Resumos
O hipercortisolismo crônico é a causa mais freqüente de osteoporose secundária, acometendo principalmente o osso trabecular. Aproximadamente 30-35% dos pacientes com síndrome de Cushing apresentam fraturas de vértebras por compressão e o risco de fraturas de colo de fêmur é aumentado em 50% nessa população. Vários mecanismos têm sido propostos para explicar a ocorrência de osteoporose nessa condição, como a ação direta dos glicocorticóides nas paratireóides e nas células ósseas, alterações na produção de prostaglandinas, citocinas, interleucinas, alterações na secreção do hormônio do crescimento (GH), do fator insulina símile-I (IGF-I) e esteróides gonadais. Resultados controversos têm sido apresentados quanto à alteração na secreção do PTH nesta situação, onde níveis normais e elevados têm sido descritos. A elevação da secreção de PTH pode ser secundária a distúrbios do metabolismo mineral induzidos pelo hipercortisolismo, como diminuição na absorção intestinal, aumento da excreção renal de cálcio, diminuição no número de receptores paratireoideanos para a 1,25(OH)2D3, anormalidades no limiar de sensibilidade do cálcio (set point) para a secreção do PTH e alteração na sua atividade. Nesta revisão, são discutidos diversos aspectos fisiopatológicos e possíveis mecanismos envolvidos na associação entre hipercortisolismo e osteoporose.
Osteoporose; Hipercortisolismo; Síndrome de Cushing; Paratormônio; Calcitonina; Cálcio; Magnésio
Chronic hypercortisolism is the most frequent cause of secondary osteoporosis involving mainly trabecular bone. Approximately 30-35% of the patients with Cushing's syndrome present with compression fractures of vertebrae, and the risk of femoral neck fracture is increased in 50% in that population. Several mechanisms have been proposed to explain the association between hypercortisolism and osteoporosis, as the direct action of glucocorticoids in parathyroid glands and bone cells, alterations in the production of prostaglandins, citokines, interleukines, growth hormone (GH), insulin like growth factor I (IGF-I) and gonadal steroids. Contradictory results have been presented in relation to PTH secretion, where normal and high levels have been described. Elevated PTH secretion can occur as a consequence of disturbances in mineral metabolism, i.e. decrease in the intestinal absortion and elevation in the renal excretion of calcium, decrease in the number of parathyroid receptors for 1,25(OH)2D3, abnormalities in the calcium set point for PTH secretion, and alteration in PTH activity. In this review, several pathophysiologic aspects and possible mechanisms involved in the association between osteoporosis and hypercortisolism are discussed.
Osteoporosis; Hypercortisolism; Cushing's syndrome; Parathyroid hormone; Calcitonin; Calcium; Magnesium
REVISÃO
Fisiopatologia da osteoporose induzida por glicocorticóide
Pathophysiology of corticosteroid-induced osteoporosis
Carla M.M. Lanna; Renan M. Montenegro Jr.; Francisco J.A. Paula
Divisão de Endocrinologia e Metabologia, Departamento de Clínica Médica, Faculdade de Medicina de Ribeirão Preto - USP, SP
Endereço para correspondência Endereço para correspondência Francisco José Albuquerque de Paula Divisão de Endocrinologia e Metabologia Departamento de Clínica Médica Faculdade de Medicina de Ribeirão Preto - USP Av. dos Bandeirantes 3.900 14048-900 Ribeirão Preto, SP Fax: (016) 633-6695 E-mail: fjpaula@fmrp.usp.br
RESUMO
O hipercortisolismo crônico é a causa mais freqüente de osteoporose secundária, acometendo principalmente o osso trabecular. Aproximadamente 30-35% dos pacientes com síndrome de Cushing apresentam fraturas de vértebras por compressão e o risco de fraturas de colo de fêmur é aumentado em 50% nessa população. Vários mecanismos têm sido propostos para explicar a ocorrência de osteoporose nessa condição, como a ação direta dos glicocorticóides nas paratireóides e nas células ósseas, alterações na produção de prostaglandinas, citocinas, interleucinas, alterações na secreção do hormônio do crescimento (GH), do fator insulina símile-I (IGF-I) e esteróides gonadais. Resultados controversos têm sido apresentados quanto à alteração na secreção do PTH nesta situação, onde níveis normais e elevados têm sido descritos. A elevação da secreção de PTH pode ser secundária a distúrbios do metabolismo mineral induzidos pelo hipercortisolismo, como diminuição na absorção intestinal, aumento da excreção renal de cálcio, diminuição no número de receptores paratireoideanos para a 1,25(OH)2D3, anormalidades no limiar de sensibilidade do cálcio (set point) para a secreção do PTH e alteração na sua atividade. Nesta revisão, são discutidos diversos aspectos fisiopatológicos e possíveis mecanismos envolvidos na associação entre hipercortisolismo e osteoporose.
Descritores: Osteoporose; Hipercortisolismo; Síndrome de Cushing; Paratormônio; Calcitonina; Cálcio; Magnésio
ABSTRACT
Chronic hypercortisolism is the most frequent cause of secondary osteoporosis involving mainly trabecular bone. Approximately 30-35% of the patients with Cushing's syndrome present with compression fractures of vertebrae, and the risk of femoral neck fracture is increased in 50% in that population. Several mechanisms have been proposed to explain the association between hypercortisolism and osteoporosis, as the direct action of glucocorticoids in parathyroid glands and bone cells, alterations in the production of prostaglandins, citokines, interleukines, growth hormone (GH), insulin like growth factor I (IGF-I) and gonadal steroids. Contradictory results have been presented in relation to PTH secretion, where normal and high levels have been described. Elevated PTH secretion can occur as a consequence of disturbances in mineral metabolism, i.e. decrease in the intestinal absortion and elevation in the renal excretion of calcium, decrease in the number of parathyroid receptors for 1,25(OH)2D3, abnormalities in the calcium set point for PTH secretion, and alteration in PTH activity. In this review, several pathophysiologic aspects and possible mechanisms involved in the association between osteoporosis and hypercortisolism are discussed.
Keywords: Osteoporosis; Hypercortisolism; Cushing's syndrome; Parathyroid hormone; Calcitonin; Calcium; Magnesium
A OSTEOPOROSE É A MAIS FREQÜENTE das doenças osteometabólicas e o seu estudo tem sido especialmente motivado em decorrência das importantes repercussões em relação à morbidade e mortalidade de indivíduos portadores dessa condição, sendo por esta razão atualmente considerada um sério problema de saúde pública.
