrenarca. O citocromo P450c17 também exerce 16-hidroxilação em 10% do substrato progesterona (92) e a partir da pregnenolona, catalisa a formação de 16-andieno, um precursor de ferormônios porcinos (94).
Zuber e cols. em 1986 expressaram, pela primeira vez, o DNA complementar do citocromo P450c17 bovino, em células não esteroidogênicas de rim de macaco (Cos 1) (95). Desde então, o estudo de atividade cinética do P450c17 humano tem sido realizado em células COS-1 (96), E.coli (93), nos microssomos extraídos de leveduras (91) e em tecidos humanos (81,92). Estes estudos permitem a comparação da atividade metabólica exercida pelas proteínas mutadas e pelo P450c17 wild type humano, na presença de substratos específicos marcados com radioisótopos. Existem três diferentes tipos de déficit enzimático (tabela 5): 1) deficiência total de 17-hidroxilação e 17,20 liase; 2) deficiência parcial combinada de 17-hidroxilação e 17,20-liase; ou 3) deficiência isolada de 17,20 liase.
Estudos com química computacional permitiram a criação de um modelo gráfico do P450c17 humano, a partir da estrutura -BMP de bactéria (92). A determinação da estrutura terciária da enzima, associada aos estudos de cinética, tem possibilitado um melhor entendimento de como as mutações afetam a ação do citocromo P450c17. A região carboxi terminal da proteína é essencial para a ligação apropriada dos substratos (94). Os últimos 48 resíduos formam parte da cobertura do anel b, essencial para ligação heme. Assim, as mutações 1, 9 e 11 (tabela 5) alteram a interação do substrato com a enzima. As mutações 21 e 26 promovem erro de splice durante o processo de transcrição do RNAm, com conseqüente deleção completa dos exons 2 e 7, e introdução de codons de parada prematuros, imediatamente antes da região de ligação heme. A mutação R440H (n. 17) e a deleção de um G na posição 438 (n. 19) são destituídas de qualquer atividade enzimática, devido o comprometimento do resíduo Cys na posição 442, essencial na doação do grupo sulfidril para a ligação heme. Os resíduos localizados na espiral B' e no anel b formam um "bolso" de ancoramento do substrato, de forma que as mutações n. 5,14,18 e 20 (tabela 5) provavelmente envolvem a interação de substrato como causa de déficit enzimático.
Recentemente o serviço de Endocrinologia da Universidade Federal de São Paulo fez um inquérito epidemiológico em centros de referência médica do Brasil, abordando o número de pacientes afetados pelas diversas formas de HAC. Avaliamos 30 pacientes brasileiros com D17OH, cujo diagnóstico laboratorial foi devidamente confirmado através da elevação dos precursores DOC, B, 18OH-B e 18OH-DOC, nos tempos basais e após estímulo com ACTH (cortrosina, 250µg EV). Observamos que a deficiência de 11b-hidroxilase não se mostrou mais prevalente do que a D17OH.(97) Dentre estes pacientes, analisamos do ponto de vista molecular, 24 pacientes pertencentes a 20 famílias (98). Este grupo apresenta mediana de idade cronológica de 16,6 anos (variação de 10,3 - 34 anos), quadro de hipertensão arterial com níveis médios de pressão sistólica de 152 ± 23,2mmHg e de pressão diastólica de 105 ± 14mmHg, além de hipocalemia (3,1 ± 0,6mEq/l). Sete novas mutações foram identificadas (tabela 6) e não se encontram descritas nos países colonizadores ou com história de imigração para o Brasil.
O estudo da expressão dessas mutações, em células COS-1 e leveduras W303B (98), demonstrou total ausência de atividade enzimática, com exceção da mutação Y329D, que retêm cerca de 5% de atividade de 17-hidroxilação e 17,20 liase (tabela 6). Este achado é compatível com o fenótipo apresentado pela paciente, que desenvolveu caracteres sexuais secundários espontâneos (Tanner V) e ciclos menstruais irregulares. Dois pacientes brasileiros, homozigotos para as mutações R347H (99) e R358Q (100), possuem dados clínicos e achados laboratoriais compatíveis com deficiência isolada de 17,20-liase (101). O estudo dessas mutações confirmou a presença de neutralização de cargas positivas nestes aminoácidos, que comprometem a interação dos elementos redox, adrenodoxina redutase e citocromo b5, com o citocromo P40c17 (90).
Hiperplasia Adrenal Congênita por Deficiência da 3bHSD
A hiperplasia adrenal congênita decorrente da deficiência da enzima 3b hidroxiesteróide desidrogenase (3bHSD) é uma doença autossômica recessiva rara, descrita em 1962 por Bongiovanni (121). O gene 3bHSD encontra-se no cromossomo 1p13.1 (122) e possui 4 exons, 3 introns e uma região 5' flanqueadora. Os genes 3bHSD do tipo 1 (HSD3B1) e do tipo 2 (HSD3B2) são os responsáveis pela codificação das isoenzimas 3bHSD do tipo 1 e do tipo 2, que possuem 93,5% de homologia (122). A enzima 3bHSD1 é expressa nos tecidos periféricos, principalmente na pele, glândulas mamárias e na placenta e a 3bHSD2 é expressa na adrenal, nos testículos e ovário (123). A enzima 3bHSD1 tem uma afinidade 10 vezes maior pelo substrato do que a enzima 3bHSD2 e pode ser estimulada após um aumento na secreção de gonadotrofina resultante de baixos níveis de andrógenos circulantes (124,125). A enzima 3bHSD catalisa a conversão dos D5 esteróides, tais como pregnenolona (Preg), 17-hidroxipregnenolona (17-Preg), deidroepiandrostenediona (DHEA) e androstenediol (D5-diol) em seus respectivos D4 esteróides, a progesterona (P), 17-hidroxiprogesterona (17-OHP), androstenediona (D4-A) e testosterona (T) (123). Essa atividade enzimática é, portanto, de fundamental importância para a síntese de todas as classes de esteróides ativos (progesterona, mineralocorticóides, glicocorticóides, andrógenos e estrógenos). Em contraste com as alterações provocadas pela deficiência da 21-hidroxilase e da 11b-hidroxilase, em que a síntese de esteróides está prejudicada apenas no córtex adrenal, a deficiência da 3bHSD impede a síntese de esteróides tanto na adrenal como nas gônadas (124,126). Como a produção de androgênios é diminuída, fetos do sexo masculino têm desenvolvimento incompleto da genitália externa com pseudo-hermafroditismo masculino, resultando em ambigüidade reconhecível ao nascimento. Recém-nascidos do sexo feminino, em geral, têm desenvolvimento normal da genitália externa ou apresentam sinais de virilização branda, como clitoromegalia, devido ao acúmulo de DHEA circulante e sua conversão, na periferia, para andrógenos mais potentes (124-126). Cerca de dois terços dos pacientes descritos com deficiência da 3bHSD são 46 XY, pois os recém-nascidos do sexo feminino não apresentam alterações na genitália externa ao nascimento, dificultando o diagnóstico (125). Existem duas formas de HAC pela deficiência da 3bHSD: uma forma clássica, reconhecida ao nascimento, que pode ou não apresentar perda de sal e uma forma não clássica, de desenvolvimento mais tardio (127-129), a qual se caracteriza pelo aparecimento de pubarca precoce em crianças e hirsutismo, acne, irregularidades menstruais em mulheres adultas.
