Abstracts
Congenital hypothyroidism affects about 1:3000-1:4000 infants. Screening programs now permit early recognition and treatment, thus avoiding the disastrous consequences of thyroid hormone deficiency on brain development. In about 85%, congenital hypothyroidism is associated with developmental defects referred to as thyroid dysgenesis. They include thyroid (hemi)agenesis, ectopic tissue and thyroid hypoplasia. Thyroid dysgenesis is usually sporadic; in only 2% it occurs in a familial fashion. It can be caused by mutations in transcription factors that are essential for the development and function of thyroid follicular cells. Thyroid hypoplasia can also result from resistance to TSH at the level of the thyrocytes. Defects in the steps required for thyroid hormone synthesis within thyroid follicular cells are referred to as dyshormonogenesis and account for about 10-15% of congenital hypothyroidism. In contrast to thyroid dysgenesis, affected patients typically present with goitrous enlargement of the thyroid. The defects leading to dyshormonogenesis typically display a recessive mode of inheritance. Careful clinical, biochemical and molecular analyses of patients with syndromic and non-syndromic forms of thyroid dysgenesis and dyshormonogenesis have significantly enhanced our understanding of the wide spectrum of pathogenetic mechanisms underlying congenital hypothyroidism and provide unique insights into the (patho)physiology of thyroid development and hormone synthesis.
Thyroid dysgenesis; Dyshormonogenesis; Congenital hypothyroidism; Thyroglobulin
Hipotireoidismo congênito afeta cerca de 1:3.000-1:4.000 recém-nascidos. Atualmente, programas de triagem neonatal permitem o reconhecimento e tratamento precoces, evitando suas conseqüências desastrosas no desenvolvimento cerebral. Em cerca de 85% dos pacientes, o hipotireoidismo congênito está associado à defeitos no desenvolvimento da tireóide referidos como disgenesia tireoideana. A disgenesia tireoideana ocorre geralmente de forma esporádica; somente 2% dos casos apresentam caráter familial. Podem ser causados por mutações nos fatores de transcrição que são essenciais para o desenvolvimento e função das células foliculares tireoideanas. Hipoplasia da tireóide pode também resultar de resistência tireoideana ao TSH. Defeitos na síntese dos hormônios tireoideanos são referidos como disormonogênese tireoideana e concorrem para 10-15% dos casos de hipotireoidismo congênito. Os pacientes usualmente apresentam bócio, ao contrário da disgenesia tireoideana. Tipicamente, os defeitos que causam disormonogênese apresentam herança recessiva. Uma série de estudos recentes aumentou significativamente nosso entendimento dos mecanismos patogênicos do hipotireoidismo congênito, oferecendo novos insights sobre a fisiopatologia do desenvolvimento e síntese hormonal tireoideanos.
Dishormogênese; Disgenesia; Tireoglobulina; Hipotireoidismo congênito
ATUALIZAÇÃO
Thyroglobulin gene mutations and other genetic defects associated with congenital hypothyroidism
Mutações no gene da tireoglobulina e outros defeitos genéticos associados com hipotireoidismo congênito
Jussara Vono-Toniolo; Peter Kopp
Division of Endocrinology, Metabolism & Molecular Medicine, Feinberg School of Medicine, Northwestern University, Chicago, USA (JV-T, PK); e Laboratório Molecular de Tireóide (LIM-25), Departamento de Endocrinologia, Hospital das Clínicas, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brasil (JV-T)
Endereço para correspondência Endereço para correspondência Peter Kopp Division of Endocrinology, Metabolism & Molecular Medicine Feinberg School of Medicine Northwestern University Tarry 15 303 East Chicago Avenue Chicago IL 60611 Fax (312) 908-9032 e.mail: p-kopp@northwestern.edu
ABSTRACT
Congenital hypothyroidism affects about 1:3000-1:4000 infants. Screening programs now permit early recognition and treatment, thus avoiding the disastrous consequences of thyroid hormone deficiency on brain development. In about 85%, congenital hypothyroidism is associated with developmental defects referred to as thyroid dysgenesis. They include thyroid (hemi)agenesis, ectopic tissue and thyroid hypoplasia. Thyroid dysgenesis is usually sporadic; in only 2% it occurs in a familial fashion. It can be caused by mutations in transcription factors that are essential for the development and function of thyroid follicular cells. Thyroid hypoplasia can also result from resistance to TSH at the level of the thyrocytes. Defects in the steps required for thyroid hormone synthesis within thyroid follicular cells are referred to as dyshormonogenesis and account for about 10-15% of congenital hypothyroidism. In contrast to thyroid dysgenesis, affected patients typically present with goitrous enlargement of the thyroid. The defects leading to dyshormonogenesis typically display a recessive mode of inheritance. Careful clinical, biochemical and molecular analyses of patients with syndromic and non-syndromic forms of thyroid dysgenesis and dyshormonogenesis have significantly enhanced our understanding of the wide spectrum of pathogenetic mechanisms underlying congenital hypothyroidism and provide unique insights into the (patho)physiology of thyroid development and hormone synthesis.
Keywords: Thyroid dysgenesis; Dyshormonogenesis; Congenital hypothyroidism; Thyroglobulin
RESUMO
Hipotireoidismo congênito afeta cerca de 1:3.000-1:4.000 recém-nascidos. Atualmente, programas de triagem neonatal permitem o reconhecimento e tratamento precoces, evitando suas conseqüências desastrosas no desenvolvimento cerebral. Em cerca de 85% dos pacientes, o hipotireoidismo congênito está associado à defeitos no desenvolvimento da tireóide referidos como disgenesia tireoideana. A disgenesia tireoideana ocorre geralmente de forma esporádica; somente 2% dos casos apresentam caráter familial. Podem ser causados por mutações nos fatores de transcrição que são essenciais para o desenvolvimento e função das células foliculares tireoideanas. Hipoplasia da tireóide pode também resultar de resistência tireoideana ao TSH. Defeitos na síntese dos hormônios tireoideanos são referidos como disormonogênese tireoideana e concorrem para 10-15% dos casos de hipotireoidismo congênito. Os pacientes usualmente apresentam bócio, ao contrário da disgenesia tireoideana. Tipicamente, os defeitos que causam disormonogênese apresentam herança recessiva. Uma série de estudos recentes aumentou significativamente nosso entendimento dos mecanismos patogênicos do hipotireoidismo congênito, oferecendo novos insights sobre a fisiopatologia do desenvolvimento e síntese hormonal tireoideanos.
Descritores: Dishormogênese; Disgenesia; Tireoglobulina; Hipotireoidismo congênito
IN ALL VERTEBRATES, THYROID hormone is essential for normal development, growth and metabolic homeostasis. This requires normal development and function of the hypothalamic-pituitary-thyroid axis, as well as an adequate nutritional intake of iodine. In iodine sufficient regions, permanent congenital hypothyroidism affects about 1:3000 to 1:4000 newborns (1-4). In about 85% of all cases, congenital hypothyroidism is associated with developmental defects that are referred to as thyroid dysgenesis. They include thyroid (hemi)agenesis, ectopic tissue and hypoplasia. Thyroid dysgenesis usually occurs in a sporadic fashion; in only 2% it is a familial disorder. In 10% to 15%, congenital hypothyroidism is caused by Inborn errors of metabolism in one of the intricate steps required for normal hormone synthesis. This category of defects, which typically displays a recessive mode of inheritance, is referred to as dyshormonogenesis.