Nas últimas décadas tem havido um crescente interesse no estudo do papel dos hormônios diretamente relacionados com o controle do metabolismo ósseo. O PTH, a vitamina D e a calcitonina são os principais hormônios envolvidos com a regulação do metabolimo mineral. O nível extracelular do íon cálcio é mantido estável por meio da ação destes hormônios em um ou mais sítios alvo (rins, ossos e trato digestivo).
O PTH é um hormônio polipeptídico com 84 aminoácidos sintetizado nas glândulas paratireóides a partir de um precursor, o pré-pró-paratormônio, sendo um dos responsáveis pelo controle da calcemia e o mais importante na regulação das alterações agudas das concentrações extracelulares de cálcio. A seqüência dos primeiros 34 aminoácidos na extremidade amino-terminal é responsável pela sua atividade biológica. Além da molécula intacta do PTH, são secretados fragmentos amino-terminais, carboxi-terminais e a sua porção média. O hormônio intacto, biologicamente ativo, representa 5 a 30% da concentração plasmática total do PTH. O PTH tem uma meia vida de aproximadamente 2 minutos, sendo que a sua metabolização ocorre na maior parte no fígado (70%), rins (20%) e em menor quantidade em outros tecidos, como o tecido ósseo. A ação do PTH se dá diretamente no rim e no osso e indiretamente no trato digestivo, sempre resultando em incremento dos níveis séricos de cálcio. No rim, através de receptores específicos nos túbulos contorcidos distais, o PTH determina a reabsorção de cálcio e magnésio e a excreção de fosfato e bicarbonato. Este órgão é particularmente responsável pelos ajustes mais rápidos da calcemia. No osso, age sobre a reabsorção, regulando a liberação de cálcio e fosfato para o líquido extracelular. Embora os osteoclastos sejam os responsáveis por essa última ação, diferentemente dos osteoblastos, essas células não possuem receptores específicos para o PTH. Portanto, a reabsorção óssea osteoclástica estimulada pelo PTH decorre da liberação de fatores parácrinos pelos osteoblastos, ou ainda pelo estímulo de expressão de proteínas de membrana nos osteoblastos, que estimulam o amadurecimento e atividade dos osteoclastos (1). As ações do PTH no osso variam de acordo com as suas concentrações plasmáticas. Em níveis fisiológicos, esse hormônio tem um efeito ósseo anabólico. A secreção intermitente do PTH associa-se com aumento do osso trabecular e cortical em ratas (2) e com a manutenção e/ou aumento da massa óssea trabecular em humanos (3). Já a sua secreção contínua e/ou aumentada apresenta efeito catabólico, ocorrendo perda de massa óssea como, por exemplo, pode ser observado no hiperparatireoidismo primário sintomático. No trato digestivo, o PTH controla indiretamente a absorção intestinal de cálcio e fósforo, através da regulação da atividade da enzima 25(OH)D-1a-hidroxilase renal e conseqüente síntese de 1,25(OH)2D. As concentrações de cálcio ionizado no líquido extracelular são os principais determinantes da secreção do PTH. Modificações dos níveis séricos de cálcio resultam em respostas rápidas da secreção desse hormônio. A detecção de variações na calcemia ocorre através de sensores de cálcio presentes nas paratireóides, que consistem em receptores protéicos localizados na superfície da membrana das células principais (4). Anormalidades nestes receptores podem alterar o limiar de sensibilidade dessas células à calcemia, podendo resultar em aumento ou diminuição dos níveis séricos de cálcio (5). Enquanto agudamente a biossíntese e secreção do PTH é determinada pelas concentrações extracelulares de cálcio ionizado, cronicamente a vitamina D passa a ser também um importante regulador desse fenômeno (6). Além desses, outros íons e hormônios participam da regulação da secreção do PTH. Níveis séricos de magnésio anormalmente elevados ou reduzidos (7) inibem ou estimulam, respectivamente, a secreção do PTH, enquanto a calcitonina (8), o cortisol (9) e a histamina (10) estimulam-na.
A calcitonina é um peptídeo com 32 aminoácidos, secretado primariamente pelas células C da tireóide. Embora conhecida há mais de 30 anos, a função desse hormônio na regulação do metabolismo mineral ósseo em humanos ainda não está totalmente esclarecida. Sabe-se que a calcitonina atua diretamente nos osteoclastos, determinando uma diminuição da reabsorção óssea. Devido a essa propriedade, a calcitonina passou a ser utilizada em distúrbios caracterizados por aumento da reabsorção óssea. As concentrações sangüíneas de cálcio ionizado são o principal regulador da secreção desse hormônio. Agudamente, o aumento e redução da calcemia resultam respectivamente em um incremento e diminuição na secreção da calcitonina (11).