O diagnóstico bioquímico da forma clássica da deficiência da 3bHSD tem sido realizado com base nas concentrações elevadas dos esteróides D5 como 17-Preg e DHEA e seus metabólitos na urina e no sangue (121). No entanto, as concentrações dos esteróides D4 também podem estar aumentadas (125,127,130), provavelmente, devido à conversão dos esteróides D5 em esteróides D4 pela enzima 3bHSD1 nos tecidos periféricos. O diagnóstico bioquímico da forma não clássica ainda apresenta dificuldades e os critérios mais aceitos eram os critérios adotados por Pang e cols. (131). Estes critérios baseavam-se em níveis de 17-Preg e DHEA, após estímulo com ACTH exógeno, maiores ou iguais a dois desvios padrões da média ( ³ 1639ng/dL e ³ 1818ng/dL, respectivamente) comparados a controles com idade e estadio puberal semelhantes. As relações 17-Preg/17-OHP e 17-Preg/cortisol (F), após estímulo com ACTH exógeno, maiores ou iguais a dois desvios padrões da média ( ³ 6,4 e ³ 52, respectivamente), também auxiliavam no diagnóstico da forma não clássica. Entretanto, estudos de crianças com pubarca precoce e mulheres hirsutas, cujas dosagens hormonais se enquadravam nos critérios acima, não apresentavam alterações moleculares compatíveis com a doença, sugerindo que esse critérios não distinguiam pacientes com ou sem mutações no gene HSD3B2. Estes autores (132) e outros estudos recentes (133,134) têm demonstrado que os valores da 17-Preg e da relação 17-Preg/F, após estímulo com ACTH exógeno, muito acima de dois desvios padrões da média têm sido os parâmetros mais válidos para o diagnóstico da forma não clássica de HAC por deficiência da 3bHSD. Marui e cols. (129), estudando crianças que apresentavam pubarca precoce, mostraram que as pacientes com mutação no gene HSD3B2 apresentaram os níveis mais elevados de 17-Preg e da relação 17-Preg/F, após estímulo com ACTH, enquanto que houve sobreposição dos níveis de DHEA e das relações 17-Preg/17-OHP e DHEA/D4-A entre as pacientes com e sem mutações. Esse trabalho sugere que a dosagem de 17-Preg e a relação 17-Preg/F, após estímulo com ACTH, são parâmetros importantes e fundamentais para o diagnóstico da forma não clássica da deficiência da 3bHSD.
Mutações têm sido descritas no gene HSD3B2 em pacientes com a forma clássica da deficiência da 3bHSD. Recentemente, também foram identificadas mutações no gene HSD3B2 na forma não clássica da doença, em meninas com pubarca precoce (129). Até hoje não foi identificada nenhuma mutação no gene HSD3B1 (132). Assim como ocorre na deficiência da 21-hidroxilase, em que mutações diferentes resultam em uma grande heterogeneidade de manifestações clínicas, o mesmo tem sido observado na deficiência da 3bHSD (125,128). Geralmente, na forma clássica da 3bHSD com perda de sal, as mutações encontradas são mutações pontuais, em homozigose ou heterozigose composta, como nonsense, frame-shift ou missense, as quais abolem completamente a atividade da enzima 3bHSD. Na forma clássica sem perda de sal, geralmente os pacientes estudados são homozigotos ou heterozigotos compostos para mutações missense, que mantêm até 10% da atividade enzimática quando esta é avaliada em estudos com células transfectadas em cultura (125). Como também se observa na 21-hidroxilase, uma atividade enzimática mínima é suficiente para sintetizar aldosterona e evitar a perda de sal e, na maioria dos pacientes, há correlação genótipo-fenótipo. Entretanto, existem exceções a essa regra, por exemplo, em pacientes que apresentam a forma não perdedora de sal foram identificadas mutações missense T259M em homozigose (129,135) cujo estudo funcional da proteína mutada mostrou ausência de atividade enzimática (125). Foi descrito (125), também, um paciente com a forma não perdedora de sal da deficiência da 3bHSD, portadora da mutação em heterozigose composta L236S / 867delG, em que o estudo funcional revelou 100% de atividade enzimática em células transfectadas com a mutação missense L236S e ausência de atividade enzimática em células contendo a mutação frame-shift 867delG.
O efeito das mutações no gene HSD3B2 sobre as proteínas codificadas e, conseqüentemente, sobre o fenótipo da doença depende da região do gene em que essa mutação ocorre (125,126,130,134, 136, 137). Mutações que ocorrem em áreas do gene conservadas em todas as espécies de mamíferos e vertebrados são mais deletérias. Exemplo disso são as mutações Y254D e Y253N, as quais levam à perda completa da atividade enzimática, pois o resíduo tirosina é muito conservado entre as espécies e se localiza no provável sítio ativo da enzima, estando envolvido na atividade isomerase desta. As mutações A10E, A10V e G15D alteram um aminoácido na região altamente conservada Gly-X-X-Gly-X-X-Gly encontrada em todos os membros da superfamília 3bHSD. No caso da mutação A10E, a alanina é conservada nos vertebrados e está localizada no provável domínio ligante de NAD, sendo que sua substituição provoca ausência de atividade enzimática na proteína mutada. Mais recentemente, o trabalho de Moisan e cols. (125) forneceu evidências de um novo mecanismo molecular para a deficiência da 3bHSD, mostrando que algumas mutações, independentemente de sua localização no gene, provocam instabilidade na proteína, levando a uma perda na atividade enzimática. Estudos posteriores têm confirmado este achado (130,136,137). A tabela 7 mostra as mutações descritas em pacientes com HAC por deficiência da 3bHSD.
