Resumos
Em 10 meninas com diagnóstico de baixa estatura idiopática (BEI), realizamos avaliação citogenética após revisão clínica. Dois cariótipos foram anormais: mos 45,X/46,XX; mos 45,X/46,X,der(Xp)/46,X,r(X), e para sua elucidação foram aplicadas técnicas de citogenética molecular e análise de microssatélites, incluindo SHOX CA repeat. Os resultados confirmaram a origem dos cromossomos anômalos e a identificação da haploinsuficiência do gene SHOX. Nos oito casos com cariótipo normal, a pesquisa de mosaicismos crípticos pela técnica FISH através da sonda centromérica (DXZ1) em células de mucosa oral (nuc ish) evidenciou a presença de dois mosaicos verdadeiros (DXZ1x2/DXZ1x1). A revisão clínica da paciente com anomalia estrutural de X e das 2 meninas com mosaicismo detectados pelo nuc ish, mostrou a presença de 3 ou mais sinais clínicos observados na síndrome de Turner (ST). Estes resultados reafirmam a importância da análise citogenética em meninas com diagnóstico de BEI e sinais clínicos da ST. Os resultados do estudo molecular para o gene SHOX confirmam sua relação com estigmas da ST. Sendo normal o cariótipo, a pesquisa de mosaicismos crípticos em outros tecidos deve ser considerada. O diagnóstico mais preciso poderá modificar a conduta terapêutica, como indicação do GH nessas meninas.
Baixa estatura idiopática; Anomalia cromossômica; FISH; SHOX; Mosaicismo; Síndrome de Turner
Ten girls with idiopathic short stature (ISS) were clinically reviewed and cytogenetic analysis performed by GTG-banding. Two abnormal karyotypes were identified: mos 45,X/46,XX and mos 45,X/46,X, der(Xp)/46,X,r(X). In the latter, FISH analysis and microsatellite investigation, including intragenic SHOX CA repeat, confirmed the origin of the abnormal structural chromosomes and revealed haploinsufficiency for the SHOX gene. In the remaining 8 patients with a normal lymphocyte karyotype FISH analysis using an alpha centromeric X probe (DXZ1) in buccal smear cells (nuc ish) revealed two further cases of true X mosaicism (DXZ1x2/DXZ1x1). Clinical review of all four abnormal patients with the exception of patient with mos 45,X/46,XX revealed three or more clinical features commonly present in Turner syndrome (TS). Our results reinforce the importance of cytogenetic investigation in all patients with ISS. The SHOX molecular result sustains its correlation with most clinical signs of TS. In those cases where a normal karyotype was observed, cryptic mosaicism should be excluded. A precise etiologic diagnosis may be relevant for the indication of GH therapy in these girls.
Idiopathic short stature; X chromosome abnormalities; Turner syndrome; SHOX; FISH; Mosaicism
ARTIGO ORIGINAL
Investigação clínica e genética em meninas com baixa estatura idiopática
Clinical and genetic studies in girls with idiopathic short stature
Rosa R.S. Martins; Hilda I.B. Ramos; Juan C. Llerena Jr.; José C.C. Almeida
Ambulatório de Crescimento e Laboratório de Citogenética do Instituto Estadual de Diabetes e Endocrinologia (RRSM); Departamento de Genética do Instituto de Biologia, Universidade Federal do Rio de Janeiro (UFRJ) (RRSM); Laboratório de Citogenética Molecular do Instituto Fernandes Figueira e Divisão de Genética do Instituto Nacional do Câncer (HIBR,JCL Jr,JCCA)
Endereço para cerrespondência Endereço para correspondência Rosa Rita dos Santos Martins Rua Marques de Olinda, 64 - Bloco B, apt. 501 22251-040 Rio de Janeiro, RJ E-mail: rosaritamartins@uol.com.br
RESUMO
Em 10 meninas com diagnóstico de baixa estatura idiopática (BEI), realizamos avaliação citogenética após revisão clínica. Dois cariótipos foram anormais: mos 45,X/46,XX; mos 45,X/46,X,der(Xp)/46,X,r(X), e para sua elucidação foram aplicadas técnicas de citogenética molecular e análise de microssatélites, incluindo SHOX CA repeat. Os resultados confirmaram a origem dos cromossomos anômalos e a identificação da haploinsuficiência do gene SHOX. Nos oito casos com cariótipo normal, a pesquisa de mosaicismos crípticos pela técnica FISH através da sonda centromérica (DXZ1) em células de mucosa oral (nuc ish) evidenciou a presença de dois mosaicos verdadeiros (DXZ1x2/DXZ1x1). A revisão clínica da paciente com anomalia estrutural de X e das 2 meninas com mosaicismo detectados pelo nuc ish, mostrou a presença de 3 ou mais sinais clínicos observados na síndrome de Turner (ST). Estes resultados reafirmam a importância da análise citogenética em meninas com diagnóstico de BEI e sinais clínicos da ST. Os resultados do estudo molecular para o gene SHOX confirmam sua relação com estigmas da ST. Sendo normal o cariótipo, a pesquisa de mosaicismos crípticos em outros tecidos deve ser considerada. O diagnóstico mais preciso poderá modificar a conduta terapêutica, como indicação do GH nessas meninas.