A 1,25(OH)2D é a forma biologicamente ativa da vitamina D. O sítio inicial de síntese endógena da vitamina D é a pele, mas esta pode também ser obtida por meio de ingestão alimentar. As fontes dietéticas ricas em vitamina D3 (colecalciferol) incluem gema de ovo, óleo de peixe e de fígado de bacalhau, enquanto que a vitamina D2 (ergocalciferol) é encontrada em plantas e leveduras e tem sido adicionada ao leite em países desenvolvidos. Na pele, o 7-dehidrocolesterol (pró-vitamina D3) está presente em grandes quantidades nas membranas celulares dos queratinócitos na epiderme. Através da ação da luz ultravioleta, o anel B do 7-dehidrocolesterol é quebrado formando a pré-vitamina D3. A pré-vitamina D3 é instável, sendo rapidamente isomerizada à vitamina D3, através de energia térmica. A síntese e a disponibilidade sistêmica da vitamina D3 é regulada pela luz solar na própria pele. A síntese de pré-vitamina D3 geralmente atinge seus níveis máximos em horas, embora o tempo necessário para isso seja diretamente relacionado à intensidade da exposição à luz e ao grau de pigmentação da pele (a melanina limita parcialmente a formação da pré-vitamina D3, através da absorção da irradiação ultravioleta). Com isso, quando há uma exposição excessiva aos raios ultravioleta ocorre um direcionamento para formação de produtos biologicamente inativos. A pré-vitamina D3 é então foto-isomerizada ao taquisterol e lumisterol. Com a redução dos níveis de pré-vitamina D3, o processo pode seguir o sentido contrário, sendo o lumisterol convertido em pré-vitamina D3. A vitamina D3 é degradada a 5,6-transvitamina D3 e suprasterol-1 e 2. Em seres humanos, a vitamina D2 tem potência biológica semelhante à vitamina D3. Na circulação, ambas são transportadas por uma a-globulina sintetizada no fígado, a proteína ligadora de vitamina D. Nesse órgão, são hidroxiladas na posição C25 resultando na 25(OH)D. Esta reação é dependente do citocromo P450 e é regulada principalmente pela quantidade de substrato. Uma segunda hidroxilação ocorre no túbulo renal, através da 25(OH)D-1a-hidroxilase, enzima também do complexo citocromo P450 mono-oxigenase, resultando na 1,25(OH)2D (calcitriol), sua forma ativa. Os níveis sangüíneos de fosfato, cálcio e PTH são os principais responsáveis pela produção de 1,25(OH)2D. Elevações ou reduções dos níveis de cálcio, respectivamente, inibem ou estimulam a 1a-hidroxilação da 25(OH)D. Isso ocorre tanto de forma indireta, através da modificação dos níveis do paratormônio (PTH), como diretamente através dos receptores de cálcio ionizado, presentes nas células produtoras de 1,25(OH)2D dos túbulos proximais renais (12). Juntamente com o PTH, a vitamina D exerce papel fundamental na regulação das concentrações extracelulares de cálcio. Sua ação primordial é no trato digestivo aumentando a absorção intestinal de cálcio e fósforo. Através de receptores nucleares encontrados no intestino delgado, a 1,25(OH)2D aumenta a síntese de proteínas envolvidas no transporte de cálcio através da mucosa intestinal. No osso, fisiologicamente atua de forma permissiva na mineralização da matriz protéica óssea. Frente a níveis reduzidos de cálcio na dieta, sua ação no osso passa a ser indutora da reabsorção. Na paratireóide, a 1,25(OH)2D age inibindo a secreção do PTH (13).
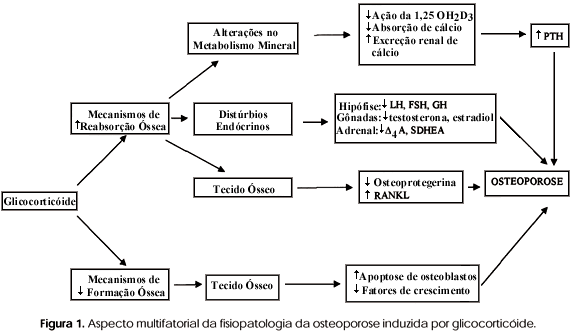
Além do PTH, da calcitonina e da vitamina D, outros hormônios também interferem com a manutenção da massa óssea. Estudos mostram que os glicocorticóides podem influenciar significativamente a massa óssea. Isto fica claramente evidenciado em situações de hipercortisolismo. Considera-se hoje que o hipercortisolismo seja a causa mais freqüente de osteoporose secundária (osteoporose induzida por glicocorticóide OIG). Tem sido demonstrada nesta situação uma rápida perda óssea que acomete principalmente o osso trabecular. Aproximadamente 30 a 35% dos pacientes com hipercortisolismo crônico apresentam fraturas (14). Em um estudo retrospectivo, Van Staa e cols. (15) verificaram que o uso de glicocorticóide exógeno potencialmente pode aumentar o risco de fratura, mesmo quando usado em baixas doses por via oral. Neste estudo, os dados de 244.235 indivíduos com história de utilização de glicocorticóide foram comparados com os de uma população de número semelhante, sem história de uso anterior de glicocorticóide. O risco relativo de fratura do primeiro grupo foi significativamente maior na coluna (rr = 2,6) e em quadril (rr = 1,6). O risco de fratura correlacionou-se diretamente com a dose utilizada, mas vale ressaltar que mesmo aqueles que usavam 2,5mg de prednisolona apresentaram maior risco de fratura que o grupo controle. Outros estudos têm verificado que a exposição a corticosteróide por via inalatória também pode induzir perda de massa óssea (16). Os mecanismos envolvidos com o surgimento da OIG ainda estão por ser melhores esclarecidos. Provavelmente, a OIG se desenvolve como conseqüência de uma série de alterações que atingem o metabolismo mineral, o componente celular do tecido ósseo e distúrbios endócrinos. A figura 1 apresenta de forma resumida os diversos fatores que contribuem para o surgimento da OIG.
Os glicocorticóides representam uma classe de drogas usadas muito freqüentemente devido às suas propriedades anti-inflamatórias e imunossupressoras. A osteoporose é a complicação iatrogênica mais freqüentemente observada no tratamento crônico com doses farmacológicas desses esteróides. A gravidade da perda óssea relaciona-se com fatores como a dose e o tempo de duração da terapia. Sabe-se que esta perda óssea é maior nos primeiros 6 meses de tratamento, podendo persistir caso o paciente esteja em uso de doses mais elevadas de glicocorticóides (17).
Diversos mecanismos têm sido propostos para explicar a ocorrência de osteoporose induzida pelos glicocorticóides.
I) AÇÃO DIRETA NAS PARATIREÓIDES
A ação dos glicocorticóides nas paratireóides é conhecida há várias décadas, a partir de estudos in vitro e in vivo. Em 1962, Raker e cols. (18) descreveram associação entre síndrome de Cushing e hiperparatireoidismo. Posteriormente, Wajchenberg e cols. (19) observaram a presença de hiperplasia de paratireóides em pacientes com Síndrome de Cushing. Em estudos in vitro, Munck & Brinck-Johnsen (20) demonstraram receptores para glicocorticóides nas paratireóides. Williams e cols. (21) e Au (9) verificaram que acetato de cortisona levava a uma hiperplasia de paratireóides de ratos e aumento dos níveis de PTH.