A fisiopatologia, os critérios hormonais e a etiopatogenia da forma clássica da HAC por deficiência da 3bHSD estão, atualmente, bem demonstrados e compreendidos. Porém, a forma não clássica, devido à heterogeneidade das manifestações clínicas e as dificuldades dos parâmetros bioquímicos, necessita de mais estudos para que estes parâmetros sejam completamente elucidados. O avanço das técnicas de biologia molecular tem possibilitado uma melhor compreensão da forma não clássica da HAC por deficiência da 3bHSD, à medida que fornece um diagnóstico mais definitivo e permite uma melhor avaliação genótipo-fenótipo dessa doença.
AGRADECIMENTOS
Dra. Maricilda Pallandi de Mello agradece Marcela de Araújo, Luciana Campos Paulino, Ivy Freitas Lau, Osmar Vaz de Carvalho Netto, Fernanda Caroline Soardi (gene CYP21); Carlos Egydio de Carvalho e Junia Yara Penachioni (gene CYP11B1); aos Profs. Drs. Sofia Helena V. de Lemos-Marini e Gil Guerra Jr. pelo acompanhamento clínico dos pacientes; a Maria Madalena V. Rosa pelo auxílio técnico. À FAPESP, processos 92/03332-6, 97/07622-2, 97/14076-4, 98/16309-9, 00/07197-4, 01/08150-4, 01/06255-3, 01/13257-2, 02/04097-4, 02/04098-0.
Dra. Tânia Bachega agradece a Dra. Ana Elisa C. Billerbeck, Prof. Dr. Ivo J.P. Arnhold e Profa. Dra. Berenice B. de Mendonça. Às Dras. Guiomar Madureira, Enecy M.L. Brenlha e Rosy Ueti pelo acompanhamento clínico dos pacientes. À FAPESP, processos 95/8325-6, 98/00243-9, 99/06468-5 e 99/12107-5 e ao CNPq, processos 501419/91-88 e 140.745/93-9.
Dra. Marivânia da Costa-Santos agradece ao Prof. Dr. Claudio Elias Kater e ao Dr. Richard Auchus.
Dra. Margaret de Castro agradece à Dra. Suemi Marui (gene HSD3B2), Natália Torres (gene CYP21); aos Profs. Drs. Ayrton C Moreira e Lucila Elias pelo acompanhamento clínico dos pacientes. À FAPESP, processos 00/00067-8 e 01/01148-4.
Endereço para correspondência:
Margaret de Castro
Departamento de Clínica Médica
Faculdade de Medicina de Ribeirão Preto - USP
14049-900 Ribeirão Preto, SP
Fax: (016) 633-1144
e.mail: castrom@fmrp.usp.br
- 1. New MI, Dupont B, Grumback K, Levine LS. Congenital adrenal hyperplasia and related conditions. In Stambury JB, Wyngaarden JB, Fredreickson DS, Goldstein JL; Brown MS. editors. The Metabolic Bases of Inherited Disease McGraw Hill, New York, 1982;973-1000.
- 2. Miller WL, Levine LS. Molecular and clinical advances in congenital adrenal hyperplasia. J Pediatr 1987;11:1-17.
- 3. Pang S, Wallace MA, Hofman L, et al. Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 1988;81:866-74.
- 4. Thilén A, Larson A. Congenital adrenal hyperplasia Sweden 1969-1986. Prevalence, symptoms and age at diagnosis. Acta Paediatr Scand 1990;79:168-75.
- 5. Carrol MC, Campbell RD, Porter RR. Mapping of steroid 21-hydroxylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. Proc Natl Acad Sci USA 1985;82:521-5.
- 6. Levine LS, Zachmann M, New MI, et al. Genetic mapping of the 21-hydroxylase-deficiency gene within the HLA linkage group. N Engl J Med 1978;299:911-5.
- 7. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet 1985;37(4):650-67.
- 8. Werkmeister JW, New MI, Dupont B, White PC. Frequent deletion and duplication of the steroid 21-hydroxylase genes. Am J Human Genet 1986;39:461-9.
- 9. White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA 1986;83:5111-5.
- 10. White PC, Gromberger D, et al. Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci USA 1985;82:1089-93.
- 11. Morel Y, Bristow J, Gitelman SE, Miller WL. Transcript encoded on the opposite strand of the human 21-hydroxylase/C4 locus. Proc Natl Acad Sci USA 1989a;86:6582-6.
- 12. Morel Y, André J, Lambert BU, et al. Rearrangements and point mutations of P450c21 genes are distinguished by five restriction endonuclease haplotypes identified by a new probing strategy in 57 families with congenital adrenal hyperplasia. J Clin Invest 1989b;83:527-36.
- 13. Tajima T, Fujieda K, Natuyama K, Fujii-Kuriyama Y. Molecular analysis of patients and carrier genes with congenital steroid 21-hydroxylase deficiency by using polymerase chain reaction and single-strand conformational polymorphism. J Clin Invest 1993;92:2182-90.
- 14. Tusié-Luna MT, Ramirez-Jimenez S, Ordóñez-Sanchez ML, et al. Low frequency of deletion alleles in patient steroid 21-hydroxylase. Hum Genet 1996;98:376-9.
- 15. Dardis A, Bergada I, Bergada C, Rivarola M, Belgorosky A. Mutations of the steroid 21-hydroxylase gene in an Argentinean population of 36 patients with classical congenital adrenal hyperplasia. J Pediatrc Endocrinol Metab 1997;10:55-61.
- 16. Araujo M, Sanches MR, Suzuki LA, Guerra Jr G, Farah SB, De Mello MP. Molecular analysis of CYP21 and C4 genes in Brazilian families with the classical form of steroid 21-hydroxylase deficiency. Braz Med Biol Res 1996;29:1-13.
- 17. Bachega TASS, Billerbeck AEC, Madureira G, et al. Low frequency of CYP21B deletions in Brazilian patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Hered 1999;49:9-14.