This review provides an overview of the multiple genetic defects causing congenital hypothyroidism with particular emphasis on the consequences of mutations in the thyroglobulin gene (figure 1).
DEVELOPMENTAL DEFECTS OF THE THYROID
Thyroid follicular cells depend on the concomitant presence of three transcription factors for normal development and gene expression: PAX8, TTF1/ NKX2.1 and TTF2/FOXE1 (5,6). Mutations in these transcription factors have been identified in human patients with syndromic and non-syndromic forms of thyroid dysgenesis. The importance of these transcription factors for normal thyroid development and function is further illustrated by murine knockout models that result in similar phenotypes.
PAX8
Heterozygous mutations in PAX8, a paired domain transcription factor involved in thyroid development and expression of the thyroperoxidase (TPO) and thyroglobulin (TG) genes, have been documented and characterized in sporadic and familial patients with thyroid hypoplasia or ectopy (7-10). These mutations may be inherited in an autosomal dominant fashion (7,9). The phenotype of carriers of the same mutation within a family may, however, be very variable. For example, a Brazilian girl with thyroid hypoplasia and overt congenital hypothyroidism was found to harbor a mutation in the PAX8 DNA binding domain inactivating its DNA binding and transactivation properties (Q40P) (9). Surprisingly, her mother was found to have the same mutation, but she only had mild, adult-onset autoimmune hypothyroidism. This finding suggests that the phenotype may be variable in carriers of the mutation (variable expressivity), or, alternatively, that not all carriers of the mutation develop an abnormal phenotype (incomplete penetrance) (9).
In contrast to humans with monoallelic PAX8 mutations, mice heterozygous for a disrupted Pax8 allele do not display an abnormal phenotype (11). Homozygous knockout mice have severe congenital hypothyroidism with a hypoplastic gland that is located in the correct position.
TTF1/NKX2.1
Thyroid transcription factor 1 is a homeobox domain transcription factor of the NKX2 family. Patients heterozygous for chromosomal deletions of 1q12-13.2 or point mutations in the TTF1/NKX2.1 gene present with mild congenital hypothyroidism with a thyroid of normal size and location (12-15). They present with neonatal respiratory distress and develop neurological alterations that include ataxia/choreoathetosis, truncal apraxia, and mental retardation. The fact that one normal TTF1 allele is insufficient for normal neurological and thyroid function is an example of haploinsufficiency, a commonly observed phenomenon associated with mutations in transcription factors (16). Heterozygous TTF-1 mutations have also been identified as the molecular cause of hereditary chorea (17). It is currently unclear whether these patients also have subtle alterations in their thyroid function tests.
Homozygous Ttf1 knockout mice survive throughout gestation, but die at birth from respiratory failure (18). They have a severe phenotype that includes an absent forebrain and pituitary gland. The lung is severely hypoplastic and consists of a sac-like structure without bronchioli, alveoli or lung parenchyma. The thyroid gland is absent (18). Mice that are heterozygous for a disrupted Ttf1 allele have a phenotype that is similar to humans with only one functioning copy of this gene (14). Compared with wild type mice, Ttf1(+/-) mice display an abnormal coordination and elevated TSH levels.
TTF2/FOXE1
Homozygosity for recessive mutations in the forkhead/winged-helix domain transcription factor FOXE1, traditionally referred to as thyroid transcription factor 2 (TTF2), results in a syndromic form of thyroid dysgenesis with the eponym Bamforth-Lazarus syndrome (19,20). This phenotype includes thyroid agenesis, cleft palate, choanal atresia, bifid epiglottis and spiky hair.
Ttf2 knockout mice die within 48 hours after birth, probably because of respiratory failure secondary to cleft palate (21). Their thyroid glands are either sublingual or completely absent suggesting that agenesis and ectopy can be caused by the same molecular defect.
HYPOTHALAMIC AND PITUITARY DEFECTS
TRH Deficiency
A few patients with congenital hypothyroidism and isolated TRH deficiency without destructive hypothalamic lesions have been reported (22). The molecular defect underlying these cases remains elusive and could affect synthesis or secretion of TRH.
TRH Receptor
Resistance to TRH in pituitary thyrotrophs was discovered in a boy with isolated central hypothyroidism (23). Mutational analysis of the TRH receptor (TRHR) gene revealed compound heterozygous point mutations that inactivate the TRH receptor.
TSH ß Subunit
TSH is formed of an a subunit that is common to the other glycoprotein hormones and a specific ß subunit. Isolated hereditary TSH deficiency is a rare cause of central hypothyroidism and can be caused by recessive mutations in the TSHß chain (24-26). In these patients, TSH is unmeasurable or very low, and the administration of TRH does not result in a rise in serum TSH. The levels and the function of the other pituitary hormones are normal, including an adequate rise of prolactin in response to TRH. Among the five currently known mutations, some are recurrent in certain populations suggesting a founder effect, while others have been found independently in sporadic and familial patients from different ethnic origins. A subset of these mutations is predicted to disrupt heterodimerization with the glycoprotein hormone a-chain, while others lead to premature truncations (25).
Combined Pituitary Hormone Deficiency
Genetic defects in the development and function of the pituitary gland can result in various forms of Combined Pituitary Hormone Deficiency (CPHD) (27). Patients with CPHD present with impaired production and secretion of one or several anterior pituitary hormones that may include TSH. CPHD has been documented in patients with mutations in several transcription factors involved in pituitary development and hormone expression, specifically POU1F1 (PIT1), PROP1, LHX3, LHX4, and HESX1 (for review: (27)). Among these defects, PROP1 mutations are by far the most common and they have been reported in several Brazilian families (28-32).
RESISTANCE TO TSH
The response to bioactive TSH may be impaired at the level of the thyrocyte (figure 2) (33-35). Total insensitivity to TSH results in a small hypoplastic thyroid gland and reduced synthesis and secretion of thyroid hormones. It has to be emphasized that a similar morphologic and biochemical phenotype may occur in patients with mutations in PAX8, a fact that illustrates the limitations of morphologic criteria for the classification of these defects. Resistance to TSH may be caused by various molecular mechanisms that include mutations in the TSH receptor and the GNAS1 (Gsa subunit) gene. Unresponsiveness to TSH can also be inherited as an autosomal dominant trait, but the molecular defect remains to be defined (36).
TSH Receptor
In a subset of patients with insensitivity to TSH the molecular cause consists of recessive mutations in the TSH receptor that are partially or completely inactivating. In partial resistance, TSH levels are elevated, but the peripheral hormones are normal, a constellation referred to as euthyroid hyperthyrotropinemia (33). In these patients, the size of the thyroid is normal or enlarged. More severe homozygous or compound heterozygous inactivating mutations in the TSHR have been found in several patients with overt hypothyroidism and thyroid hypoplasia (35,37).
Gs a Subunit
Resistance to TSH, in combination with resistance to PTH, LH, FSH and the morphologic features of Albright's hereditary osteodystrophy (short stature, brachydactyly, ectopic calcifications), also occurs in Pseudohypoparathyroidism Ia (PHP Ia) (38). The molecular basis consists of inactivating mutations in the maternal copy of the GNAS1 (Gsa subunit) gene, which is imprinted in a tissue-specific manner (38).