II) ALTERAÇÕES NA SECREÇÃO DO PTH
Vários autores sugerem que a perda óssea apresentada pelos pacientes com hipercortisolismo seja secundária às alterações na secreção do PTH. Em alguns estudos, níveis plasmáticos aumentados de PTH têm sido encontrados em pacientes com síndrome de Cushing (22-24), enquanto que em outros tem-se demonstrado concentrações plasmáticas normais desse peptídeo (23,25,26). Estas divergências podem decorrer da utilização de métodos não específicos para dosagem do PTH e/ou de seleção inadequada dos controles, principalmente no que concerne à idade, raça, doenças associadas ou deficiência subclínica de vitamina D (27).
Em estudo recente (28), observamos que, embora os pacientes com hipercortisolismo crônico tenham apresentado níveis basais de PTH normais, mostraram um aumento significativamente maior que indivíduos normais na secreção de PTH durante o teste de estímulo realizado por meio de infusão endovenosa de solução de EDTA (figura 2). Estes dados sugerem que as dosagens basais de PTH não são adequadas para avaliar a secreção deste hormônio no hipercortisolismo crônico.
A elevação nos níveis plasmáticos de PTH observada no hipercortisolismo crônico pode ser secundária a fatores como a diminuição na absorção intestinal ou na reabsorção renal de cálcio, diminuição no número de receptores paratireoideanos para a 1,25(OH)2D3, anormalidades no limiar do cálcio (set point) para a secreção do PTH ou alteração na atividade do PTH.
Gennari e cols. (27) descreveram uma diminuição na absorção intestinal de cálcio com a utilização de glicocorticóides, com aumento secundário nos níveis do PTH. A absorção intestinal de cálcio requer a presença das proteínas transportadoras através da mucosa intestinal, que são fundamentais para o seu transporte ativo pelas alças intestinais. Kimberg e cols. (29) demonstraram que a vitamina D participa na síntese destas proteínas na mucosa intestinal. A 1,25(OH)2D3 se liga ao seu receptor e induz à expressão da calbindina-D28K no intestino (30). Os glicocorticóides apresentam uma ação variável na indução da expressão do gene da calbindina-D28K. Em baixas concentrações, eles aumentam diretamente a indução provocada pela 1,25(OH)2D3 na produção da calbindina-D28K. Entretanto, em altas concentrações, observa-se o efeito contrário, resultando na diminuição do transporte de cálcio.
Alguns estudos encontraram concentrações séricas diminuídas de vitamina D no hipercortisolismo (31,32). Esta redução pode interferir com a síntese das proteínas transportadoras de cálcio na mucosa intestinal e a redução da absorção de cálcio funcionaria como estímulo para secreção de PTH. No entanto, a redução de concentração de vitamina D pode levar a aumento de secreção de PTH por vários mecanismos, visto que a 1,25(OH)2D3 apresenta um efeito inibidor sobre a transcrição do gene e secreção do PTH (33). Outro mecanismo foi proposto por Karmali e cols. (34). Segundo estes autores, a administração de cortisol às células paratireoideanas causaria uma diminuição no número de receptores para 1,25(OH)2D3, com isso alterando a resposta supressiva da 1,25(OH)2D3 sobre a secreção do PTH.
Os glicocorticóides poderiam interferir na ação da vitamina D, competindo com este hormônio nos tecidos-alvo, alterando a sua metabolização ou facilitando a sua degradação. Neste sentido, Luckert e cols. (1973) não verificaram redução nos níveis séricos de vitamina D no hipercortisolismo e, apesar disto, observaram uma redução na absorção intestinal de cálcio (35). Outros estudos verificaram que a resposta à administração de vitamina D no trato intestinal está prejudicada no hipercortisolimo, sugerindo a existência de resistência à ação da vitamina D neste sítio (36,37). Desta forma, são amplas as possibilidades de interferência do glicocorticóide sobre a ação da vitamina D no trato intestinal. A implicação prática deste aspecto é que o Colégio Americano de Reumatologia, em revisão recente, fez a recomendação de se utililizar vitamina D como medida preventiva de perda de massa óssea induzida por glicocorticóide (38).
Um outro tecido importante para a manutenção do equilíbrio do cálcio, o tecido renal, provavelmente também sofre influência dos glicocorticóides. É descrito que estes esteróides provocam uma diminuição da reabsorção tubular de cálcio, aumentando a perda deste íon. Fuller e cols. (39) descreveram receptores para os glicocorticóides em rins humanos, capazes de modificar o transporte renal de cálcio e outros íons. Findling e cols. (40) encontraram uma correlação direta entre a excreção urinária de cálcio e o cortisol livre urinário, sugerindo que a hipercalciúria pode estar diretamente relacionada com o hipercortisolismo.
Bikle e cols. observaram que no hipercortisolismo a calcemia não se correlacionava com o padrão de secreção do PTH (41). Com isso questionou-se uma possível alteração na regulação da secreção do PTH nessa condição, onde haveria uma redução da inibição provocada pelo cálcio na secreção do PTH.
Anormalidades na supressibilidade da secreção do PTH pelo cálcio foi também evidenciada durante nossos estudos em pacientes com hipercortisolismo crônico. Observamos que os pacientes com hipercortisolismo crônico exógeno apresentaram uma calcemia significativamente mais elevada e uma menor supressão do PTH que os indivíduos normais. Isso sugere uma anormalidade nos sensores do cálcio com uma elevação do limiar de sensibilidade ao cálcio para a inibição do PTH (42).
Além de alterar a secreção do PTH, os glicocorticóides são capazes de promover aumento no número dos receptores para o PTH, aumento da ligação do PTH aos seus receptores e aumento na expressão genética do receptor do PTH (43,44). Estes esteróides também aumentam o nível de adenilciclase e diminuem a fosfodiesterase, aumentando a ação do PTH, visto que algumas das ações do PTH são realizadas via AMPc (14).
Este aumento nos níveis plasmáticos do PTH, juntamente com uma redução nos níveis da calcitonina, presentes no hipercortisolismo, poderiam ser responsáveis pela osteoporose apresentada por estes pacientes, porém a participação da calcitonina na determinação da massa óssea é controversa. Segundo Heath e Sizemore (11), este hormônio é responsável por uma diminuição da reabsorção óssea e a sua deficiência pode estar relacionada com uma redução da massa óssea. Entretanto, em estudos mais recentes em que foram avaliados pacientes portadores de carcinoma medular de tireóide, condição em que ocorre aumento da secreção de calcitonina, não foi demonstrado aumento ou diminuição da massa óssea (45). Paula e cols. (46), estudando pacientes com deficiência de calcitonina, portadores de hipotireoidismo congênito, também não encontraram diminuição de massa óssea.