Descritores: Baixa estatura idiopática; Anomalia cromossômica; FISH; SHOX; Mosaicismo; Síndrome de Turner
ABSTRACT
Ten girls with idiopathic short stature (ISS) were clinically reviewed and cytogenetic analysis performed by GTG-banding. Two abnormal karyotypes were identified: mos 45,X/46,XX and mos 45,X/46,X, der(Xp)/46,X,r(X). In the latter, FISH analysis and microsatellite investigation, including intragenic SHOX CA repeat, confirmed the origin of the abnormal structural chromosomes and revealed haploinsufficiency for the SHOX gene. In the remaining 8 patients with a normal lymphocyte karyotype FISH analysis using an alpha centromeric X probe (DXZ1) in buccal smear cells (nuc ish) revealed two further cases of true X mosaicism (DXZ1x2/DXZ1x1). Clinical review of all four abnormal patients with the exception of patient with mos 45,X/46,XX revealed three or more clinical features commonly present in Turner syndrome (TS). Our results reinforce the importance of cytogenetic investigation in all patients with ISS. The SHOX molecular result sustains its correlation with most clinical signs of TS. In those cases where a normal karyotype was observed, cryptic mosaicism should be excluded. A precise etiologic diagnosis may be relevant for the indication of GH therapy in these girls.
Keywords: Idiopathic short stature; X chromosome abnormalities; Turner syndrome; SHOX; FISH; Mosaicism
O CRESCIMENTO APRESENTA UM PADRÃO de herança multifatorial em que interagem fatores genéticos e ambientais. Várias mutações gênicas envolvendo secreção, liberação e resposta ao hormônio de crescimento, fatores de crescimento e seus receptores, secreção e liberação de outros hormônios têm sido descritas associadas à baixa estatura (1,2). Anomalias cromossômicas numéricas e estruturais, principalmente as que envolvem os cromossomos sexuais, estão freqüentemente associadas à baixa estatura. Entretanto, cerca de 3% das crianças que apresentam estatura 2 ou mais desvios padrão (DP) abaixo da média populacional para idade e sexo não têm um fator etiológico reconhecido e são classificadas como portadoras de baixa estatura idiopática (BEI) (1,2).
O conhecimento de que perdas terminais dos braços curtos dos cromossomos sexuais X e Y apresentam baixa estatura como um sinal clínico constante levou vários pesquisadores a concentrarem nessa região a pesquisa de um ou mais genes envolvidos na etiopatogenia da baixa estatura (3-7). Em 1997, foi seqüenciado o gene SHOX (short stature homeobox containing gene), localizado na região pseudoautossômica dos braços curtos dos cromossomos X (Xp22.3) (figura 1) e Y (Yp11.3) (5,8). Mutações e deleções do gene SHOX vêm sendo descritas em 1 a 2,4% dos portadores de BEI (8-12) e com grande freqüência em uma forma de displasia óssea, discondrosteose de Leri Weill, em que se associam à baixa estatura, displasia óssea mesomélica e deformidade de Madelung (10,13-18).
A deficiência completa do gene SHOX, em homozigose, é responsável por uma forma mais rara e mais grave de displasia óssea mesomélica, conhecida como Síndrome de Langer (19,20).
Pesquisas recentes têm demonstrado que a haploinsuficiência do gene SHOX está associada à etiopatogenia das alterações esqueléticas descritas na Síndrome de Turner (ST), como palato ogival, quarto metacarpo curto, cúbito valgo e a deformidade de Madelung (17,21-24).
Crianças com mosaicismo celular, com baixa contagem de linhagem 45,X e/ou anomalias estruturais dos cromossomos sexuais, podem ter pouca expressão clínica, não exibindo os sinais clínicos conhecidos na ST como linfedema congênito, ptose palpebral, pescoço alado, tórax largo, podendo, por isso, ser diagnosticadas como BEI simples (25). Além disso, quando a freqüência da linhagem 45,X é muito baixa, esta pode não ser detectada no cariótipo de rotina em sangue periférico, sendo necessário extender a investigação citogenética com técnicas de biologia molecular e, preferencialmente, em mais de um tipo de tecido (26-28). Tais exemplos podem ser considerados como mosaicos crípticos.
Entre as técnicas de Citogenética Molecular de particular utilidade para estes diagnósticos, a de FISH (Fluorescense In Situ Hybridization) é, geralmente, a de escolha. Esta técnica utiliza sondas de DNA marcadas com haptenos, que identificam segmentos cromossômicos de cópia única, segmentos com seqüências repetitivas (como as regiões centroméricas) ou todo o cromossomo (whole chromosome painting - wcp) (29-31). A técnica permite ainda detectar anomalias numéricas e estruturais dos cromossomos em metáfases, assim como pode ser aplicada em células interfásicas, sem necessidade de cultura (nuc ish - nuclear in situ hybridization) e em preparações citológicas diversas (32-34), como as células de mucosa oral. Em crianças com baixa estatura e com cariótipo normal em sangue periférico, o estudo com sondas alfa centroméricas de X e Y nas preparações de mucosa oral permite identificar mosaicismos crípticos (32,34).
Entre as técnicas de Genética Molecular para o estudo de BEI, a análise do microssatélite SHOX CA repeat, localizado na região 5 UTR (untranslated region) do exon 1 do gene SHOX, tem sido empregada na pesquisa de microdeleções da região pseudoautossômica onde está mapeado o gene SHOX e tem-se mostrado útil na identificação da haploinsuficiência do SHOX (9,13,41) (figura 1).
Neste trabalho, dez meninas com diagnóstico inicial de BEI foram selecionadas para revisão clínica e estudo citogenético convencional e molecular. A técnica FISH em células de mucosa oral foi indicada na pesquisa de mosaicismos crípticos. A análise do microssatélite SHOX CA repeat, e de dois outros marcadores da região pseudoautossômica, foi também complementar ao estudo de uma das meninas.