- 18. Donohue PA, Van Dop C, McLean RH, White PC, Jospe N, Migeon CJ. Gene conversion in salt losing congenital adrenal hyperplasia with absent complement C4B protein. J Clin Endocrinol Metab 1986;62:995-1002.
- 19. Higashi Y, Hiromasa T, Tanae A. Effects of individual mutations in the P-450 (C21) pseudogene on the P-450 (C21) activity and their distribution in the patient genomes of congenital steroid 21-hydroxylase deficiency. J Biochem 1991;109:638-44.
- 20. Speiser PW, Dupont J, Zhu D, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest 1992;90:584-95.
- 21. Wedell A, Thilén A, Ritzén EM, Stengler B, Luthman H. Mutational spectrum of the steroid 21-hydroxylase gene is Sweden: implications for genetic diagnosis and association with disease manifestation. J Clin Endocrinol Metab 1994; 78:1145-52.
- 22. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2001;21(3):245-91.
- 23. Miller WL. Gene conversions, deletions and polymorphisms in congenital adrenal hyperplasia. Am J Hum Genet 1988;42:4-7.
- 24. Morel Y, Murena M, Nicolino M, Forest MG. Molecular genetics of the congenital adrenal hyperplasia due to 21-hydroxylase deficiency. In JM Saez; AC Brownie; A Capponi; EM Chambaz; F Mantero editors. Cellular and Molecular Biology of the Adrenal Cortex Coloque INSERM/John Libbey Eurotex LTD 1992;222:123-36.
- 25. Bachega TASS, Billerbeck AEC, Madureira G, et al. Molecular genotyping in Brazilian patients with the classical and nonclassical forms of 21-hydroxylase deficiency. J Clin Endocrinol Metab 1998;83:4416-9.
- 26. Paulino LC, De Araujo M, Guerra Jr G, Marini SNVL, De Melo MP. Mutation distribution and CYP21/C4 locus variability in Brazilian families with the classical form of the 21-hydroxylase deficiency. Acta Paediatr 1999;88:275-83.
- 27. Billerbeck AEC, Bachega TASS, Frazzatto ET, et al. A novel missense mutation, GLY424SER, in Brazilian patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab 1999; 84:2870-2.
- 28. Lau IF, Soardi FC, Lemos-Marini SHV, Guerra Jr G, Baptista MTM, De Melo MP. H28+C insertion in the CYP21 gene: A novel frameshift mutation in a Brazilian patient with the classical form of 21-hydroxylase deficiency. J Clin Endocrinol Metab 2001;86:5877-80.
- 29. Bachega TASS, Billerbeck AEC, Madureira G, et al. 21-hydroxylase in Brazil. Braz J Med Biol Res 2000;52(5):601-7.
- 30. Bachega TASS, Billerbeck AEC, Marcondes JAM, Madureira G, Arnhold IJP, Mendonça BB. Influence of different genotypes on 17-hydroxyprogesterone levels in patients with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2000;52:601-7.
- 31. Bachega TASS, Brenlha EML, Billerbeck AEC, et al. Variable ACTH-stimulated 17-hydroxyprogesterone values in 21-hydroxylase deficiency carriers are not related to the different CYP21 gene mutations. J Clin Endocrinol Metab 2002;87(2):786-90.
- 32. Tusié-Luna MT, Traktman P, White PC. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem 1990;265:20916-22.
- 33. Wilson RC, Mercado AB, Cheng KC, New MI. Steroid 21-hydroxylase deficiency: genotype may not predict phenotype. J Clin Endocrinol Metab 1995;80:2322-9.
- 34. Tajima T, Okada T, Ma XM, Ramsey W, Bronstein S, Aguilera G. Restoration of adrenal steroidogenesis by adenovirus-mediated transfer of human cytochrome P450 21-hydroxylase into the adrenal gland of 21-hydroxylase-deficiency mice. Gene Ther 1999;6(11):1898-903.
- 35. Jeffcoate TNA, Fleigner JRH, Russel SH, Davies JC, Wade AP. Diagnosis of the adrenal genital syndrome before birth. Lancet 1965;2:553.
- 36. Pollack MS, Maurer D, Levine LS, New MI, et al. HLA typing of amniotic cells: The prenatal diagnosis of congenital adrenal hyperplasia (21-OH-deficiency type). Transplant Proc 1979;11:1726.
- 37. Hughes IA, Diaz J, Riad-Fahmy D, Laurence KM. Prenatal diagnosis of congenital adrenal hyperplasia: reliability of amniotic fluid analysis. J Med Genet 1987;24:344-7.
- 38. Speiser PW, White PC, Dupont J, Zhu D, Mercado AB, New MI. Prenatal diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency by allele-specific-hybridization and Southern blot. Hum Genet 1994;93:424-8.
- 39. Fitness J, Dixit N, Webster D, et al. Genotyping of CYP21, linked chromosome 6p markers, and a sex-specific gene in neonatal screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab 1999;84:960-6.
- 40. Pang S, Pollack MS, Marshall RN, Immken L. Prenatal treatment of congenital adrenal hyperplasia due to 21 hydroxylase deficiency. New Engl J Med 1990;322:111-5.
- 41. Bachega TASS, Madureira G, Brenlha EML, et al. Tratamento da hiperplasia supra-renal congênita por deficiência da 21-hidroxilase. Arq Bras Endocrinol Metab 2001:45/1:64-72.
- 42. Zachmann M, Tassinari D, Prader A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11 b-hydroxylase deficiency. A study of 25 patients. J Clin Endocrinol Metab 1983;56:222-9.
- 43. Rösler A, Leiberman E, Cohen T. High frequency of congenital adrenal hyperplasia (classic 11b-hydroxylase deficiency) among Jews from Morocco. Am J Med Genet 1992;42:827-34.
- 44. Al-Jurayyan N. Congenital adrenal hyperplasia due to 11b-hydroxylase deficiency in Saudi Arabia: clinical and biochemical characteristics. Acta Paediatr 1995;84:651-4.
- 45. Erdmann B, Denner K, Gerst H, Lenz D, Bernhardt R. Human Adrenal CYP11B1: Localization by in situ-Hybridization and Functional Expression in Cell Cultures. Endocr Res 1995;21:425-35.