A secreção de calcitonina no hipercortisolismo crônico também foi avaliada recentemente por nós (42). Neste estudo, não observamos diminuição na secreção da calcitonina nos pacientes com hipercortisolismo crônico, sugerindo que este hormônio não se encontra envolvido com o desenvolvimento da OIG.
Especificamente no tecido ósseo, os glicocorticóides também são responsáveis pela manutenção dos receptores para a 1,25(OH)2D3 e pela inibição da sua degradação, colaborando assim para o aumento na reabsorção óssea, visto que a 1,25(OH)2D3 tem ação óssea reabsortiva (47).
III) AÇÃO DIRETA NAS CÉLULAS ÓSSEAS
Embora as alterações do metabolismo mineral possam contribuir para o surgimento da osteoporose induzida por glicocorticóide (OIG), é improvável que este seja o seu componente mais importante. A perda de massa óssea induzida pelo PTH ocorre apenas quando há aumento significativo e persistente da secreção de PTH. Estudos recentes indicam que os níveis circulantes basais da molécula biologicamente ativa de PTH são normais no hipercotisolismo crônico e apenas a secreção estimulada por hipocalcemia é aumentada neste pacientes (28). Além disto, o padrão histomorfométrico presente na OIG difere significativamente do apresentado por pacientes com hiperparatireoidismo primário (HPTP) (48). No HPTP ocorre principalmente redução da espessura cortical óssea e observa-se manutenção do volume do osso trabecular. Na OIG oberva-se redução tanto da espessura da cortical óssea quanto do volume do osso trabecular. À histomorfometria dinâmica, o HPTP caracteriza-se por aumento da atividade de formação e reabsorção óssea, enquanto que na OIG o padrão dominante é de redução de atividade osteoblástica, sendo que o aumento de reabsorção óssea ocorre principalmente na fase inicial de exposição ao esteróide (48).
As células ósseas também apresentam receptores para os glicocorticóides (49). Em concentrações fisiológicas estes esteróides são importantes para a diferenciação e função das células ósseas. Em doses farmacológicas, porém, alteram a atividade dessas células, favorecendo a perda de massa óssea. A identificação de receptores de glicocorticóide (RG) em osteoblasto é mais uma evidência de que estas células centralizam o controle de remodelação óssea, não apenas produzindo fatores e expressando receptores de substâncias que estimulam sua própria atividade (controle autócrino) mas, também, produzindo fatores que inibem ou estimulam a atividade de osteoclastos (controle parácrino).
O RG pertence à superfamília de receptores nucleares, composta pelos receptores de todos os outros hormônios esteróides, dos hormônios tireoidianos, do ácido retinóico e dos chamados receptores nucleares orfãos. O RG inativo, não ligado a hormônio, encontra-se no citoplasma associado a um complexo protéico formado, entre outras, pelas proteínas de choque 90 (hsp 90) e 70 (hsp 70). A associação do RG a hsp 90 aparentemente tem a função de facilitar a ligação do hormônio ao RG, aumentando a efetividade da resposta ao glicocorticóide. Após a associação ao ligante, o RG é liberado do complexo protéico, ocorrendo dimerização, localização nuclear, ligação ao DNA e ativação de funções específicas. Dentro do núcleo, o receptor ativado pelo hormônio interage com seqüências específicas de DNA, os elementos responsivos aos glicocorticóides (ERG), presentes, geralmente, na região promotora dos genes responsivos aos glicocorticóides. Estes homodímeros, ligados diretamente aos ERGs, atuam na maquinaria da transcrição gênica, provavelmente através da estabilização do complexo de iniciação da polimerase II, e estimulam a transcrição de genes responsivos aos glicocorticóides (50). Em alguns promotores, como no gene da pró-opiomelanocortina (POMC), têm sido identificados ERGn (elementos negativos responsivos aos glicocorticóides), onde os glicocorticóides atuam causando inibição da transcrição deste gene. Outro mecanismo de ação dos glicocorticóides envolve interação entre proteínas, como a interação do RG com outros fatores de transcrição como o sítio AP-1, entre outros. Estes fatores de transcrição, mesmo na ausência de uma ligação DNA-específica, modulam a transcrição dos genes responsivos aos glicocorticóides. Este mecanismo é responsável pelos efeitos anti-inflamatórios e imunossupressivos dos glicocorticóides, os quais envolvem uma regulação negativa da transcrição gênica.
Hofbauer e cols. (1999) demonstraram que os glicocorticóides reduzem os níveis de RNAm de osteoprotegerina (OPG), em várias linhagens de osteoblastos, por meio de inibição da transcrição do gene de OPG (51). Adicionalmente, estes autores verificaram que os glicocorticóides aumentam os níveis de RNAm do ligante de osteoprotegerina (OPG-L) nestas células. A OPG é um receptor solúvel - recentemente descrito -, que tem efeito inibitório sobre a reabsorção óssea. Por outro lado, o OPG-L é uma citocina pró-reabsortiva. Os mecanismos moleculares decorrentes da sinalização da ligação do glicocorticóide com o seu receptor no tecido ósseo ainda não são completamente conhecidos.
Canalis & Avioli (52) demonstraram que os glicocorticóides suprimem a função osteoblástica, inibindo a replicação celular, a síntese de colágeno e de proteínas não-colágenas. Estas alterações na atividade dos osteoblastos ficam evidentes durante a avaliação dos diferentes marcadores de formação óssea. Oikarinen e cols. (53) observaram que, quando glicocorticóides são administrados a pacientes normais ou em situações patológicas, os níveis séricos de osteocalcina, de fosfatase alcalina e de pró-colágeno I diminuem, evidenciando uma redução da atividade osteoblástica e, portanto, da formação óssea, a qual é restabelecida após a retirada da medicação.
A ação negativa dos glicocorticóides sobre o processo de remodelação óssea pode ser verificada também por sua influência sobre os osteoclastos. Estes esteróides podem estimular a reabsorção óssea diretamente ou ainda indiretamente, através do aumento na secreção do PTH (54). O aumento na excreção urinária de metabólitos de degradação do colágeno (hidroxiprolina, deoxipiridinolina, e telopeptídeos N e C terminal de colágeno tipo I) é uma evidência bioquímica do incremento de reabsorção óssea induzida pelos glicocorticóides (55-57).