CASUÍSTICA E MÉTODOS
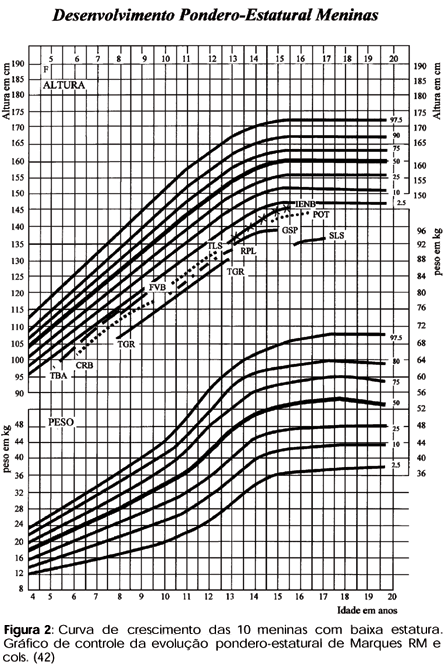
De 486 crianças acompanhadas no ambulatório de crescimento do IEDE - Instituto Estadual de Diabetes e Endocrinologia, 15 (3,1%) tinham diagnóstico de BEI por apresentarem dois ou mais DP abaixo da média populacional da altura, segundo índices referidos por Marques, Marcondes e cols. para crianças e adolescentes brasileiros (42) (figura 2). Destas, dez meninas foram selecionadas para reavaliação clínica e radiológica e submetidas a avaliação genética. Todas apresentavam dosagens séricas basais normais de T4 livre, TSH, IGF1 e IGFBP3.
Este trabalho foi aprovado pelo Comitê de Ética em Pesquisa do IEDE e consentimento escrito foi obtido do responsável de cada paciente (Registro CEP-IEDE:16).
Avaliação Clínica, Radiológica e Laboratorial
O estudo clínico incluiu história familiar, peso e estatura ao nascimento, altura e pesos atuais e exame físico.
A curva de crescimento adotada e o cálculo do DP da altura foram os descritos por Marques, Marcondes e cols., por serem baseados em dados de uma população brasileira (42). A altura dos pais de todas as meninas estava acima do percentil 2,5 da curva específica para os sexos masculino (altura igual ou superior a 160cm) e feminino (altura igual ou superior a 150cm).
Foram realizadas radiografias de mãos e punhos para avaliação de idade óssea e presença de 4º. metacarpo curto, radiografias de antebraços para pesquisa da deformidade de Madelung, radiografias de joelho antero-posterior para avaliar assimetria dos côndilos femurais e rebaixamento do platô tibial interno descritos por Kosowicz em portadoras de disgenesia gonadal (43) e para detectar a presença de exostoses e região tibial interna protuberante, observadas na discondrosteose de Leri Weil (21,44).
Análise Citogenética
A análise do cariótipo foi realizada em culturas de sangue periférico, segundo a técnica descrita por Moorhead, modificada por Bender em 1965 (45). A técnica de bandeamento GTG, com padrão aproximado de 400 a 550 bandas, segundo o protocolo descrito por Seabright em 1971 (46), foi realizada em todos os pacientes. A técnica de bandeamento CBG, descrita por Summer em 1972 (47), foi aplicada quando indicada. Das mesmas culturas foram reservadas lâminas para técnicas FISH.
Análise pela Técnica FISH em Metáfases de Sangue Periférico
Sondas centroméricas de X (DXZ1) (Oncor, Inc.) marcadas com biotina e uma sonda de seqüência única da região Xq13, locus XIST (X-inactive specific transcript) (Oncor, Inc.) marcada com digoxigenina, foram aplicadas em metáfases de cultura de sangue periférico. A sonda wcp X, cedida pelo Dr. R. Stanyon (48), marcada com biotina, foi usada para identificação do cromossomo X.
As técnicas de FISH com aplicação da sonda centromerica DXZ1 e da sonda de cópia única XIST foram baseadas no protocolo descrito por Ramos (34). Para pintura completa (wcp) dos cromossomos X, foi aplicado o protocolo indicado por Stanyon (48).
A detecção e amplificação das sondas foram realizadas com anticorpos específicos conjugados à fluoresceina. A identificação e contraste dos sinais fluorescentes foi realizada com 10µl de iodeto de propideo/antifade (0,2mg/µl).
Análise pela Técnica FISH em Células de Mucosa Oral
A mesma sonda (Oncor, Inc.) alfa-satélite, centromérica (DXZ1), foi aplicada em esfregaço de mucosa oral feito em lâminas silanizadas. O protocolo usado foi o citado por Ramos (34). Como controle feminino, usamos as lâminas de uma menina com cariótipo normal 46,XX.
As lâminas foram examinadas em microscópio de epifluorescência Axiophot Carl Zeiss equipado com conjunto de filtros apropriados para sondas moleculares conjugadas ao FITC. As lâminas das preparações com sondas wcp do cromossomo X foram analisadas em um microscópio Olympus, acoplado a um sistema de captação de imagens Power Macintosh 63.
Análise de Microssatélites
A extração de DNA genômico foi realizada em sangue periférico, segundo o protocolo de Miller e cols. (49), e a amplificação da região intragênica SHOX CA repeat foi realizada, segundo a técnica descrita por Belin e cols. (13), com primer ''sense'' 5' CATGTCATATATATATGTGATCC 3' marcado com 6-FAM e primer ''reverse'' não marcado 5' GACACAGAAATCCTTCATAAA 3'. Foram realizadas também com primers ''sense'' marcados com 6-FAM a amplificação dos microssatélites DXYS233 (5' TTGGGAATTCGAGGCT 3' e 5' TGATTTCCATCCTGGGGGT 3') e DXYS234 (5' CCCAGATCGCGCCCATT 3' e 5' ATGGCTCTGAGGCGGG 3'), localizados, respectivamente, a 0 e 2cM da região telomérica do braço curto de X. A análise dos fragmentos corados com ROX foi realizada na máquina ABI prisma 377, e o tamanho dos alelos determinado pelo programa Gene Scan.
Na figura 1, no ideograma do cromossomo X (850 bandas), segundo ICSN (50), delimitamos as regiões investigadas pelas técnicas FISH e análise de microssatélites.
RESULTADOS
Avaliação Clínica, Radiológica e Laboratorial
As informações clínicas estão sumarizadas na tabela 1. Os resultados das dosagens hormonais estão descritos na tabela 2. Retardo de crescimento intra-útero foi relatado em 3/10 crianças (POT, SLS, TBR).