- 46. Kirita S, Morohashi K-I, Hashimoto T, Yoshioka H, Fujii-Kuriyama Y, Omura T. Expression of two kinds of cytochrome P-450(11beta) mRNA in bovine adrenal cortex. J Biochem 1988;104:683-6.
- 47. Nonaka Y, Matsukawa N, Ying Z, Ogihara T, Okamoto M. Molecular Nature of Aldosterone Synthase, A Member of Cytochrome P-45011b Family. Endocr Res 1991;17:151-63.
- 48. Müller J, Schmid C, Boni-Schnetzler M, Lauber M. Two Forms of Cytochrome P45011b in Rat Glomerulosa Cells - a Short Review. Endocr Rev 1991;17:165-84.
- 49. Okamoto M, Nonaka Y, Ohta M, Takemori H, Krishna Halder S, Zhi-Nong W, et al. Cytochrome P450(11b): Structure-Function Relationship of the Enzyme and its Involvement in Blood Pressure Regulation. J Steroid Biochem Molec Biol 1995;53:89-94.
- 50. Pascoe L, Curnow KM, Slutsker L, Connell LMC, Speiser PW, New MI, et al. Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal crossovers between CYP11B1 and CYP11B2. Proc Natl Acad Sci USA 1992;89:8327-31.
- 51. Lifton RP, Dluhy RG, Powers M, Rich GM, Gutkin M, Fallo F, et al. Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nature Genet 1992;2:66-74.
- 52. Curnow KM, Slutsker L, Vitek J, Cole T, Speiser PW, New MI, et al. Mutations in CYP11B1 causing Congenital Adrenal Hyperplasia and Hypertension Cluster in Exons 6,7 and 8. Proc Natl Acad Sci USA 1993;90:4552-6.
- 53. White PC, Curnow KM, Pascoe L. Disorders of Steroid 11beta-Hydroxylase Isozymes. Endocr Rev 1994;15:421-38.
- 54. Hampf M, Dao NT, Hoan NT, Bernhardt R. Unequal crossing-over between aldosterone synthase and 11beta-hydroxylase genes causes congenital adrenal hyperplasia. J Clin Endocrinol Metab 2001;86:4445-52.
- 55. Portrat S, Mulatero P, Curnow KM, Chaussain JL, Morel Y, Pascoe L. Deletion hybrid genes, due to unequal crossing over between CYP11B1 (11beta-hydroxylase) and CYP11B2(aldosterone synthase) cause steroid 11beta-hydroxylase deficiency and congenital adrenal hyperplasia. J Clin Endocrinol Metab 2001;86:3197-201.
- 56. White PC, Dupont B, New MI, Leiberman E, Hochberg Z, Rösler A. A Mutation in CYP11B1 (Arg-448ÞHis) Associated with Steroid 11b-Hydroxylase Deficiency in Jews of Moroccan Origin. J Clin Invest 1991;87:1664-7.
- 57. Helmberg A, Ausserer B, Kofler R. Frame Shift by Insertion of 2 Basepairs in Codon 394 of CYP11B1 Causes Congenital Adrenal Hyperplasia due to Steroid 11b-Hydroxylase Deficiency. J Clin Endocrinol Metab 1992;75:1278-81.
- 58. Naiki Y, Kawamoto T, Mitsuuchi Y, Miyahara K, Toda K, Orii T, et al. A Nonsense Mutation (TGG [Trp116]ÞTAG [Stop] in CYP11B1 Causes Steroid 11b-Hydroxylase Deficiency. J Clin Endocrinol Metab 1993;77:166-82.
- 59. Skinner CA, Rumsby G. Steroid 11b-Hydroxylase Deficiency Caused by a Five Base Pair Duplication in the CYP11B1 Gene. Hum Mol Genet 1994;3:377-8.
- 60. Nakagawa Y, Yamada M, Ogawa H, Igarashi Y. Missense mutation in CYP11B1 (CGA[Arg-384] - GGA [Gly]) causes steroid 11b-hydroxylase deficiency. Eur J Endocrinol 1995;132:286-9.
- 61. Geley S, Kapelari K, Johrer K, Peter M, Glatzl J, Vierhapper H, et al. CYP11B1 Mutations Causing Congenital Adrenal Hyperplasia due to 11b-Hydroxylase Deficiency. J Clin Endocrinol Metab 1996;81:2896-901.
- 62. Rosler A, Cohen H. Absence of steroid biosynthetic defects in heterozygote individuals for classic 11 beta-hydroxylase deficiency due to a R448H mutation in the CYP11B1 gene. J Clin Endocrinol Metab 1995;80:3771-3.
- 63. Rösler A, White PC. Mutations in Human 11b-Hydroxylase Genes: 11b-Hydroxylase Deficiency in Jews of Morocco and Corticosterone Methyl-Oxidase II Deficiency in Jews of Iran. J Steroid Biochem Molec Biol 1993;45:99-106.
- 64. De Carvalho CE, Penachioni JY, Castro M, Moreira AC, De Mello MP. CYP11B1 Intragenic Polymorphisms Give Evidences For A Different Q356X Allele In An African-Brazilian Patient. J Endocr Gen 1999;1:79-86.
- 65. Penachioni JY, De Carvalho CE, Castro M, Moreira AC, Kater CE, Mendonça BB, et al. Efeito Fundador da Mutação Q356X no Gene CYP11B1 em Pacientes com Deficiência de 11-Beta Hidroxilase. Gen Mol Biol 1999;22:200.
- 66. Merke DP, Tajima T, Chhabra A, Barnes K, Mancilla E, Baron J, et al. Novel CYP11B1 mutations in congenital adrenal hyperplasia due to steroid 11 beta-hydroxylase deficiency. J Clin Endocrinol Metab 1998;83:270-3.
- 67. Cooper DN, Youssoufian H. The CpG dinucleotide and human disease. Hum Genet 1988;78:151-5.
- 68. Chabre O, Portrat-Doyen S, Vivier J, Morel Y, Defaye G. Two novel mutations in splice donor sites of CYP11B1 in congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. Endocr Res 2000;26:797-801.
- 69. De Mello MP, Penachioni JY, Castro M, Bachega TASS, Mendonça BB. A Novel Mutation (G267S) on the CYP11B1 Gene in a Patient with 11beta-Hydroxylase Deficiency Causing Complete Virilization. Pediatric Research 2001;49:119A.