IV) ALTERAÇÕES NA PRODUÇÃO DE PROSTAGLANDINAS, CITOCINAS, INTERLEUCINAS 1 e 6, HORMÔNIO DO CRESCIMENTO (GH) E FATOR DE CRESCIMENTO INSULINA SÍMILE (IGF)
As prostaglandinas (PG) agudamente inibem a função osteoclástica no osso normal, mas cronicamente estimulam a reabsorção óssea por aumentar a replicação e diferenciação dos novos osteoclastos (58). Como os glicocorticóides inibem a produção das PGs, é pouco provável que estes ecosanóides sejam responsáveis pela diminuição de massa óssea observada no hipercortisolismo crônico.
A hipótese de que o efeito osteolítico dos glicocorticóides poderia ser mediado pelas interleucinas (IL) 1 e 6 tem sido descartada, porque a produção de IL pelos linfócitos também é inibida pelos glicocorticóides (59). Estudos recentes têm sugerido a existência de controle da osteoclastogênese por meio da osteoprotegerina (OPG). A OPG é um receptor solúvel de membrana que inibe a maturação e atividade de osteoclastos, neutralizando a osteoprotegerina ligante (OPG-L), que é considerada um efetor final de osteoclastogênese. Os osteoblastos secretam tanto a OPG como o OPG-L. Nos últimos anos, diversos estudos têm verificado que os glicocorticóides podem estimular a reabsorção óssea, de um lado inibindo a produção de osteoprotegerina, e de outro estimulando a produção da OPG-L (51,60). O aumento da expressão de OPG-L vai estimular a osteoclastogênese por meio de interação entre osteoblastos e as células precursoras de osteoclastos, as quais expressam o receptor de membrana RANK, induzindo a formação de células maduras. A atividade dos osteoclastos maduros também é estimulada pelo mesmo mecanismo.
O efeito ósseo dos glicocorticóides pode, ainda, estar relacionado a alterações nas concentrações de GH e de IGF-I. Estes hormônios exercem efeitos tróficos sobre o osso, sendo considerados importantes reguladores da formação e manutenção óssea. O GH e o PTH são os maiores estimuladores da produção de IGF-I. Este peptídeo, que é sintetizado pelas células ósseas, é capaz de estimular a replicação e síntese de cólageno (61). Sabe-se que doses farmacológicas de glicocorticóides inibem a síntese de IGF-I, como também podem afetar as suas proteínas transportadoras, as IGFBPs, comprometendo assim a massa óssea (62).
V) ALTERAÇÕES NA PRODUÇÃO DOS ESTERÓIDES GONADAIS
Outro mecanismo proposto para a ocorrência de osteoporose induzida pelos glicocorticóides é que esta seja secundária à diminuição na produção dos esteróides gonadais.
Receptores para os andrógenos em células semelhantes aos osteoblastos humanos foram descritos por Colvard e cols. (63). Estes esteróides têm importante efeito anabólico sobre a massa óssea. São capazes de estimular a proliferação e diferenciação dos osteoblastos, como também de inibir o aumento do AMPc estimulado pelo PTH (64).
A importância dos estrógenos na mineralização óssea tem sido também amplamente demonstrada. Sabe-se que os osteoblastos têm receptores para estrógenos e que estes hormônios são capazes de diminuir a reabsorção óssea, possivelmente diminuindo a estimulação do AMPc provocada pelo PTH (65,66).
O hipogonadismo no hipercortisolismo tem sido associado à ação supressiva direta sobre a esteroidogênese gonadal, inibição da liberação do GnRH ou ainda supressão da sensibilidade da hipófise ao GnRH (67). Além destes mecanismos, sabe-se que os glicocorticóides sintéticos são capazes de suprimir o ACTH, levando a uma atrofia adrenal com conseqüente diminuição dos andrógenos (68).
No entanto, é pouco provável que a redução nos níveis dos andrógenos seja um fator isolado responsável pela perda óssea no hipercortisolismo crônico. Recentemente, demonstramos que pacientes com hipercortisolismo crônico endógeno, apesar dos níveis séricos de andrógenos elevados, apresentam massa óssea reduzida, quando comparados aos controles (28).
Em conclusão, a osteoporose induzida pelos glicocorticóides é uma condição onde provavelmente vários fatores estão envolvidos na sua patogênese. As evidências atuais indicam que, em grande parte, o efeito catabólico dos glicocorticóides se deve à ação direta destes esteróides sobre a atividade de células ósseas, com comprometimento predominante da formação óssea, além de aumento na reabsorção óssea. A relevância deste distúrbio, como problema em saúde pública, certamente servirá como mantenedor de interesse de pesquisa sobre os seus diversos aspectos fisiopatológicos e propostas terapêuticas mais eficientes. Tendo em vista que os benefícios terapêuticos dos glicocorticóides justificam o seu uso em diversos distúrbios, é necessário que se esteja atento à necessidade de minimizar o efeito indutor de perda de massa óssea destes esteróides. Em publicação recente (38), o Colégio Americano de Reumatologia estabeleceu critérios de indicação de prevenção/tratamento da OIG, os quais devem ser seguidos tendo em vista que cerca de metade dos pacientes que usa este medicamento não recebe nenhum tipo de orientação quanto à prevenção/tratamento da OIG.
Recebido em 19/06/06
Revisado em 21/10/02
Aceito em 27/11/02
- 1.Potts JT. Parathyroid hormone and parathyroid hormone related peptide in the regulation of calcium homeostasis and bone development. In: DeGroot LJ, Jameson JL, ed. Endocrinology, 4th ed., Philadelphia, Saunders, 2001, 969-98.
- 2.Guiness-Hey M, Hock JM. Increased trabecular bone mass in rats treated with human synthetic parathyroid hormone. Metab Bone Dis Relat Res 1984;5:177-81.
- 3.Rosen CJ, Donahue LR. Parathyroid hormone and osteoporosis. Curr Opin Endocrinol Diab 1996;3:532-9.
- 4.Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 1993;366:575-80.
- 5.Brown EM. Physiology and pathophysiology of the extracellular calcium-sensing receptor. Am J Med 1999;106:238-53.