DYSHORMONOGENESIS
Mutations in any of the steps involved in thyroid hormone synthesis may result in compensated or overt congenital hypothyroidism (figure 2). In contrast to developmental defects of the thyroid, absence or bioinactivity of TSH, or TSH resistance, patients with mutations in thyroidal genes involved in thyroid hormone synthesis typically present with goitrous enlargement of the thyroid gland because of TSH-mediated growth stimulation of thyroid follicular cells.
Sodium Iodide Symporter (NIS)
Normal iodide uptake at the basolateral membrane by the perchlorate-sensitive sodium/iodide symporter (NIS) is an essential and rate-limiting step in thyroid hormone synthesis (39). Several individuals with hypothyroidism associated with impaired iodide uptake were found to be homozygous or compound heterozygous for inactivating mutations in the NIS gene (39). If untreated, these patients present with a diffuse or nodular goiter. Functional testing reveals little or no uptake of radioiodine, and a decreased saliva/serum radioiodine ratio.
Pendrin
Efflux of iodide at the apical membrane of thyroid follicular cells is at least in part mediated by pendrin (SCL26A4), a member of the Solute Carrier Family 26A (40). Mutations in the PDS/SCL26A4 gene cause Pendred's syndrome, an autosomal recessive disorder traditionally defined by the triad of sensorineural congenital deafness, goiter, and a partially positive perchlorate test (40). The partial discharge of radioiodine after the administration of perchlorate indicates that the gland has an impaired ability to organify iodide. Although some patients with Pendred's syndrome present with congenital hypothyroidism, the majority of individuals are clinically and biochemically euthyroid. It may be difficult to establish the diagnosis on clinical grounds since phenocopies, i.e. individuals with an identical phenotype caused by other etiologies, do exist. This has, e.g., been illustrated by a very large, highly inbred kindred from Northeastern Brazil (41). Multiple individuals presented with deafness and goiter. Molecular analyses revealed that a subset of these individuals were homozygous for an inactivating mutation in the PDS/SCL26A4 gene. Others only had only one affected allele or were homozygous for the wild type allele indicating that deafness and goiter were the consequence of distinct pathogenetic mechanisms (41).
Thyroperoxidase
Thyroperoxidase, a glycosylated hemoprotein located at the apical membrane facing the follicular lumen, iodinates tyrosine residues in thyroglobulin (TG), and the coupling of iodinated tyrosines to generate T4 and catalyzes T3. TPO defects are among the most frequent causes of inborn errors of thyroid hormone synthesis. Homozygous or compound heterozygous mutations in the TPO gene have been reported in numerous families with a partial or total organification defect (42,43). Total iodide organification defects occur in ~1:66,000 neonates, and the majority of these infants have a defective TPO gene (43).
H2O2-Generating System, THOX2 Gene Mutations
The iodination and coupling reactions are dependent on H2O2 as an essential cofactor. Recently, two NADPH oxidases that are part of the H2O2-generating system, THOX1 and THOX2, have been cloned (44-46). Heterozygous loss of function mutations in the THOX2 gene result in a mild and transient form of congenital hypothyroidism (47). Biallelic THOX2 mutations are associated with a severe phenotype and confirm that H2O2 is essential for iodide organification (47). As of yet, there are no reported mutations in THOX1. In two affected siblings from a Brazilian family presenting with hypothyroidism, goiter, and iodine organification defects, NADPH oxidase activity measured in tissue slices was nearly undetectable suggesting that these subjects may have a defect in H2O2 generation (48).
Thyroglobulin
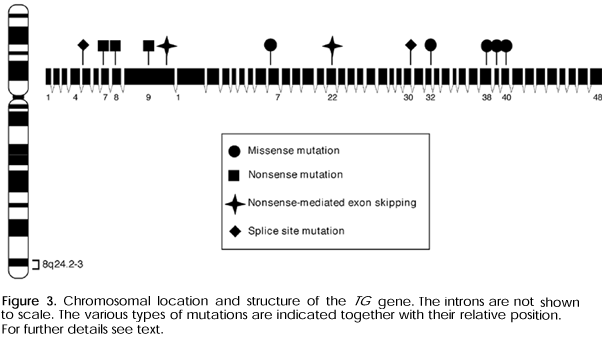
Thyroglobulin (TG) is a key element in thyroid hormone synthesis and storage. It is encoded by a very large gene spanning about 270kb and containing 48 exons (49). Recessive mutations in the TG gene have been reported in a number of human patients, as well as animal models, and are discussed in more detail in the remainder of this review (see below).
Dehalogenase
After entering the follicular cell, TG is hydrolyzed, and T4 and T3 are secreted into the blood at the basolateral membrane. The iodotyrosines MIT and DIT, which are much more abundant in the TG molecule, are deiodinated by an intrathyroidal dehalogenase and recycled for hormone synthesis. Several patients with leakage of MIT and DIT from the thyroid and urinary secretion of these metabolites have been identified (50). The disorder is recessive, but the intrathyroidal dehalogenase has not been cloned.
THYROGLOBULIN GENE AND PROTEIN STRUCTURE
Chromosomal Location and Gene Structure
The human TG gene is located on chromosome 8q24.2-8q24.3 (51-53). It is unusually large spanning about 270kg and contains 48 exons separated by introns of up to 65kb (54-56) (Gene Bank accession number: NT_ 008046). The synthesis of the TG gene is controlled by transcription factors such as TTF-1/NKX2.1, TTF-2/FOXE1 and PAX 8 (5,6). The full-length 8.5kb messenger RNA (mRNA) sequence contains a 41-nucleotide 5'-untranslated segment preceding an open reading frame of 8307 bases and a 3'-untranslated segment ranging from 101 to 120bp (Gene Bank accession number: NM 003235.2) (57,58). The TG gene contains multiple polymorphisms and recent studies established linkage of the TG locus with autoimmune thyroid disease (AITD) (49,59,60). These observations suggest that alterations in the TG gene, together with variations in other genes and environmental factors, may play a role in the pathogenesis of AITD (59,60).
Protein Structure
The TG monomer is composed of a 19-amino acid signal peptide followed by 2749 residues containing 66 tyrosines (49). It contains three distinct regions that each contains different types of repetitive elements and the carboxyterminus is highly homologous to acetylcholinesterase (56,57,61,62). This structure suggests that the TG gene arouse from the fusion of two ancestral DNA sequences (63).
After translation of the mRNA, the TG peptide is targeted to the endoplasmic reticulum (ER) by its signal peptide of 19 amino acids, and it is submitted to folding, glycosylation and dimerization. Properly folded TG dimers migrate to the Golgi apparatus where it is submitted to further glycosylation. In the follicular lumen, is found as a glycosylated dimer of 660kDa (19S dimer) (64).
Intrafollicular Hormone Synthesis
Synthesis of thyroxine (T4) and triiodothyronine (T3) occurs on TG as matrix (65). In the follicle, selected tyrosyl residues of the TG polypeptide are iodinated by TPO. This step, a reaction referred to as organification, results in the formation of monoiodotyrosines (MIT) and diiodotyrosines (DIT). The next step in thyroid hormone synthesis consists of the coupling of two DIT residues to form T4, or one DIT and one MIT to form T3. This process is also catalyzed by TPO (65). During the coupling reaction, a tyrosyl residue donates its iodinated phenyl group to become the outer ring of the iodothyronine amino acid at an acceptor site, leaving dehydroalanine or its derivative at the donor position. Four main hormonogenic sites have been identified in human TG and are located at positions 5, 1291, 2554, 2568 and 2747 in the mature peptide lacking the signal peptide (66). The most important T4 forming site is at tyrosine 5 and there is evidence that the tyrosine 130 is the dominant donor site (67).