- 70. Skinner CA, Rumsby G, Honour JW. Single strand conformation polymorphism (SSCP) analysis for the detection of mutations in the CYP11B1 gene. J Clin Endocrinol Metab 1996;81:2389-93.
- 71. Penachioni JY, Bachega TASS, Mendonça BB, Castro M, Moreira AC, De Mello MP. Descrição de uma nova mutação no éxon 8 do gene CYP11B1 em uma paciente com deficiência de 11-beta-hidroxilase. Gen Mol Biol 2000;23:591.
- 72. Joehrer K, Geley S, Strasser-Wozak EM, Azziz R, Wollmann HA, Schmitt K, et al. CYP11B1 mutations causing non-classic adrenal hyperplasia due to 11 beta-hydroxylase deficiency. Hum Mol Genet 1997;6:1829-34.
- 73. Biglieri EG, Herron MA, Brust N. 17-Hydroxylation deficiency in man. J Clin Invest 1966;45:1946-53.
- 74. Grumbach MM, Conte FA. Disorders of sex differentiation. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, editors. Williams Textbook of Endocrinology. 9th Edition. Philadelphia: WB Saunders Company, 1998;1303-426.
- 75. Biglieri EG, Kater CE. Mineralocorticoids in congenital adrenal hyperplasia. J Steroid Biochem Mol Biol 1991;40:493-9.
- 76. Yanase T, Simpson ER, Waterman MR. 17-alpha-Hydroxylase/17,20-lyase deficiency: From clinical investigation to molecular definition. Endocr Rev 1991;12:91-108.
- 77. Gupta MK, Geller DH, Auchus RJ. Pitfalls in Characterizing P450c17 Mutations Associated with Isolated 17,20-Lyase Deficiency. J Clin Endocrinol Metab 2001;86:4416-23.
- 78. Garcia-Mayor RVG, Sopeña B, Fluiters E, Páramo C, Martin FR, Galofré JC, et al. Testicular adrenal-like tissue in a patient with 17a-hydroxylase deficiency. Horm Res 1992;38:241-4.
- 79. Condom E, Villabona CM, Gómez JM, Carrera M. Adrenal myelolipoma in a woman with congenital 17-hydroxilase deficiency. Arch Pathol Lab Med 1985;109:1116-7.
- 80. Ogawa K, Hara A, Tanabe S, Tamori S, Voshida H, Pak CH, et al. A case of 17-alpha-hydroxylase deficiency syndrome associated with right adrenal tumor. Clin Exp Hypertens A 1984;6:863-77.
- 81. Gennes JL, Jambart S, Turpin G, Elkik F, Roger M. 17a-Hydroxylase deficiency syndrome associated with bilateral streak gonads and impaired development of müllerian ducts derivatives. Acta Endocrinol 1982;100:68-76.
- 82. Matteson KJ, Picado-Leonard J, Chung BC, Mohandas TK, Miller WL. Assignment of the gene for adrenal P450c17 (steroid 17-alpha-hydroxylase/17,20 lyase) to human chromosome 10. J Clin Endocrinol Metab 1986;63:789-91.
- 83. Biason A, Mantero F, Scaroni C, Simpson ER, Waterman MR. Deletion within the CYP17 Gene Together with Insertion of Foreign DNA is the Cause of Combined Complete 17a-Hydroxylase/17,20-Lyase Deficiency in an Italian Patient. Mol Endocrinol 1991;5:2038-45.
- 84. Biason-Lauber A, Kempken B, Werder E, Forest MG, Einaudi S, Ranke MB, et al. 17-a-Hydroxylase/17,20-lyase deficiency as a model to study enzymatic activity regulation: Role of phosphorylation. J Clin Endocrinol Metab 2000;85:1226-31.
- 85. Yamaguchi H, Nakazato M, Miyazato M, Kangawa K, Matsukura S. A 5'-Splice site mutation in the cytochrome P450 steroid 17a-hydroxylase gene in 17a-hydroxylase deficiency. J Clin Endocrinol Metab 1997;82:1934-8.
- 86. Suzuki Y, Nagashima T, Nomura Y, Onigata K, Nagashima K, Morikawa A. A new compound heterozygous mutation (W17X, 436+5GÆT) in the cytochrome P450c17 gene causes 17a-hydroxylase/17,20-lyase deficiency. J Clin Endocrinol Metab 1998;83:199-202.
- 87. Lin C, Martens J, Miller W. NF-1C, Sp1, and Sp3 Are Essential for Transcription of the Human Gene for P450c17 (Steroid 17_-hydroxylase/17,20 lyase) in Human Adrenal NCI-H295A Cells. Mol Endocrinol 2001;15:1277-93.
- 88. Brentano ST, Picado-Leonard J, Mellon SH, Moore CCD, Miller WL. Tissue-specific, cAMP-induced, and phorbol ester repressed expression from the human P450c17 promoter in mouse cells. Mol Endocrinol1990;4:1972-9.
- 89. Chung B-C, Picado-Leonard J, Haniu M, Bienkowski M, Hall PF, Shively JE. Cytochrome P450c17 (steroid 17a-hydroxylase/17,20-lyase): cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc Natl Acad Sci USA 1987;84:407.
- 90. Geller DH, Auchus RJ, Miller WL. P450c17 mutations R347H and R358Q selectively disrupt 17,20-lyase activity by disrupting interactions with P450 oxireductase and cytochrome b5. Mol Endocrinol 1999;13:167-75.
- 91. Auchus RJ, Lee TC, Miller WL. Cytochrome b5 augments the 17,20 lyase activity of human P450c17 without direct electron transfer. J Biol Chem 1998;273:3158-65.
- 92. Swart P, Swart AC, Waterman MR, Estabrook RW, Mason JI. Progesterone 16a-hydroxylase activity is catalyzed by human cytochrome P450 17a-hydroxylase. J Clin Endocrinol Metab 1993;77:98-102.
- 93. Lee-Robichaud P, Wright JN, Aktar ME, Aktar M. Modulation of the activity of human 17a-hydroxylase-17,20-lyase (CYP17) by cytochrome b5: endocrinological and mechanic implications. Biochem J 1995;308:901-8.
- 94. Auchus R. The Genetics, Pathophysiology, and Management of Human Deficiencies of P450c17. Endocrinol Metab Clin North Am 2001;30:101-19.