Os sinais clínicos mais observados foram palato ogival em 4/10 e cúbito valgo em 4/10. A alteração radiológica mais comum, em 4/10 crianças, foi o 4º. metacarpo curto. A maior associação de sinais clínicos foi observada nas meninas SLS e POT.
A deformidade de Madelung foi encontrada na menina SLS, como também a alteração de joelho (região tibial interna protuberante) e exostoses nas fíbulas descritas na síndrome de Discondrosteose de Leri Weill (figura 3). A menina TBR apresentou, na radiografia de mãos e punhos, sinostose dos ossos do carpo (fusão do semilunar com o piramidal à direita e do grande osso com o ganchoso bilateralmente) e subluxação do cotovelo direito.
Avaliação da Citogenética Convencional e Molecular
Foram analisadas de 50 a 100 metáfases nas lâminas de cada paciente por técnicas de bandeamento GTG (tabela 3). Dois cariótipos foram anormais: a menina SLS, com mosaicismo celular: 27 metáfases com ausência de um cromossomo do par X (45,X); 13 metáfases com um cromossomo X apresentando uma anomalia estrutural envolvendo braço curto, 46,X, der(X); e 10 metáfases com um cromossomo X em anel, 46,X, r(X) (figura 4a,b). A análise pela técnica CBG demonstrou que, tanto o cromossomo derivado de X, como o cromossomo X em anel eram monocêntricos. A análise pela técnica FISH com sondas centroméricas (DXZ1) confirmou que ambos os cromossomos anormais eram derivados de X (figura 4c,d). A aplicação da sonda XIST, região Xq13, em metáfases afastou a possibilidade de duplicação completa do braço longo de X, já que havia apenas uma marcação fluorescente nos dois cromossomos anômalos e no cromossomo X normal (figura 4e,f). A aplicação do sonda wcp X afastou a presença de um rearranjo cromossômico entre o X e algum autossomo (figura 4g,h). Os pais desta menina apresentaram cariótipo normal.
A amplificação de 3 microssatélites: DXYS 233, SHOX CA repeat e DXYS 234 a 0cM, 0,7cM e 2cM do telômero do braço curto de X mostrou um único alelo para a região dos microssatélites SHOX CA repeat e um único alelo para a região DXYS 234, ambos de origem materna, com perda dos alelos paternos; o DXYS 233 não foi informativo porque ambos os pais apresentaram o mesmo alelo (tabela 4). Estes resultados confirmaram a perda da região do gene SHOX nos cromossomos X anômalos, levando à haploinsuficiência do gene SHOX.
O segundo cariótipo anormal foi da menina TGR, com mosaicismo celular com duas linhagens celulares: uma metáfase com monossomia do cromossomo X (45,X) e 49 com cariótipo normal (46,XX). Este mosaicismo não foi observado em células de mucosa oral, na análise pela técnica FISH com aplicação da sonda de DNA centromérica (DXZ1).
Pesquisa de Mosaicismos Celulares nas Meninas com Cariótipo Normal
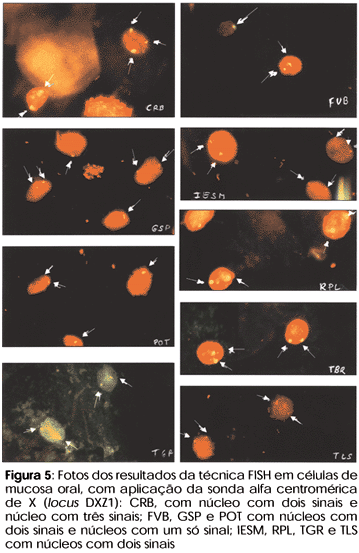
A pesquisa de mosaicismos crípticos, através da técnica de FISH em esfregaço de mucosa oral com sondas centroméricas (DXZ1), foi realizada nas 8 meninas com cariótipo normal. Foi também realizado um controle feminino.
Cinco meninas e o controle feminino apresentaram resultado normal (tabela 3), incluindo a menina portadora do mosaicismo 45,X/46,XX. Quatro apresentaram mosaicismo celular: 3 mosaicos (DXZ1x2/ DXZ1x1) com freqüências de 4, 6 e 15% de núcleos com um único sinal centromérico de X, 1 mosaico (DXZ1 x 2/DXZ1 x 3), com 30% dos núcleos com 3 sinais fluorescentes (tabela 3) (figura 5). Na menina com mosaicismo (DXZ1x2/DXZ1x3), os núcleos com 3 sinais apresentavam dois com mesma intensidade e tamanho e o 3º. menor e igualmente brilhante. A aplicação da mesma sonda em lâminas de cultura de sangue periférico confirmou a presença de três sinais fluorescentes em 15 metáfases, enquanto 35 apresentavam dois sinais para DXZ1. A 3º. marcação fluorescente foi evidenciada na região pericentromérica de um dos cromossomos do par 17. A técnica CBG mostrou que o par X e o par 17 eram monocêntricos. A aplicação da sonda wcp de X hibridou com ambos os cromossomos X e nenhuma marcação nos cromossomos 17. Os pais apresentavam cariótipo normal pela técnica de bandeamento GTG. O estudo das metáfases maternas pela técnica FISH, com sondas alfacentroméricas de X, evidenciou o mesmo padrão de hibridação cruzada. Tais resultados permitiram afastar uma anomalia numérica e estrutural envolvendo o cromossomos X na menina CRB.
DISCUSSÃO
A importância da análise do cariótipo em meninas com baixa estatura idiopática ficou evidenciada em nosso estudo, ao mostrar a variabilidade da expressão clínica em portadoras de mosaicismo celular tendo a baixa estatura como sinal clínico constante.