Further processing of TG requires its reentry into the thyroid cell, through vesicular internalization with subsequent fusion with lysosomes resulting in breakdown of the TG-iodothyroxine complexes, releasing thyroid hormones (64,68). Vesicular internalization is initiated by nonselective fluid phase uptake and by receptor-mediated endocytosis (69).
MUTATIONS IN THE HUMAN THYROGLOBULIN GENE
Clinical Presentation
TG gene defects are transmitted in an autosomal recessive manner (70,71). Affected individuals are therefore homozygous or compound heterozygous for mutations in the TG gene. Not surprisingly, mutations have been documented more commonly in the offspring of consanguineous parents. Thyroid dyshormonogenesis caused by TG mutations may be associated with congenital goiter or lead to formation of goiter later in life unless treatment with levothyroxine occurs early. Goiters are often remarkably large and display continuous growth. Symptoms caused by compression of adjacent neck structures can occur. The degree of hypothyroidism is variable; it can be severe, but in a few instances peripheral thyroid hormone concentrations may be normal. The radioiodine uptake is elevated indicating an upregulation of NIS secondary to the chronic TSH stimulation. In patients evaluated with a perchlorate discharge test, there is no increased release of radioiodine after administration of the competitor since the organification process itself is not affected. The serum TG levels are usually low or in the low normal range (71).
TG Gene Mutations in Humans
In a patient presenting with hypothyroidism, congenital goiter, and a marked impairment of TG synthesis, Ieiri and cols. documented for the first time a naturally occurring human TG gene mutation in 1991 (70). The parents were first degree cousins and two of her five siblings presented with a similar phenotype. After demonstrating linkage to the TG gene, analysis of the TG mRNA obtained from the goitrous tissue demonstrated that it was reduced in size and sequencing of the cDNA revealed that exon 4 was missing. Sequence analysis of genomic DNA then revealed a mutation in the splice acceptor of intron 3 consisting of a transversion of cytosine to guanine at position 3 (IVS3-3C>G). This splice site mutation leads to skipping of exon 4, a region that coded for one of the important donor tyrosines, without affecting the remainder of the reading frame (70).
An outbred Brazilian family with two affected siblings presenting with congenital goiter and hypothyroidism due to abnormal TG synthesis was studied extensively at the molecular level (72,73). Studies of TG transcripts indicated that one of the alleles was found to lack exon 22. Exon 22 was then found to contain a nonsense mutation at position 4626 (4246C>T) that introduces a premature stop codon at position 1530 of the TG precursor (C1530X; this mutation was originally described as C1510X. The original description was based on the amino acid numbering without signal peptide. According to new nomenclature recommendations, numbering should start at methionine 1 and both numberings are indicated in this review (74)). The mutation in the other allele was not identified in these reports. The explanation for the shortened mRNA on the allele carrying the missense mutation is provided by the phenomenon of nonsense-mediated altered splicing (75). Nonsense-mediated altered splicing is defined by excision of the exon harboring the mutation and can result in the generation of a transcript that may have a partial function. As discussed below, this phenomenon has also been documented in the Afrikander cattle (76).
In a consanguineous family with two affected individuals, the TG mRNA was found to lack 138 nucleotides corresponding to exon 30 (77). Analysis of genomic DNA showed a point mutation in the splice donor site of intron 30 consisting of a guanine to thymine transversion at position +1 (IVS30+1G>T). The skipping of exon 30 does not affect the reading frame of the resulting mRNA and generates a TG polypeptide chain that is shortened by 46 residues (78). Immunohistochemical and electron microscopic analyses demonstrate that the mutated TG is retained in the ER of thyroid follicular cells (79). The folding process is submitted to a quality control in the ER that is exerted by molecular chaperones (80). Misfolded TG is accumulated in the ER and then translocated back into the cytoplasm to undergo degradation by the proteasome system, a process referred to as ER-associated degradation or ERAD (80,81).
Three offspring of a consanguineous marriage presenting with mild hypothyroidism associated with defective TG synthesis were found to have a premature stop codon in exon 7 (886C>T; R296X; originally described as p.R277X) (82). This truncated TG still contains the main hormonogenic sites in the mature polypeptide (acceptor tyrosine 5 and donor tyrosine 149) and appears to retain partial hormone synthesis. The partially retained hormone synthesis of this mutant is in agreement with a study using a 26kDa TG aminoterminal peptide, which is also able to allow T4 synthesis (83).
Two unrelated patients with congenital goiter were found to be homozygous for a point mutation (c.3790T>C; originally described as c.3787T>C) leading to substitution of cysteine 1264 in the precursor by arginine (84,85). One of these patients was euthyroid, the other individual had mild hypothyroidism, but both had undetectable TG levels. As demonstrated by sensitivity to digestion with endoglycosidase H and formation of high molecular aggregates, this mutation is retained in the ER (84,85).
Two sisters presenting with euthyroid adenomatous goiter and increased serum TG levels, were found to harbor a thymine to adenine substitution at nucleotide 5986 of the TG cDNA (c.5986T>A; originally described as c.5983T>A) resulting in an amino acid substitution from cysteine to serine at codon 1996 (p.C1996S; originally described as p.C1995S) (85). The p.C1996S TG is only partially resistant to endoglycosidase H treatment, and a fraction of the protein is transported to the Golgi and, as reflected by the slightly increased serum levels, secreted into the circulation (85).
The coincidental intrauterine detection of a fetal goiter by ultrasound led to the detection of compound heterozygous TG gene mutations in the index patient and her younger brother in a recent study by Caron et al. (86). The goiter was first observed at six month of gestation and cordocentesis revealed severe hypothyroidism of the fetus. Despite repeated intraamniotic injections of 200µg levothyroxine, the neonate had a TSH of 284mU/l. Similar findings were observed in a second pregnancy. During the second gestation, intraamniotic injection of 500µg levothyroxine at 32 and 36 weeks led to a significant reduction of the TSH, which was 472mU/l at 29 weeks and 39mU/l at birth. The clinical findings and a very low serum TG led to the suspicion that a TG defect could be the cause of the goitrous hypothyroidism. Sequence analysis of genomic DNA obtained from the two siblings revealed a paternal TG gene deletion 1143delC in exon 9 resulting in a frameshift beginning at residue 381 that generates a stop codon at position 401 (G381fsX401 originally described as G362fsX382) and a 6725G>A transition in the maternal allele that leads to a substitution of arginine 2242 by histidine (R2242H; originally described as R2223H) (86).