- 95. Zuber MX, Simpson ER, Waterman MR. Expression of Bovine 17-alpha-hydroxylase cytochrome P450 cDNA in nonsteroidogenic (COS 1) cells. Science 1986;234:1258-61.
- 96. Lin D, Black SM, Nagahama Y, Miller WL. Steroid 17a-hydroxylase and 17,20 lyase activities of P450c17: contributions of serine106 and P450 reductase. Endocrinology 1993;132:2498-506.
- 97. Santos MC, Kater CE, Grupo Multicêntrico Brasileiro de Estudo de Hiperplasia Adrenal Congênita. Estudo de Freqüência da Hiperplasia Adrenal Congênita em Centros de Referência Médica do Brasil. Arq Bras Endocrinol Metab 1998; 42:385-92.
- 98. Costa-Santos M, Kater CE, Auchus RJ. Molecular-Genetic Study of 20 Brazilian Families Affected By 17-OH-Deficiency. In: Program & Abstracts. The Endocrine Society's 83rd Annual Meeting. Denver, 2001:108.
- 99. Fardella CE, Hum DW, Homoki J, Miller W. Point Mutation of Arg 440 to His Cytochrome P450c17 Causes Severe 17a-Hydroxylase Deficiency. J Clin Endocrinol Metab 1994; 79:160-4.
- 100.Yanase T. 17a -Hydroxylase / 17,20-lyase defects. J Steroid Biochem Mol Biol 1995;53:153-7.
- 101.Geller DH, Auchus RJ, Mendonça BB, Miller WL. The genetic and functional basis of isolated 17,20 lyase deficiency. Nature Genet 1997;17:201-5.
- 102.Kagimoto M, Winter JSD, Kagimoto K, Simpson ER, Waterman MR. Structural characterization of normal and mutant human steroid 17a-Hydroxylase genes: molecular basis of one example of combined 17a-Hydroxylase/17,20-Lyase deficiency. Mol Endocrinol 1988;2:564-70.
- 103.Yanase T, Kagimoto M, Matsui N, Simpson ER, Waterman MR. Combined 17a-Hydroxylase /17,20-Lyase Deficiency due to a stop codon in the N-Terminal region of 17a-Hydroxylase cytochrome P-450. Mol Cel Endocrinol 1988;59:249-53.
- 104.Yanase T, Kagimoto M, Suzuki S, Hashiba K, Simpson ER, Waterman MR. Deletion of a Phenylalanine in the N-Terminal Region of Human Cytochrome P-450 17a Results in Partial Combined 17a-Hydroxylase /17,20-Lyase Deficiency. J Biol Chem 1989;264:18076-82.
- 105.Yanase T, Sanders D, Shibata A, Matsui N, Simpson ER, Waterman MR. Combined 17a-Hydroxylase /17,20-Lyase Deficiency due to a 7-Basepair Duplication in the N-Terminal Region of the Cytochrome P450 17a (CYP17) Gene. J Clin Endocrinol Metab 1990;70:1325-9.
- 106. Lin D, Harikrishna JA, Moore CCD, Jones KL, Miller WL. Missense Mutation Serine 106ÆProline Causes 17a-Hydroxylase Deficiency. J Biol Chem 1991;266:15992-38.
- 107.Fardella CE, Zhang L-H, Mahachoklertwattana P, Lin Dog, Miller W. Deletion of Amino Acids Asp487-Ser488-Phe489 in Human Cytochrome P450c17 Causes Severe 17a-Hydroxylase Deficiency. J Clin Endocrinol Metab 1993;77:489-93.
- 108.Toyata N, Monno S, Kobayashi M. Two-bp deletion in exon 5 of CYP 17 gene in a family with 17a-hydroxylase deficiency. Folia Endocr Japon 1993;69:295(
- 109.Imai Tsuneo, Globerman H, Gertner JM, Kagawa N, Waterman MR. Expression and Purification of Functional Human 17a-Hydroxylase/17,20-Lyase (P450c17) in Escherichia coli. J Biol Chem 1993;268:19681-9.
- 110.Rumbsy G, Skinner C, Lee HA, Honour JW. Combined 17a-Hydroxylase/17,20-lyase deficiency caused by heterozygous stop codons in the cytochrome P450 17a-hydroxylase gene. Clin Endocrinol 1993;39:483-5.
- 111.Monno S, Ogawa H, Date T, Fujioka M, Miller W, Kobayashi M. Mutation of Histidine 373 to Leucine in Cytochrime P40c17 Causes 17a-Hydroxylase Deficiency. J Biol Chem 1993;268:25811-7.
- 112.Oshiro C, Takasu N, Wakugami T, Komiya I, Yamada T, Eguchi Y, et al. Seventeen a-hydroxylase deficiency with one base pair deletion of the cytochrome P45c17 (CYP17) gene. J Clin Endocrinol Metab 1995;80:2526-9.
- 113.Laflamme N, Leblanc J-F, Mailloux J, Faure N, Lalnie F, Simard J. Mutation R96W in cytochrome P450c17 gene causes combined 17a-hydroxylase/17,20-lyase deficiency in two French Canadian patients. J Clin Endocrinol Metab 1996;81:264-8.
- 114.Biason-Lauber A, Leiberman E, Zachmann M. A single amino acid substitution in the putative redox partner-binding site of P45c17 as cause of isolated 17,20-lyase deficiency. J Clin Endocrinol Metab 1997;82:3807-12.
- 115.Yamaguchi H, Nakazato M, Miyazato M, Toshimori H, Oki S, Shimizu K, et al. Identification of a novel splicing mutation and 1-bp deletion in the 17a-hydroxylase gene of Japanese patients with 17a-hydroxylase deficiency. Hum Genet 1998;102:635-9.
- 116.Satoh J, Kuroda Y, Nawata H, Yanase T. Molecular basis of hypokalemic myopathy caused by 17-hydroxylase/17,20-lyase deficiency. Neurology 1998;51:1748-51.
- 117.Ching-Wan Lam, Wiebke Arlt, Chi-Kwok Chan, John W. Honour, Chin Jia Lin, Sui-Fan Tong, et al. Mutation of Proline 409 to Arginine in the Meander Region of Cytochrome P450c17 Causes Severe 17a-Hydroxylase Deficiency. Mol Gen Met 2001;72:254-9.