As portadoras de mosaicismo 45,X/46,XX apresentaram baixa freqüência da monossomia X, podendo, por consegüinte, não ser diagnosticadas no cariótipo em sangue periférico, necessitando da análise de um outro tecido por técnicas de citogenética molecular.
O cariótipo anormal, mos 45,X/46,XX, na menina TGR não teve seu mosaicismo observado nas células de mucosa oral por FISH. É conhecido na literatura que perdas espontâneas dos cromossomos sexuais podem ocorrer em culturas in vitro mesmo em preparações sem cultura celular (32). Seu crescimento vem se mantendo com 2,9 DP abaixo da média de altura para meninas da mesma idade, independente do desenvolvimento puberal normal para a idade. A presença de 4º. metacarpo curto com único e a ausência de estirão puberal ainda não nos permitiu afastar, com certeza, o diagnóstico de mosaicismo verdadeiro.
Nas oito meninas com cariótipo normal, a pesquisa de mosaicismo celular por FISH, em células de mucosa oral, foi uma opção para o estudo de mosaicismo por originarem-se do ectoderma, diferentemente dos linfócitos. Além disso, permitiu estudar células em intérfase, sem necessidade de cultura celular. Outra vantagem foi a possibilidade de, em uma única lâmina, poder ser analisado um maior número de células.
Três meninas apresentaram mosaicismo celular (DXZ1x2/DXZ1x1), com células com dois sinais fluorescentes (correspondentes aos centrômeros dos 2 cromossomos X) e células com um único sinal fluorescente (correspondendo a um único cromossomo X na célula), sendo que as freqüências das células com um único sinal foram de 4% (FVB), 6% (POT) e 15% (GSP), respectivamente.
Para diferenciar o mosaicismo verdadeiro do falso, nos baseamos no trabalho de Schad e cols. (32). Estes autores, estudando células de mucosa oral em controles masculinos e femininos e em portadores de mosaicismo envolvendo cromossomos sexuais X e Y com sondas centroméricas dos cromossomos sexuais, consideraram como mosaicismos verdadeiros freqüências iguais ou acima de 6%, já que uma variação de até 5,2% de perdas espontâneas, esporádicas dos cromossomos sexuais em indivíduos normais foi encontrada.
Das 3 portadoras de mosaicismo (DXZ1x2/ DXZ1x1), a menina FBV apresentou uma freqüência baixa (4% dos núcleos com um sinal fluorescente), e um único estigma (nevus), sendo, por nós, considerada, portanto, como pseudomosaicismo. As duas outras meninas com mosaicismo com percentual de 6% (GSP) e 15% (POT) de células com um único sinal fluorescente apresentaram, respectivamente, 3 e 5 sinais clínicos descritos na Síndrome de Turner. Ambas têm história de irregularidade menstrual, queixa clínica freqüente em portadoras de mosaicismo 45,X/46,XX (56).
Nas duas meninas (POT e GSP) com mosaicismos crípticos e na menina SLS com anomalia estrutural do X, o acentuado comprometimento do crescimento ocorreu, provavelmente, pela ausência de estirão puberal normal (figura 2), como ocorre nas meninas com ST.
Uma 4ª. menina (CRB) apresentou mosaicismo celular com 2 e 3 sinais fluorescentes. A aplicação da técnica FISH mostrou que o 3º. sinal fluorescente correspondia a uma marcação justacentromérica de um cromossomo do par 17. O emprego da sonda wcp afastou a possibilidade de uma translocação entre as regiões justacentroméricas de X e 17 pela ausência de material de X no cromossomo 17. A possibilidade de hibridação cruzada entre regiões centroméricas do X e autossomos já foi citada na literatura por Crolla & Llerena (51). Em estudo sobre mosaicismo de anomalias numéricas e estruturais do cromossomo X, estes autores relataram que 10% das metáfases hibridadas com sondas alfa centroméricas de X mostravam pequenas hibridações cruzadas para os centrômeros dos cromossomos 11 e 17. Esta variação, provavelmente, não tem relação com a baixa estatura apresentada pela paciente, já que a mesma hibridação cruzada foi observada nas metáfases de sua mãe.
A menina SLS, com cariótipo mos 45,X/ 46,X,der(Xp)/46,X,r(X), teve desenvolvimento puberal normal para idade e ciclos menstruais normais, raramente observados na ST. Entretanto, apresentava cinco sinais clínicos comuns à ST e dois sinais radiológicos descritos em portadores da haploinsuficiência do gene SHOX, como a deformidade de Madelung e as alterações dos joelhos (figura 3, tabela 1). Tem sido descrita uma variabilidade clínica nas portadoras de anel de X. Geralmente o diagnóstico é tardio, como aconteceu na nossa paciente, porque estão ausentes os sinais clínicos característicos como linfedema congênito, pescoço curto e largo e infantilismo sexual (51-53). SLS teve desenvolvimento motor normal sem dificuldade de escolaridade. É provável que a conservação do centro de inativação do cromossomo X, comprovada pela aplicação da sonda XIST (figura 4e,f) tenha mantido a integridade desta região e a inativação normal desse cromossomo, evitando o comprometimento do desenvolvimento mental e cognitivo descrito em algumas das portadoras de cromossomo X em anel nas quais o centro de inativação do X está ausente (51,53).
A análise do microssatélite SHOX CA repeat localizado na região intragênica, na porção não traduzida (5UTR) do exon 1 e do microssatélite DXYS234 localizado a 2cM do telômero de Xp (figura 1), confirmou a haploinsuficiência do SHOX, pela ausência dos alelos paternos. A análise do DXYS233 não foi informativa, porque ambos os pais apresentaram o mesmo tamanho de um dos alelos.