Recently, we have studied five patients from four unrelated Brazilian families with goiter and variable degrees of thyroid function alterations, but low basal and TSH-stimulated TG levels suggesting the possibility of a TG gene defect (87,88). In addition, their radioiodine uptake was elevated indicating an activation of the iodine concentration mechanism, and the perchlorate test was negative indicating a normal organification process. The index patient of the first family, an inbred kindred from the state of Bahia, presented with congenital goiter, but was euthyroid (total T3 100ng/dl; total T4 7.0mg/dl; free T4 1.1ng/dl; TG 3.9ng/ml; 131I uptake 54% at 2h and 79% at 24h) (87). At age 22, he underwent thyroidectomy because of his large goiter that had a volume of ~70 cm3 on ultrasound. One of his first-degree cousins had a similar phenotype. Total thyroidal RNA was reverse transcribed and the whole TG cDNA was then amplified by PCR generating 20 overlapping fragments covering the complete coding region of 8307bp. Direct sequence analysis revealed the presence of three novel nucleotide substitutions that were present in both alleles. The first one, a transition of guanine to adenine at position 113 in exon 2 results in a substitution of arginine 38 by lysine in the precursor (R38K), respectively R19K in the mature protein. Although this substitution results in a rather discrete amino acid change, it is in close vicinity to the tyrosine residue 5, which forms the preferential acceptor site, involved in T4 formation. At position 2561, a transition of guanine to adenine in exon 10 leads to a substitution of arginine 854 by glutamine (R854Q). Interestingly, the bovine wild type TG contains a glutamine at this position suggesting that this alteration may reflect a polymorphism. Lastly, a transversion of guanine to cytosine at position 7414 in exon 43 results in the substitution of valine 2471 by leucine (V2471L). This conservative amino acid change may also be a polymorphism. Analysis of genomic DNA confirmed the presence of these alterations and mutational analysis of his relatives indicates that the substitutions segregate with the phenotype. We hypothesize that the R38K mutation may be causing a partial impairment of TG synthesis, secretion or function, and that R854Q and V2471L could be simple polymorphisms. Further elucidation of the molecular consequences of the three TG alterations requires in vitro analyses (87).
Sequence analysis of reverse transcribed mRNA obtained from the propositi of the three other families led to the detection of the same homozygous nucleotide substitution 6701C>A in exon 38, which results in substitution of alanine by asparagine at position 2234 (A2234N), in all three of index patients (88).
The phenotype of these patients was very similar. In Family 1, the propositus presented with euthyroid congenital goiter with a low serum TG level (TSH 4.0mU/L; T4 7.8ng/dl; TG 9.6ng/ml). At age 16, he underwent thyroidectomy because of a large multinodular goiter. In Family 2, the two affected brothers presented with goiter and mild thyroid failure (TSH 10.0 and 4.0; T4 4.0 and 6.0ng/dl; TG 9.5 and 29.2ng/ml). In the inbred Family 3, the index patient had goiter and mild hypothyroidism (TSH 12.7mU/L; T4 6.0ng/dl; TG 3.0ng/ml). Analysis of genomic DNA of the index patients and their relatives confirmed segregation of the 6701C>A alteration with the abnormal phenotype. These findings suggest that the substitution A2234N may be associated with impaired secretion and/or function of TG, a notion that requires experimental confirmation using expression studies of the mutant in transfected cells (88).
Human TG Gene Mutations in Simple Goiter
Given its physiological importance and the observations that mild TG defects can result in mildly hypothyroid or euthyroid goiter, it is reasonable to speculate that TG variants could play a role in the development of simple goiter. Addressing this hypothesis, a monoallelic TG alteration has been associated with non-endemic simple goiter (89). Analyzing 56 individuals, a 2610G>T transversion in exon 10 substituting glutamine 870 by histidine (Q870H, originally described as Q851H) was found in 14 individuals with simple goiter from three different families and the authors proposed an autosomal dominant inheritance of the defect (89). However, it has to be emphasized that 11 unaffected individuals also carried the same allele (89). Subsequently, the same authors reported the Q870H mutation in 1 of 36 patients with endemic goiter and proposed an association of this allele with goiter development (90). Given that the TG gene contains multiple polymorphisms, and in the absence of any functional data, it remains unclear whether this alteration is indeed causally involved in the development of the abnormal phenotype. In another series of 50 patients with simple euthyroid goiter a monoallelic TG deletion encompassing the promoter and the first eleven exons was found in a single patient (91). It remains questionable whether this alteration has any significance for the development of the abnormal phenotype given that heterozygous individuals with inactivating TG mutations do not display an abnormal phenotype (91).
TG GENE MUTATIONS IN ANIMALS
The phenotype of the Afrikander cattle is characterized by euthyroid congenital goiter with TG deficiency (92). At the molecular level, this strain has a cytosine to thymine transition (2146C>T) in exon 9 of the TG gene that creates a stop codon (R716X, originally described as R697X). Rather than generating a truncated protein, alternative splicing removes the exon harboring the premature stop codon by nonsense-mediated altered splicing (76,92). This truncated TG protein still contains the amino-terminal hormonogenic site and appears to be sufficient for hormone synthesis at the expense of a large, compensatory goiter.
The inbred Dutch goats, a strain presenting with congenital goiter and hypothyroidism, have no 19S TG and only a small 7S protein with a molecular weight of a ~35kDa (71). The truncated aminoterminal protein is able to make some thyroid hormone and under conditions of high iodine intake these animal are euthyroid, but still have a large goiters (93,94). At the molecular level, a transition of cytosine to thymine at position 945 of the TG cDNA results in a stop codon in exon 8 (945C>T, Y315X, originally described as Y296X) (95,96).
The cog/cog mouse, an inbred mouse strain with autosomal recessive hypothyroidism, congenital goiter, and a TG with abnormal immunological and sedimentation properties (97,98) has a thymine to cytosine substitution at position 6848 of the TG cDNA. This missense mutation results in the substitution of leucine by proline at position 2263 (6848T>C, L2283P, originally described as L2263P) (99). This mutation is localized in the region of TG that is homologous to acetylcholinesterase, a domain that is important for structural properties (61). Analogous to some of the TG mutations identified in humans, the mutated TG of the cog/cog mouse retained in the ER causing an ER storage disease (ERSD) (80,81,99).
The phenotype of the rdw rat is defined by dwarfism and hypothyroidism (100). In contrast to the animal strains and human patients discussed above, the rdw rat has no goiter and low levels of TG in the follicular lumen. However, these animals have detectable TG in the dilated ER, suggesting that the export of TG is impaired, and increased expression of molecular chaperones (101,102). The molecular defect has been unraveled independently by two groups and consists of a transversion of guanine to cytosine at position 6958. This transversion results in the substitution of glycine by arginine at position 2320 (6958G>C, G2320R) (103,104). The identification of a mutation in the Tg gene as a cause of non-goitrous hypothyroidism in the rdw/rdw rat is important since it challenges the previously held view that non-goitrous congenital hypothyroidism is caused by thyroid dysgenesis or defects in TSH-signaling.
CONCLUSIONS
The isolation and the identification of genes controlling thyroid development and thyroid hormone synthesis continues to provide unique insights into the ontogenesis and physiology of the hypothalamic-pituitary-thyroid axis, as well as a more precise understanding of (congenital) disorders at the molecular level. It has become apparent that thyroid dysgenesis is, at least in part, a genetic disorder (3,7,19). However, the molecular defects known to date only account for a minority of cases of thyroid dysgenesis. It is likely that a further subset of patients with thyroid dysgenesis have defects in other transacting proteins that remain to be discovered. In other instances, thyroid dysgenesis may be a polygenic disease or have a multifactorial basis. Genetic testing is currently of limited importance in patients with congenital hypothyroidism. However, analyses at the molecular level may be useful and informative in familial cases and selected sporadic patients.
A thorough understanding of the molecular pathophysiology often has unexpected and important ramifications. For example, expression of functional NIS has been reported in numerous other tissues, among them in breast cancer tissue (105,106). Gene therapy with NIS and subsequent radioiodine therapy have been successful in several tumors in vitro (107). NIS expression under the control of tissue-specific promoters such as the PSA promoter (prostate-specific antigen) may become an efficacious strategy in the therapy of selected cancers in vivo (108).