- 118.Takeda Y, Yoneda T, Demura M, Furukawa K, Koshida H, Miyamori I, et al. Genetic analysis of the cytochrome P-450c17a (CYP17) and aldosterone synthase (CYP11B2) in Japanese patients with 17-a-Hydroxylase deficiency. Clin Endocrinol 2001;54:751-8.
- 119.Katsumata N, Satoh M, Mikami A, Mikami S, Nagashima-Miyokawa A, Sato N, et al. New Compound Heterozygous Mutation in the CYP17 Gene in a 46,XY Girl with 17-a-Hydroxylase/17,20-Lyase Deficiency. Horm Res 2001;55:141-6.
- 120.Di Cerbo A, Biason-Lauber A, Savino M, Piemontese MR, Di Giorgio A, Perona M, et al. Combined 17alpha-Hydroxylase/17,20-lyase deficiency caused by Phe93Cys mutation in the CYP17 gene. J Clin Endocrinol Metab 2002;87:898-905.
- 121.Bongiovanni AM. The adrenogenital syndrome with deficiency of 3b-hydroxysteroid dehydrogenase. J Clin Invest 1962;41:2086-92.
- 122.Luu The V, Lachance Y, Labrie C, Leblanc G, Thomas J, Strickler R, et al. Full-length cDNA structure and deduced amino acid sequence of human 3b-hydroxy-5-ene-steroid dehydrogenase. Mol Endocrinol 1989;3:1310.
- 123.Simard J, Durocher F, Mebarki F, Turgeon C, Sanchez R, Labrie Y, et al. Molecular biology and genetics of the 3b-hydroxysteroid dehydrogenase/D5 D4 -isomerase gene family. J Endocrinol 1996;150:S189-207.
- 124.Rhéaume E, Simard J, Morel Y, Mebarki F, Zachmann M, Forest MG, et al. Congenital adrenal hyperplasia due to point mutations in the type II 3b-hydroxysteroid dehydrogenase gene. Nat Genet 1992;1:239-45.
- 125.Moisan AM, Ricketts ML, Tardy V, Desrochers M, Mébarki F, Chaussain JL, et al. New insight into the molecular basis of 3b-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3B2 gene in eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab 1999;84:4410-25.
- 126.Simard J, Rhéaume E, Sanchez R, Laflamme N, Launoit Y, Luu-The V, et al. Molecular basis of congenital adrenal hyperplasia due to 3b-hydroxysteroid dehydrogenase deficiency. Mol Endocrinol 1993;7:716-28.
- 127.Rosenfield RL, Rich BH, Wolfsdorf, Cassorla F, Parks JS, Bongiovanni AM, et al. Pubertal presentation of congenital D5-3b-hydroxysteroid dehydrogenase. J Clin Endocrinol Metab 1980;51:345-53.
- 128.Mendonça BB, Russel AJ, Vasconcelos-Leite M, Arnhold IJ, Bloise W, Wajchenberg BL, et al. Mutation in 3b-hydroxysteroid dehydrogenase type II associated with pseudohermaphroditism in males and premature pubarche or cryptic expression in females. J Mol Endocrinol 1994;12:119-22.
- 129.Marui S, Castro M, Latronico AC, Elias LL, Arnhold IJ, Moreira AC, et al. Mutations in the type II 3b-hydroxysteroid dehydrogenase (HSD3B2) gene can cause premature pubarche in girls. Clin Endocrinol 2000;52(1):67-75.
- 130.Simard J, Ricketts ML, Moisan AM, Tardy V, Peter M, Vliet GV, et al. A new insight into the molecular basis of 3b-hydroxysteroid dehydrogenase deficiency. Endocr Res 2000; 26(4):761-70.
- 131.Pang S, Lerner AJ, Stoner E, Oberfield SE, Engel I, New MI. Late-onset adrenal steroid 3b-hydroxysteroid dehydrogenase deficiency I: A cause of hirsutism in pubertal and postpubertal women. J Clin Endocrinol Metab 1985;60:428-39.
- 132.Pang S. Congenital adrenal hyperplasia owing to 3b-hydroxysteroid dehydrogenase deficiency. Endocrinol Metab Clin North Am 2001;30(1):81-99.
- 133.Rhéaume E, Sanchez R, Simard J, Chang YT, Wang J, Pang S, et al. Molecular basis of congenital adrenal hyperplasia in two siblings with classical nonsalt 3b-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab 1994; 79:1012.
- 134.Sanchez R, Rheaume E, Laflamme N, Rosenfield RL, Labrie F, Simard J. Detection and functional characterization of the novel missense mutation Y254D in the type II 3b-hydroxysteroid dehydrogenase (3bHSD) gene of a female patient with nonsalt-losing 3b-HSD deficiency. J Clin Endocrinol Metab 1994;78:561.
- 135.Paula FJA, Dick-De-Paula I, Pontes A, Schimitt FCL, Mendonça BB, Foss MC. Hyperandrogenism due to 3b-hydroxysteroid dehydrogenase deficiency with accessory adrenocortical tissue: a hormonal and metabolic evaluation. Braz J Med Biol Res 1994;27:1149-58.
- 136.Alos N, Moisan AM, Ward L, Desrochers M, Legault L, Leboeuf G, et al. A novel A10E homozygous mutation in the HSD3B2 gene causing severe salt-wasting 3b-hydroxysteroid dehydrogenase deficiency in 46,XX and 46,XY French-Canadians: Evaluation of gonadal function after puberty. J Clin Endocrinol Metab 2000;85(5):1968-74.
- 137.Zhang L, Mason JI, Naiki Y, Copeland KC, Magana MC, Gordon-Walker TT, et al. Characterization of two novel homozygous missense mutations involving codon 6 and 259 of type II 3b-hydroxysteroid dehydrogenase (3bHSD) gene causing, respectively, nonsalt-wasting and salt-wasting 3bHSD deficiency disorder. J Clin Endocrinol Metab 2000;85(4):1678-85.
- 138.Cartin MS, Russell AJ, Fisher RA, Wallace AM, Arnhold IJP, Mason JI, et al. Phenotypic variability and origins of mutations in the gene encoding 3b-hydroxysteroid dehydrogenase type II. J Mol Endocrinol 2000;24:75-82.
Datas de Publicação
-
Publicação nesta coleção
20 Nov 2002 -
Data do Fascículo
Ago 2002
Histórico
-
Recebido
29 Jul 2002 -
Aceito
02 Ago 2002