Esta paciente está a menos 3,9 DP da média de altura para adolescentes brasileiras, com um comprometimento maior na estatura do que observado em meninas portadoras de mosaicismo 45,X/46,XX. Segundo Ogata (19,54), o efeito da maturação dos estrogênios gonadais nos tecidos ósseos ocorre de uma maneira desbalanceada nas meninas com haploinsuficiência do gene SHOX. Nestas circunstâncias, ocorre fusão prematura das placas de crescimento, sugerindo que o gene SHOX tenha uma função repressora na maturação óssea. Portanto, o comprometimento maior da estatura nas meninas com haploinsuficiência do SHOX está provavelmente relacionada a uma susceptibilidade aumentada à ação que os estrogênios exercem nos tecidos ósseos (19,54). Nos membros superiores, pela haploinsuficiência do SHOX, a ação maior dos estrogênios leva ao encurtamento dos ossos do antebraço, mais evidente durante a puberdade (11,15,54). Ogata (19) ainda cita em seu trabalho uma paciente que tinha diagnóstico de BEI antes da puberdade e que manifestou sinais radiológicos da discondrosteose de Leri Weill durante a puberdade. A mesma evolução foi observada na paciente SLS, na qual a deformidade dos antebraços não havia sido notada pelos pais nem pelo médico acompanhante, por se tornar mais evidente após a puberdade.
As anomalias dos ossos do carpo e do cotovelo foram observadas na menina TBR, com avaliação citogenética normal e sem mosaicismo críptico. A sinostose dos ossos do carpo e alterações da articulação do cotovelo são descritas em displasias ósseas mesomélicas como a síndrome de Mesomelia e Sinostose (OMIM 600383), Síndrome de Sinostose Múltipla (OMIM 186400) e Displasia Mesomélica tipo Kantaputra (OMIM 156233) (55). Em todas estas síndromes, as alterações clínico-radiológicas são mais graves do que as apresentadas pela paciente, que deverá ser investigada para mutações do gene SHOX.
Desde a descrição do papel do gene SHOX no crescimento e desenvolvimento ósseo e do reconhecimento que ele codifica um fator de transcrição expressado em células osteogênicas e em outros tecidos, vários trabalhos têm sido descritos associando mutações e deleções desse gene em 1 a 2,4% de crianças diagnosticadas como portadoras de baixa estatura idiopática (8,11,12,17,21,23,41).
A expressão desse gene no desenvolvimento do esqueleto foi estudada por Clement-Jones e cols. (23) em células de embriões humanos. Estes autores demonstraram a expressão do gene SHOX em membros superiores (cotovelo e punho), nos joelhos e nos primeiro e segundo arcos faríngeos, de onde se originam a maxila, mandíbula e elementos ósseos do ouvido médio e externo. A revisão, por estes autores, de duas pacientes descritas em trabalhos anteriores (8) que apresentavam cariótipo normal e mutações do gene SHOX, demonstrou que, além da baixa estatura, estas pacientes apresentavam no estudo radiológico alterações descritas na ST. Essa associação de sinais radiológicos observados na ST com haploinsuficiência do gene SHOX já havia sido citada por Kosho e cols. em 1999 (21).
Outros autores também têm reforçado a importância de complementar o exame clínico na busca de sinais clínicos e radiológicos achados na ST nas crianças com diagnóstico de BEI (15,19,41).
Concluindo, neste grupo de 10 meninas com diagnóstico inicial de BEI, nós observamos uma menina com uma anomalia estrutural do cromossomo X e um mos 45,X/46,XX e entre as 8 crianças com cariótipo normal, consideramos mosaicismo verdadeiro em duas delas, diagnosticadas por técnicas de citogenética molecular em células de mucosa oral.
Assim, na investigação da BEI, o exame citogenético por técnicas de bandeamento deve ser indicado em todas os casos, principalmente nos que apresentam sinais clínicos descritos na ST como palato ogival, cúbito valgo, deformidade de Madelung, 4º. metacarpo curto e alterações de joelho. Persistindo ainda a dúvida, as portadoras de cariótipo normal em sangue periférico devem ter a investigação citogenética complementada pela técnica de citogenética molecular FISH em outro tecido; as células de mucosa oral são um ótimo alvo.
Como é conhecido que meninas com ST apresentam resposta favorável ao tratamento com hormônio de crescimento, o diagnóstico de uma anomalia numérica ou estrutural do X no grupo de baixa estatura idiopática modificaria a conduta terapêutica, permitindo que, frente a um diagnóstico precoce, pudessem ser incluídas em programas de terapia com hormônio de crescimento (9,28,41). Se existem controvérsias quanto ao ganho de estatura final em pacientes com ST, não há dúvidas que o início do GH no período escolar antecipa um ganho de estatura numa faixa etária importante para a melhora do comportamento psicossocial destas crianças (57).
AGRADECIMENTOS
Os autores agradecem às pacientes e seus responsáveis por aceitarem participar deste trabalho; aos Drs. Cláudio Hoineff e Jane Silveira, do Ambulatório de Crescimento do IEDE pelo apoio e colaboração no acompanhamento das pacientes; à Dra. Ieda Oriolli do Departamento de Genética do Instituto de Biologia da UFRJ pelo incentivo na realização e revisão deste trabalho e pelo apoio na técnica de extração de DNA; aos Drs. Hector N. Seuanez e Miguel Angelo M. Moreira da Divisão de Genética do Instituto Nacional do Câncer do RJ, pelo apoio na realização das técnicas FISH com sonda wcp X e análise de microssatélites; à Dra. Suely R. dos Santos pela realização das técnicas CBG, e à FAPERJ pelo apoio financeiro (E - 26/170. 793/2000-APQ1).
Recebido em 09/06/03
Revisado em 05/09/03
Aceito em 07/09/03
- 1. Ranke MB. Towards a consensus on the definition of idiopathic short stature. Horm Res 1996;45(suppl 2):64-6.
- 2. Attie KMA. Genetic studies in idiopathic short stature. Curr Opin Pediatr 2000;12:400-4.