The "experiments of nature" told by naturally occurring mutations are frequently particularly informative. For example, it is now clear that inactivating human PAX8 mutations can be transmitted in an autosomal dominant fashion and cause thyroid hypoplasia (7,9). The phenotype may, however, be variable either due to incomplete penetrance or altered expressivity (see above). In the case of TG, it is apparent that TG missense mutations can be associated with a classic ERSD (80). Mutations which cause alterations in the protein structure give rises to intracellular retention of the altered proteins, emphasizing that a correct conformation is essential for protein transport and biological activity. The goitrous phenotype can be explained by the accumulation of misfolded proteins in the cells affected by ERSD with expansion and dilatation of the ER (Kim, 1998 #110). Some of the nonsense mutations in the TG gene are of particular interest because of the plasticity generated by the mechanism of nonsense-mediated exon skipping (72,75,76). Lastly, it appears that certain very short TG molecules can be secreted and are sufficient for partial thyroid hormone synthesis (82,93,94).
In conclusion, these examples illustrate that the study of Inborn Errors of Metabolism continues to be an important approach in the quest for a more complete understanding of human disease and the development of novel preventative and therapeutic strategies.
ACKNOWLEDGMENTS
JV-T has been supported by grants from the Brazilian Research Council (CNPq - 201571/88-9). This work has been supported, in part, by grant DO2FE-14 from the Morris Animal Foundation and 1R01DK63024-01 from NIH/NIDDK to PK.
Recebido em 30/11/03
Aceito em 15/12/03
- 1. Gillam MP, Kopp P. Genetic regulation of thyroid development. Curr Opin Pediatr 2001;13:358-63.
- 2. Gillam MP, Kopp P. Genetic defects in thyroid hormone synthesis. Curr Opin Pediatr 2001;13:364-72.
- 3. Van Vliet G. Development of the thyroid gland: lessons from congenitally hypothyroid mice and men. Clin Genet 2003;63:445-55.
- 4. Knobel M, Medeiros-Neto G. An outline of inherited disorders of the thyroid hormone generating system. Thyroid 2003;13:771-801.
- 5. Damante G, Di Lauro R. Thyroid-specific gene expression. Biochim Biophys Acta 1994;1218:255-66.
- 6. Kambe F, Seo H. Thyroid-specific transcription factors. Endocr J 1997;44:775-84.
- 7. Macchia PE, Lapi P, Krude H, Pirro MT, Missero C, Chiovato L, et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet 1998;19:83-6.
- 8. Vilain C, Rydlewski C, Duprez L, Heinrichs C, Abramowicz M, Malvaux P, et al. Autosomal dominant transmission of congenital thyroid hypoplasia due to loss-of-function mutation of PAX8. J Clin Endocrinol Metab 2001;86:234-8.
- 9. Congdon T, Nguyen LQ, Nogueira CR, Habiby RL, Medeiros-Neto G, Kopp P. A novel mutation (Q40P) in PAX8 associated with congenital hypothyroidism and thyroid hypoplasia: evidence for phenotypic variability in mother and child. J Clin Endocrinol Metab 2001;86:3962-7.
- 10. Komatsu M, Takahashi T, Takahashi I, Nakamura M, Takada G. Thyroid dysgenesis caused by PAX8 mutation: the hypermutability with CpG dinucleotides at codon 31. J Pediatr 2001;139:597-9.
- 11. Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet 1998;19:87-90.
- 12. Devriendt K, Vanhole C, Matthis G, De Zegher F. Deletion of thyroid transcription factor-1 gene in an infant with neonatal thyroid dysfunction and respiratory failure. N Engl J Med 1998;338:1317-8.
- 13. Iwatani N, Mabe H, Devriendt K, Kodama M, Miike T. Deletion of NKX2.1 gene encoding thyroid transcription factor-1 in two siblings with hypothyroidism and respiratory failure. J Pediatr 2000;137:272-6.
- 14. Pohlenz J, Dumitrescu A, Zundel D, Martine U, Schonberger W, Koo E, et al. Partial deficiency of thyroid transcription factor 1 produces predominantly neurological defects in humans and mice. J Clin Invest 2002;109:469-73.
- 15. Krude H, Schutz B, Biebermann H, von Moers A, Schnabel D, Neitzel H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Invest 2002;109:475-80.
- 16. Seidman JG, Seidman C. Transcription factor haploinsufficiency: when half a loaf is not enough. J Clin Invest 2002;109:451-5.
- 17. Kleiner-Fisman G, Rogaeva E, Halliday W, Houle S, Kawarai T, Sato C, et al. Benign hereditary chorea: clinical, genetic, and pathological findings. Ann Neurol 2003;54:244-7.
- 18. Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, et al. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev 1996;10:60-9.
- 19. Clifton-Bligh RJ, Wentworth JM, Heinz P, Crisp MS, John R, Lazarus JH, et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet 1998;19:399-401.
- 20. Castanet M, Park SM, Smith A, Bost M, Leger J, Lyonnet S, et al. A novel loss-of-function mutation in TTF-2 is associated with congenital hypothyroidism, thyroid agenesis and cleft palate. Hum Mol Genet 2002;11:2051-9.
- 21. De Felice M, Ovitt C, Biffali E, Rodriguez-Mallon A, Arra C, Anastassiadis K, et al. A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat Genet 1998; 19:395-8.
- 22. Persani L. Hypothalamic thyrotropin-releasing hormone and thyrotropin biological activity. Thyroid 1998;8:941-6.
- 23. Collu R, Tang J, Castagné J, Lagacé G, Masson N, Huot C, et al. A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab 1997;82:1361-5.
- 24. Hayashizaki Y, Hiraoka Y, Tatsumi Y, Hashimoto T, Furuyama J, Miyai K, et al. Deoxyribonucleic acid analysis of five families with familial inherited thyroid stimulating hormone deficiency. J Clin Endocrinol Metab 1990;71:792-6.
- 25. Medeiros-Neto G, Heodotou DT, Rajan S, Kommareddi S, de Lacerda L, Sandrini R, et al. A circulating, biologically inactive thyrotropin caused by a mutation in the beta subunit gene. J Clin Invest 1996;97:1250-6.
- 26. Pohlenz J, Dumitrescu A, Aumann U, Koch G, Melchior R, Prawitt D, et al. Congenital secondary hypothyroidism caused by exon skipping due to a homozygous donor splice site mutation in the TSHbeta-subunit gene. J Clin Endocrinol Metab 2002;87:336-9.
- 27. Cohen LE, Radovick S. Molecular basis of combined pituitary hormone deficiencies. Endocr Rev 2002;23: 431-42.
- 28. Cogan JD, Wu W, Phillips JA 3rd, Arnhold IJ, Agapito A, Fofanova OV, et al. The PROP1 2-base pair deletion is a common cause of combined pituitary hormone deficiency. J Clin Endocrinol Metab 1998;83:3346-9.
- 29. Nogueira CR, Sabacan L, Jameson JL, Medeiros-Neto G, Kopp P. Combined pituitary hormone deficiency in an inbred Brazilian kindred associated with a mutation in the PROP-1 gene. Mol Genet Metab 1999;67:58-61.
- 30. Mendonça BB, Osório MG, Latrônico AC, Estefan V, Lo LS, Arnhold IJ. Longitudinal hormonal and pituitary imaging changes in two females with combined pituitary hormone deficiency due to deletion of A301,G302 in the PROP1 gene. J Clin Endocrinol Metab 1999;84:942-5.