- 6.Chertow BS, Baker GR, Henry HL, Norman AW. Effects of vitamin D metabolites on bovine parathyroid hormone release in vitro. Am J Physiol 1980;238:E384-8.
- 7.Anast CS, Mohs JM, Kaplan SL, Burns TW. Evidence for parathyroid failure in magnesium deficiency. Science 1972;177:606-8.
- 8.Fischer JA, Oldham SB, Sizemore GW, Arnaud CD. Calcitonin stimulation of parathyroid hormone secretion in vitro. Horm Metab Res 1971;3:223-4.
- 9.Au WY. Cortisol stimulation of parathyroid hormone secretion by rat parathyroid glands in organ culture. Science 1976;193:1015-7.
- 10.Brown EM. Histamine receptors on dispersed parathyroid cells from pathological human parathyroid tissue. J Clin Endocrinol Metab 1980;51:1325-9.
- 11.Heath H III, Sizemore GW. Plasma calcitonin in normal men: differences between men and women. J Clin Invest 1977;60:1135-40.
- 12.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 2001;81:239-97.
- 13.Miller WL, Portale AA. Genetic disorders of vitamin D biosynthesis. Endocrinol Metab Clin North Am 1999;28:825-40.
- 14.Adinoff AD, Hollister JR. Steroid-induced fractures and bone loss in patients with asthma. N Engl J Med 1983;309:265-8.
- 15.Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J Bone Miner Res 2000;15:993-1000.
- 16.Israel E, Banerjee TR, Fitzmaurice GM, Kotlov TV, LaHivie K, Leboff MS. Effects of inhaled glucocorticoids on bone density in premenopausal women. N Engl J Med 2001;345:941-7.
- 17.Adachi JD. Corticosteroid-induced osteoporosis. Am J Med Sci 1997;313:41-9.
- 18. Raker JW, Henneman PH, Grafi WS. Coexisting primary hyperparathyroidism and Cushing's syndrome. J Clin Endocrinol Metab 1962;22:273-80.
- 19.Wajchenberg BL, Quintão ER, Liberman B. Antagonism between adrenal steroids and parathyroid hormone. J Clin Endocrinol Metab 1965;25:1677-81.
- 20.Munck A, Brinck-Johnsen T. Specific and nonspecific physicochemical interactions of glucocorticoids and related steroids with rat thymus cells in vitro. J Biol Chem 1968;243:5556-65.
- 21.Williams GA, Peterson WC, Bowser N, Henderson WJ, Hargis GK, Martinez NJ. Interrelationship of parathyroid and adrenocortical function in calcium homeostasis in the rat. Endocrinology 1974;95:707-12.
- 22.Hahn JT, Halstead LR, Baran DT. Effects of short-term glucocorticoid administration on intestinal calcium absorption and circulating vitamin D metabolite concentrations in man. J Clin Endocrinol Metab 1981;52:111-5.
- 23.Bikle DD, Pillai S. Vitamin D, calcium and epidermal differentiation. Endocr Rev 1993;14:3-19.
- 24.Hattersley AT, Meeran K, Burrin, Hill P, Shiner R, Ibbertson K. The effect of long- and short-term corticosteroids on plasma calcitonin and parathyroid hormone levels. Calcif Tissue Int 1994;54:198-202.
- 25.Seeman E, Kumar R, Hunder GG, Scott M, Heath III, Riggs BL. Production, degradation, and circulation levels of 1,25-dihydroxyvitamin D in health and chronic glucocorticoid excess. J Clin Invest 1980;66:664-9.
- 26.Slovik DM, Neer RM, Ohman JL, Lowell FC, Clark MB, Segre GV, et al. Parathyroid hormone and 25-hydroxyvitamin D levels in glucocorticoid-treated patients. Clin Endocrinol 1980;12:243-8.
- 27.Gennari C, Imbimbo B, Montagnani M, Bernini M, Nardi P, Avioli V. Effects of prednisone and deflazacort on mineral metabolism and parathyroid hormone activity in humans. J Clin Invest 1984;36:245-52.
- 28.Lanna CM, Paula FJ, Montenegro RM Jr, Moreira AC, Foss MC. Parathyroid hormone secretion in chronic human endogenous hypercortisolism. Braz J Med Biol Res 2002;35:229-36.
- 29.Kimberg DV. Effects of vitamin D and steroid hormones on the active transport of calcium by the intestine. N Engl J Med 1969;258:1396-405.
- 30.Corradino RA, Fullmer CB. Positive cotranscriptional regulation of intestinal calbindin-D28K gene expression by 1,25-dihydroxy-vitamin D3 and glucocorticoids. Endocrinology 1991;128:944-50.
- 31.Klein GR, Arnaud SB, Gallagher JC, DeLuca H, Riggs BL. Intestinal calcium absorption in exogenous hypercortisolism. J Clin Invest 1977;60:253-9.
- 32.Nordin BEC, Crilly RG, Smith DA. Osteoporosis. In: Nordin BEC (ed). Metabolic bone and stone disease Churchill Livingstone, Edinburgh, 1984, 1-70.
- 33.Silver J, Russel J, Sherwood LM. Regulation by vitamin D metabolites of messenger ribonucleic acid for preproparathyroid hormone glue transcription in vivo in rat. Proc Natl Acad Sci USA 1985;82:4270-3.
- 34.Karmali R, Farrow S, Hewison M. Effects of 1,25 dihydroxyvitamin D3 and cortisol on bovine and human parathyroid cells. J Endocrinol 1989;123:137-42.
- 35.Lukert BP, Stanbury SW, Mawer EB. Vitamin D and intestinal transport of calcium: effects of prednisone. Endocrinology 1973;93:718-21.
- 36.Colette C, Monnier L, Herbute P, Blotman F, Mirouze J. Calcium absorption in corticoid treated subjects of a single oral dose of calcitriol. Horm Metab Res 1987;19:335-8.
- 37.Favus MJ, Walling MW, Kimberg DV. Effects of 1,25-dihydroxycholecalciferol on intestinal calcium transport in cortisone-treated rats. J Clin Invest 1973;52:1680-5.
- 38.American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Rheum 2001;44:1496-503.
- 39.Fuller PL, Funder JW. Mineralcorticoid and glucocorticoid receptors in human kidney. Kidney Int 1976;10:154-7.