- 3. Ballabio A, Bardoni B, Carrozzo R, Andria G, Bick D, Campbell L, et al. Contiguous genes syndromes due to deletions in the distal short arm of human X chromosome. Proc Nat Acad Sci USA 1989;86:10001-5.
- 4. Ogata T, Yoshizawa A, Muroya, K, Matsuo N, Fujushima Y, Rappold G, et al. Short stature in a girl with partial monosomy of the pseudoautosomal region distal to DXYS15: further evidence for the assignment of the critical region for a pseudoautosomal growth gene(s). J Med Genet 1995;32:831-4.
- 5. Ellison JW, Wardak, Z, Young MF, Robey PG, Laig-Webster M, Chiong W. PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Mol Genet 1997;6:1341-7.
- 6. Rao E, Weiss B, Fukami M, Merts A, Meder J, Ogata T, et al. FISH deletion mapping defines a 270-kb short stature critical interval in pseudoautosomal region PAR1 on human sex chromosomes. Hum Genet 1997;100:236-9.
- 7. Zinn AR, Tonk VS, Chen Z, Flejter WL, Gardner HA, et al. Evidence for a Turner Syndrome locus at Xp11.2-p22.1. Am J Hum Genet 1998;63:1757-66.
- 8. Rao E, Weiss B, Fukami M, Niesler B, Merts A, Muroya K, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner Syndrome. Nat Genet 1997;16:54-63.
- 9. Binder G, Schwarze CP, Ranke MB. Identification of short stature caused by SHOX deletions and therapeutic effect of recombinant human growth hormone. J Clin Endocrinol Metab 2000;85:245-9.
- 10. Bernasconi S, Maruabu S, Falcinelli C, Milioli S, Iughetti L, Forabosco A. SHOX gene in Leri-Weill syndrome and idiopathic short stature. J Endocrinol Invest 2001;24:737-41.
- 11. Rappold GA, Fukami M, Niesler H, Schiller S, Zumkeller W, Bettendorf M, et al. Deletions of the homeobox gene SHOX (Short Stature Homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab 2002;87:1402-6.
- 12. Ogata T. SHOX defects in idiopathic short stature. J Pediatric Endocrinol Metab 2002;15:1439-40.
- 13. Belin V, Cusin V, Viot G, Girlich D, Toutain S, Moncla A, et al. SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat Genet 1998;19:67-9.
- 14. Cormier-Daire V, Belin V, Cusin V, Viot G, Girlich D, Toutain A, et al. SHOX gene mutations and deletions in dyschondrosteosis or Leri-Weill syndrome. Acta Paediat 1999;433:55-9.
- 15. Müsebeck J, Mohnike K, Beye P, Tönnies H, Neitzel H, Schnabel D, et al. Short stature homeobox-containing gene deletion screening by fluorescence in situ hybridization in patients with short stature. Eur J Pediat 2001;160:561-5.
- 16. Shears DJ, Vassal HJ, Goodman FR, Palmer RW, Reardon W, Superti-Furga A, et al. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat Genet 1998;19:70-3.
- 17. Ross JL, Scott C, Marttlila P, Kowal K, Nass A, Papenhausen P, et al. Phenotypes associated with SHOX deficiency. J Clin Endocrinol Metab 2001;86:5674-80.
- 18. Grigelione G, Schoumans AS, Neumeyer L, Ivarson SA, Eklöf O, Enkvist O, et al. Analysis of short stature homeobox containing gene (SHOX) and auxological phenotype in dyschondrosteosis and isolated Madelung. Hum Genet 2001;109:551-8.
- 19. Ogata T, Muroya K, Sasaki G, Nishimura G, Kitoh H, Hattori T. SHOX nullyzygosity and haploinsufficiency in a Japanese family: implication for the development of Turner skeletal features. J Clin Endocrinol Metab 2002;87:1390-4.
- 20. Zinn AR, Wei F, Zhang L, Elder FF, Scott CI, Marttila P, et al. Complete SHOX deficiency causes Langer mesomelic dysplasia. Am J Med Genet 2002;110:158-63.
- 21. Kosho T, Muroya K, Nagai T, Fujimoto M, Yokoya S, Sakamoto H, et al. Skeletal features and growth patterns in 14 patients with haploinsufficiency of SHOX: implications for the development of Turner Syndrome. J Clin Endocrinol Metab 1999;84:4613-21.
- 22. Blaschke JR, Rappold GA. Growth, Leri-Weill and Turner Syndrome. Trends Endocrinol Metab 2000;11:227-30.
- 23. Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, et al. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet 2000;9:695-702.
- 24. Ogata T. SHOX haploinsufficiency and its modifying factors. J Pediatric Endocrinol Metab 2002;15:1289-94.
- 25. Rimoin DL, Graham J Jr. Short Stature. In: Rimoin DL, editor. Principles and practice of medical genetics, 2nd ed. London:Churchil Livingstone, 1990p.225-34.
- 26. Gicquel C, Gaston V, Cabrol S, Le Bouc Y. Assessment of Turners Syndrome by molecular analysis of the X chromosome in growth-retarded girls. J Clin Endocrinol Metab 1998;83:1472-6.
- 27. Azcona C, Bareille P, Stanhope R. Turners syndrome mosaicism in patients with a normal blood lymphocyte karyotype. Br Med J 1999;318:856-7.
- 28. Saenger P. Growth promoting strategies in Turners Syndrome. J Clin Endocrinol Metab 1999;84:4345-8.
- 29. Blancato JK. Fluorescence in situ hybridization. In: Gersen LG, Keagle MB, editors. The principles of clinical cytogenetics. New Jersey:Humana Press,1999p.443-71.
- 30. Nath J, Johnson KL. A review of fluorescence in situ hybridization (FISH): current status and future prospects. Biotech Histochem 1999;75:54-78.