- 31. Osorio MG, Kopp P, Marui S, Latronico AC, Mendonça BB, Arnhold IJ. Combined pituitary hormone deficiency caused by a novel mutation of a highly conserved residue (F88S) in the homeodomain of PROP-1. J Clin Endocrinol Metab 2000;85:2779-85.
- 32. Vieira TC, Dias da Silva MR, Cerutti JM, Brunner E, Borges M, Arnaldi LT, et al. Familial combined pituitary hormone deficiency due to a novel mutation R99Q in the hot spot region of Prophet of Pit-1 presenting as constitutional growth delay. J Clin Endocrinol Metab 2003;88:38-44.
- 33. Sunthornthepvarakul T, Hayashi Y, Refetoff S. Polymorphism of a variant human thyrotropin receptor (hTSHR) gene. Thyroid 1994;4:147-9.
- 34. Kopp P. Resistance to TSH. In: Jameson J, editor. Hormone resistance syndromes Totowa: Humana Press, 1999p.111-44.
- 35. Kopp P. The TSH receptor and its role in thyroid disease. Cell Mol Life Sci 2001;58:1301-22.
- 36. Xie J, Pannain S, Pohlenz J, Weiss RE, Moltz K, Morlot M, et al. Resistance to thyrotropin (TSH) in three families is not associated with mutations in the TSH receptor or TSH. J Clin Endocrinol Metab 1997;82:3933-40.
- 37. Abramowicz MJ, Duprez L, Parma J, Vassart G, Heinrichs C. Familial congenital hypothyroidism due to inactivating mutation of the thyrotropin receptor causing profound hypoplasia of the thyroid gland. J Clin Invest 1997;99:3018-24.
- 38. Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 2001;22:675-705.
- 39. Dohan O, De la Vieja A, Paroder V, Riedel C, Artani M, Reed M, et al. The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocr Rev 2003;24:48-77.
- 40. Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nature Genet 1997;17:411-22.
- 41. Kopp P, Karamanoglu Arseven O, Sabacan L, Kotlar T, Dupuis J, Cavaliere H, et al. Phenocopies for deafness and goiter development in a large inbred kindred with Pendred's syndrome associated with a novel mutation in the PDS gene. J Clin Endocrinol Metab 1999;84:336-41.
- 42. Abramowicz MJ, Targovnik HM, Varela V, Cochaux P, Krawiec L, Pisarev MA, et al. Identification of a mutation in the coding sequence of the human thyroid peroxidase gene causing congenital goiter. J Clin Invest 1992;90:1200-4.
- 43. Bakker B, Bikker H, Vulsma T, de Randamie JS, Wiedijk BM, De Vijlder JJ. Two decades of screening for congenital hypothyroidism in The Netherlands: TPO gene mutations in total iodide organification defects (an update). J Clin Endocrinol Metab 2000;85:3708-12.
- 44. Dupuy C, Ohayon R, Valent A, Noel-Hudson MS, Deme D, Virion A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cDNAs. J Biol Chem 1999;274:37265-9.
- 45. De Deken X, Wang D, Many MC, Costagliola S, Libert F, Vassart G, et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem 2000;275:23227-33.
- 46. De Deken X, Wang D, Dumont JE, Miot F. Characterization of ThOX proteins as components of the thyroid H2O2-generating system. Exp Cell Res 2002;273:187-96.
- 47. Moreno JC, Bikker H, Kempers MJ, van Trotsenburg AS, Baas F, de Vijlder JJ, et al. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med 2002;347:95-102.
- 48. Figueiredo MD, Cardoso LC, Ferreira AC, Campos DV, da Cruz Domingos M, Corbo R, et al. Goiter and hypothyroidism in two siblings due to impaired Ca+2/NAD(P)H-dependent H2O2-generating activity. J Clin Endocrinol Metab 2001;86:4843-8.
- 49. van de Graaf SA, Ris-Stalpers C, Pauws E, Mendive FM, Targovnik HM, de Vijlder JJ. Up to date with human thyroglobulin. J Endocrinol 2001;170:307-21.
- 50. Medeiros-Neto G, Stanbury JB. The iodotyrosine deiodinase defect. In: Medeiros-Neto G, Stanbury JB, editors. Inherited disorders of the thyroid system Boca Raton: CRC Press, 1994p.139-59.
- 51. Baas F, Bikker H, Geurts van Kessel A, Melsert R, Pearson PL, de Vijlder JJ, et al. The human thyroglobulin gene: a polymorphic marker localized distal to C-MYC on chromosome 8 band q24. Hum Genet 1985;69:138-43.
- 52. Rabin M, Barker PE, Ruddle FH, Brocas H, Targovnik H, Vassart G. Proximity of thyroglobulin and c-myc genes on human chromosome 8. Somat Cell Mol Genet 1985;11:397-402.
- 53. Berge-Lefranc JL, Cartouzou G, Mattei MG, Passage E, Malezet-Desmoulins C, Lissitzky S. Localization of the thyroglobulin gene by in situ hybridization to human chromosomes. Hum Genet 1985;69:28-31.
- 54. Mendive FM, Rivolta CM, Vassart G, Targovnik HM. Genomic organization of the 3' region of the human thyroglobulin gene. Thyroid 1999;9:903-12.
- 55. Moya CM, Mendive FM, Rivolta CM, Vassart G, Targovnik HM. Genomic organization of the 5' region of the human thyroglobulin gene. Eur J Endocrinol 2000;143:789-98.
- 56. Mendive FM, Rivolta CM, Moya CM, Vassart G, Targovnik HM. Genomic organization of the human thyroglobulin gene: the complete intron-exon structure. Eur J Endocrinol 2001;145:485-96.
- 57. Malthiery Y, Lissitzky S. Primary structure of human thyroglobulin deduced from the sequence of its 8448-base complementary DNA. Eur J Biochem 1987;165:491-8.
- 58. van de Graaf SA, Pauws E, de Vijlder JJ, Ris-Stalpers CR. The revised 8307 base pair coding sequence of human thyroglobulin transiently expressed in eukaryotic cells. Eur J Endocrinol 1997;136:508-15.
- 59. Sakai K, Shirasawa S, Ishikawa N, Ito K, Tamai H, Kuma K, et al. Identification of susceptibility loci for autoimmune thyroid disease to 5q31-q33 and Hashimoto's thyroiditis to 8q23-q24 by multipoint affected sib-pair linkage analysis in Japanese. Hum Mol Genet 2001;10:1379-86.
- 60. Tomer Y, Ban Y, Concepcion E, Barbesino G, Villanueva R, Greenberg DA, et al. Common and unique susceptibility loci in Graves and Hashimoto diseases: results of whole-genome screening in a data set of 102 multiplex families. Am J Hum Genet 2003;73:736-47.
- 61. Swillens S, Ludgate M, Mercken L, Dumont JE, Vassart G. Analysis of sequence and structure homologies between thyroglobulin and acetylcholinesterase: possible functional and clinical significance. Biochem Biophys Res Commun 1986;137:142-8.
- 62. Mercken L, Simons MJ, De Martynoff G, Swillens S, Vassart G. Presence of hormonogenic and repetitive domains in the first 930 amino acids of bovine thyroglobulin as deduced from the cDNA sequence. Eur J Biochem 1985;147:59-64.