- 40.Findling JW, Adams ND, Lemann J, Gray RW, Thomas CJ, Tyrrell B. Vitamin D metabolites and parathyroid hormone in Cushing's Syndrome: Relationship to calcium and phosphorus. J Clin Endocrinol Metab 1982;54:1039-44.
- 41.Bikle DD, Pillai S. Vitamin D, calcium and epidermal differentiation. Endocr Rev 1993;14:3-19.
- 42.Lanna CMM. Estudo da secreção do paratormônio (PTH) e calcitonina no hipercortisolismo crônico humano endógeno e exógeno. Ribeirão Preto, 1999 Tese (Doutorado) - Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo.
- 43.Urena P, Iida-Klein A, Kong XF. Regulation of parathyroid hormone (PTH)/PTH-related peptide receptor messeger ribonucleic acid by glucocorticoids and PTH in ROS17/2.8 and OK cells. Endocrinology 1994;134:451-6.
- 44.McSheehy PMG, Chambers TJ. Osteoblastic cells mediate osteoclastic responsiveness to parathyroid hormone. Endocrinology 1986;118:824-8.
- 45.Hurley DL, Tiegs RD, Wahner HW, Heath H. Axial and appendicular bone mineral density in patients with long-term deficiency or excess of calcitonin. N Engl J Med 1987;317:537-42.
- 46.Paula FJA, Daripa M, Rufino ACTBF, Iazigi N, Foss MC. Densidade mineral óssea (DMO) e secreção de calcitonina em indivíduos normais e com defeito de gêneses de tireóide (DGT). Arq Bras Endocrinol Metab 2000;44(suppl.2):S185.
- 47.Manolagas SC, Anderson DC, Lumb GA. Glucocorticoids regulate the concentration of 1,25-dihydroxycholecalciferol receptors in bone. Nature 1979;277:314-5.
- 48.Rubin MR, Bilezikian JP. The role of parathyroid hormone in pathogenesis of glucocorticoi-induced osteoporosis: a re-examination of the evidence. J Clin Endocrinol Metab 2002;4033-41.
- 49.Subramaniam M, Colvard D, Keeting PE. Glucocorticoid regulation of alkaline phosphatase, osteocalcin and proto-oncogenes in normal human osteoblast-like cells. J Cell Biochem 1992;50:411-24.
- 50.Hollenberg S, Guiguère V, Segui P, Evans RM. Colocalization of DNA-binding and transcriptional activation functions in the human glucocorticoid receptor. Cell 1987;49:39-46.
- 51.Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, et al. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology 1999;140:4382-9.
- 52.Canalis E, Avioli L. Effects of deflazacort on aspects of bone formation in cultures of intact calvariae and osteoblast-enriched cells. J Bone Miner Res 1992;7:1085-92.
- 53.Oikarinen A, Autio P, Vuori J. Systemic glucocorticoid treatment decreases serum concentration of carboxyterminal propeptide of type I procollagen and aminoterminal propeptide of type III pro-collagen. Br J Dermatol 1992;126:172-8.
- 54.Reid DM, Nicoll JJ, Smith M. Corticosteroids and bone mass in asthma: comparisons with rheumatoid arthritis and polymyalgia rheumatica. Br Med J 1986;293:1463-6.
- 55. Chiodini I, Carnevale V, Torlontano M, Fusilli S, Guglielmi G, Pileri M, et al. Alterations of bone turnover and bone mass at different skeletal sites due to pure glucocorticoid excess: study in eumenorrheic patients with Cushing's syndrome. J Clin Endocrinol Metab 1998;83:1863-7.
- 56.Ali NJ, Capewell S, Ward MJ. Bone turnover during high dose inhaled corticosteroid treatment. Thorax 1991;46:160-4.
- 57.Bornefalk E, Dahlén I, Michaëlsson K, Ljunggren Ö, Ljunghall S. Age-dependent effect of oral glucocorticoids on markers of bone resorption in patients with acute asthma. Calcif Tissue Int 1998;63:9-13.
- 58.Bhyun YS, Raisz LG. Opposing effects of prostaglandin E2 and cortisol in bone growth in organ culture. Clin Res 1982;30:387A.
- 59.Marusic A, Raisz L. Cortisol modulates the actions of interleukin-1 alpha on bone formation, resorption and prostaglandin production in cultured mouse parietal bones. Endocrinology 1991;129:2699-706.
- 60. Hofbauer LC, Heufelder AE. The role of receptor activator of nuclear factorkB ligand and osteoprotegerin in the pathogenesis and treatment of metabolic bone diseases. J Clin Endocrinol Metab 2000;85:2355-63.
- 61.Ernst M, Froesch ER. Growth hormone dependent stimulation of osteoblast-like cells in serum-free cultures via local synthesis of insulin-like growth factor. Biochem Biophys Res Commun 1988;151:142-7.
- 62.Mc Carthy TL, Centrella M, Canalis E. Cortisol limits the synthesis of insulin-like growth factor-I in skeletal cells. Endocrinology 1990;126:1569-75.
- 63.Colvard DS, Eriksen EF, Keeting PE. Identification of androgen receptors in normal human osteoblast-like cells. Proc Natl Acad Sci USA 1989;86:854-7.
- 64.Fukayama S, Tashjian AH. Direct modulation by androgens of the response of human bone cells (SaOS-2) to human parathyroid hormone (PTH) and PTH-related protein. Endocrinology 1989;125:1789-94.
- 65.Eriksen EF, Colvard DS, Berg NJ. Estrogen building receptor mRNA and biologic response in osteoblast-like osteosarcoma cells. Science 1988;241:84-6.
- 66.Faustini-Fustini M, Rochira V, Carani C. Oestrogen deficiency in men: Where are we today? Eur J Endocrinol 1999;140:111-29.
- 67.Veldhuis JD, Lizarralde G, Iranmanesh A. Divergent effects of short-term glucocorticoid excess on the gonadotropic and somatotropic axes in normal men. J Clin Endocrinol Metab 1992;4:96-102.
- 68.Doerr P, Pirke KM. Cortisol induced supression of plasma testosterone in normal adult males. J Clin Endocrinol Metab 1976;43:622-9.
Endereço para correspondência
Datas de Publicação
-
Publicação nesta coleção
09 Jun 2003 -
Data do Fascículo
Fev 2003
Histórico
-
Aceito
27 Nov 2002 -
Recebido
19 Jun 2002