- 31. Ogata T, Matsuo N, Fukushima Y, Saito M, Nose O, Mihaaru N, et al. FISH analysis for apparently simple terminal deletions of the X chromosome: identification of hidden structural abnormalities. Am J Med Genet 2001;104:307-11.
- 32 Schad CR, Kuffel DG, Wyatt WA, Zinsmeister AR, Jenkins RB, Dewald GW, et al. Application of fluorescent in situ hybridization with X and Y chromosome specific probes to buccal smear analysis. Am J Med Genet 1996;66:187-92.
- 33. Werner M, Wilkens L, Aubele M, Nolte M, Zitzelsberger H, Komminoth P. Interphase cytogenetics in pathology: principles, methods, and applications of fluorescence in situ hybridization (FISH). Histochem Cell Biol 1997;108:381-90.
- 34. Ramos HIB. Estudo citogenético-molecular pela técnica de hibridizaçăo in situ fluorescente (FISH) na investigaçăo etiológica de malformaçőes congęnitas e patologias pediátricas correlatas. Tese (Doutorado) - Universidade Federal Fluminense, 1999
- 35. Migeon BR, Luo S, Jani M, Jeppesen P. The severe phenotype of females with tiny ring X chromosomes is associated with inability of these chromosomes to undergo X inactivation. Am J Hum Genet 1994;55:497-504.
- 36. Koch J, Hindkjćr J, Křlvraa S, Bolund L. Construction of a panel of chromosome-specific oligonucleotide probes (PRINS-primers) useful for the identification of individual human chromosomes in situ Cytogen Cell Genetic 1995;71:142-7.
- 37. Pellestor F, Giardet A, Lefort G, Andréo B, Charlieu JP. Use of primed in situ labeling (PRINS) technique for a rapid detection of chromosomes 13, 16, 18, 21, X and Y. Hum Genet 1995;95:12-7.
- 38. Musio A, Baroli P, Sbrana I. Primed in situ labeling (PRINS): a method for rapid identification and quantification of human chromosomes in both lymphocytes and sperm nuclei. Genome 1998;41:739-41.
- 39. Cervantes A, Guevara-Yanes R, Lopes M, Monroy N, Aguinaga M, Valdez H, et al. PCR-RINS-FISH analysis of abnormal sex chromosomes, in eight patients with Turners phenotype. Clin Genet 2001;60:385-92.
- 40. Harrer T, Schwinger E, Memmocle L. A new technique for cyclic in situ amplification and a case report about amplification of a single copy gene sequence in human metaphase chromosomes through PCR-PRINS. Hum Mutation 2001;17:131-40.
- 41. Ezquieta B, Cueva E, Oliver A, Gracia R. SHOX intragenic microsatellite analysis in patients with short stature. J Pediatric Endocrinol Metab 2002;15:139-48.
- 42. Marques RM, Marcondes E, Berquó E, Prani R, Yunes J. Crescimento e desenvolvimento pubertário em crianças e adolescentes brasileiros. Citado por: Monte O, Longui CA, Callliari LP, editores. Endocrinologia para o pediatra, 2Ş ed. Săo Paulo:Editora Atheneu, 1998p.24-47; 536-8.
- 43. Kosowicz J. The deformity of the medial tibial condyle in nineteen cases of gonadal dysgenesis. J Bone Joint Surgery 1960;42-A:600-4.
- 44. Spranger JW, Langer LO, Wiedemann HR. Skeletal dysplasia with predominant involvement of single sites or segments. In: Spranger JW, Langer LO, Wiedemann HR, editors. Bone dysplasias. An atlas of constitutional disorders of skeletal development, New York: Saunders, 1973
- 45. Bender MA. Methods in human cytogenetics. Arq Bras Endocrinol Metab 1965;14:47-72.
- 46. Seabright M. A rapid banding technique for human chromosomes. Lancet 1971;2:971-2.
- 47. Summer AT. A simple technique for demonstrating centromeric heterochromatine. Exper Cell Res 1972;75:304-6.
- 48. Wienberg J, Stanyon R. Comparative panting of mammalian chromosomes. Curr Opin Genet Develop 1997;7:784-91.
- 49. Miller AS, Dykes SS, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16:1215-9.
-
50ICSN 1995. An international system for human cytogenetics nomenclature. Report of the standing committee on human cytogenetic nomenclature publication Basel: Karger AG, 1995
- 51. Crolla JA, Llerena JC. A mosaic 45,X/46,X,r(X) karyotype investigated with X and Y centromere-specific probes using a non-autoradiographic in situ hybridization technique. Hum Genet 1988;81:81-4.
- 52. Boucher CA, Sargent CA, Ogata T, Affara NA. Breakpoint analysis of Turner patients with partial Xp deletions: implications for the lymphoedema gene location. J Med Genet 2001;38:591-8.
- 53. Kunstsi J, Skuse D, Elgar K, Morris E, Turner C. Ring-X chromosomes: their cognitive and behavioral phenotype. Ann Hum Genet 2000;64:295-305.
- 54. Ogata T, Matsuo N, Nishimura G. SHOX haploinsufficiency and overdosage: impact of gonadal function status. J Med Genet 2001;38:1-6.
-
55OMIM. Online Mendelian Inheritance in Man, 2003 http://www.ncbi.nlm.nih.gov/OMIM
- 56. Elsheikh M, Dunger DB, Conway GS, Wass JAH. Turner's syndrome in adulthood. Endocr Rev 2002;23:120-40.
- 57. Siegel PT, Coppeer R, Stabler B. The psychological consequences of Turner syndrome and review of the National Cooperative Growth Study Psychological Substudy. Pediatr 1998;102:488-93.
Datas de Publicação
-
Publicação nesta coleção
10 Mar 2004 -
Data do Fascículo
Dez 2003
Histórico
-
Revisado
05 Set 2003 -
Recebido
09 Jun 2003 -
Aceito
07 Set 2003