- 63. Parma J, Christophe D, Pohl V, Vassart G. Structural organization of the 5' region of the thyroglobulin gene. Evidence for intron loss and "exonization" during evolution. J Mol Biol 1987;196:769-79.
- 64. Dunn JT, Dunn AD. Thyroglobulin: chemistry, biosynthesis, and proteolysis. In: Braverman LE, Utiger RD, editors. Werner & Ingbar's the thyroid: A fundamental and clinical text, 8th ed. Philadelphia: Lippincott Williams & Wilkins, 2000p.91-104.
- 65. Taurog A. Hormone synthesis. In: Braverman LE, Utiger RD, editors. The thyroid: a fundamental and clinical text, 8th ed. Philadelphia: Lipincott-Raven, 2000p.61-85.
- 66. Lamas L, Anderson PC, Fox JW, Dunn JT. Consensus sequences for early iodination and hormonogenesis in human thyroglobulin. J Biol Chem 1989;264:13541-5.
- 67. Dunn AD, Corsi CM, Myers HE, Dunn JT. Tyrosine 130 is an important outer ring donor for thyroxine formation in thyroglobulin. J Biol Chem 1998;273:25223-9.
- 68. Dunn JT, Dunn AD. Update on intrathyroidal iodine metabolism. Thyroid 2001;11:407-14.
- 69. Marino M, Pinchera A, McCluskey RT, Chiovato L. Megalin in thyroid physiology and pathology. Thyroid 2001;11:47-56.
- 70. Ieiri T, Cochaux P, Targovnik HM, Suzuki M, Shimoda S, Perret J, et al. A 3' splice site mutation in the thyroglobulin gene responsible for congenital goiter with hypothyroidism. J Clin Invest 1991;88:1901-5.
- 71. Medeiros-Neto G, Targovnik HM, Vassart G. Defective thyroglobulin synthesis and secretion causing goiter and hypothyroidism. Endocr Rev 1993;14:165-83.
- 72. Targovnik HM, Medeiros-Neto G, Varela V, Cochaux P, Wajchenberg BL, Vassart G. A nonsense mutation causes human hereditary congenital goiter with preferential production of a 171-nucleotide-deleted thyroglobulin ribonucleic acid messenger. J Clin Endocrinol Metab 1993;77:210-5.
- 73. Targovnik HM, Frechtel GD, Mendive FM, Vono J, Cochaux P, Vassart G, et al. Evidence for the segregation of three different mutated alleles of the thyroglobulin gene in a Brazilian family with congenital goiter and hypothyroidism. Thyroid 1998;8:291-7.
- 74. den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet 2001;109:121-4.
- 75. Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002;3:285-98.
- 76. Ricketts MH, Simons MJ, Parma J, Mercken L, Dong Q, Vassart G. A nonsense mutation causes hereditary goitre in the Afrikander cattle and unmasks alternative splicing of thyroglobulin transcripts. Proc Natl Acad Sci USA 1987;84:3181-4.
- 77. Targovnik HM, Vono J, Billerbeck AE, Cerrone GE, Varela V, Mendive F, et al. A 138-nucleotide deletion in the thyroglobulin ribonucleic acid messenger in a congenital goiter with defective thyroglobulin synthesis. J Clin Endocrinol Metab 1995;80:3356-60.
- 78. Targovnik HM, Rivolta CM, Mendive FM, Moya CM, Vono J, Medeiros-Neto G. Congenital goiter with hypothyroidism caused by a 5' splice site mutation in the thyroglobulin gene. Thyroid 2001;11:685-90.
- 79. Medeiros-Neto G, Kim PS, Yoo SE, Vono J, Targovnik HM, Camargo R, et al. Congenital hypothyroid goiter with deficient thyroglobulin. Identification of an endoplasmic reticulum storage disease with induction of molecular chaperones. J Clin Invest 1996;98:2838-44.
- 80. Kim PS, Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev 1998;19:173-202.
- 81. Rutishauser J, Spiess M. Endoplasmic reticulum storage diseases. Swiss Med Wkly 2002;132:211-22.
- 82. van de Graaf SA, Ris-Stalpers C, Veenboer GJ, Cammenga M, Santos C, Targovnik HM, et al. A premature stopcodon in thyroglobulin messenger RNA results in familial goiter and moderate hypothyroidism. J Clin Endocrinol Metab 1999;84:2537-42.
- 83. Rawitch AB, Mercken L, Hamilton JW, Vassart G. The structure of a naturally occurring 10K polypeptide derived from the amino terminus of bovine thyroglobulin. Biochem Biophys Res Commun 1984;119:335-42.
- 84. Hishinuma A, Kasai K, Masawa N, Kanno Y, Arimura M, Shimoda SI, et al. Missense mutation (C1263R) in the thyroglobulin gene causes congenital goiter with mild hypothyroidism by impaired intracellular transport. Endocr J 1998;45:315-27.
- 85. Hishinuma A, Takamatsu J, Ohyama Y, Yokozawa T, Kanno Y, Kuma K, et al. Two novel cysteine substitutions (C1263R and C1995S) of thyroglobulin cause a defect in intracellular transport of thyroglobulin in patients with congenital goiter and the variant type of adenomatous goiter. J Clin Endocrinol Metab 1999;84:1438-44.
- 86. Caron P, Moya CM, Malet D, Gutnisky VJ, Chabardes B, Rivolta CM, et al. Compound heterozygous mutations in the thyroglobulin gene (1143delC and 6725G>A [R2223H]) resulting in fetal goitrous hypothyroidism. J Clin Endocrinol Metab 2003;88:3546-53.
- 87. Vono-Toniolo J, Medeiros-Neto G, Kopp P. Three novel homozygous nucleotide substitutions in the TG gene in an inbred Brazilian kindred with congenital goiter and defective thyroglobulin synthesis. 84th Meeting of the Endocrine Society 2002, San Francisco Abstract P3-185:536.
- 88. Vono-Toniolo J, Medeiros-Neto G, Kopp P. Three Brazilian families with congenital goiter and defective thyroglobulin synthesis associated with a novel homozygous mutation (A2234N) in the thyroglobulin gene. 74th Annual Meeting of the American Thyroid Association 2002, Los Angeles, CA Abstract 162:190.
- 89. Corral J, Martin C, Perez R, Sanchez I, Mories MT, San Millan JL, et al. Thyroglobulin gene point mutation associated with non-endemic simple goitre. Lancet 1993;341:462-4.
- 90. Perez-Centeno C, Gonzalez-Sarmiento R, Mories MT, Corrales JJ, Miralles-Garcia JM. Thyroglobulin exon 10 gene point mutation in a patient with endemic goiter. Thyroid 1996;6:423-7.
- 91. Gonzalez-Sarmiento R, Corral J, Mories MT, Corrales JJ, Miguel-Velado E, Miralles-Garcia JM. Monoallelic deletion in the 5' region of the thyroglobulin gene as a cause of sporadic nonendemic simple goiter. Thyroid 2001; 11:789-93.
- 92. Ricketts MH, Pohl V, de Martynoff G, Boyd CD, Bester AJ, Van Jaarsveld PP, et al. Defective splicing of thyroglobulin gene transcripts in the congenital goitre of the Afrikander cattle. EMBO J 1985;4:731-7.
- 93. Van Voorthuizen WF, Dinsart C, Flavell RA, DeVijlder JJ, Vassart G. Abnormal cellular localization of thyroglobulin mRNA associated with hereditary congenital goiter and thyroglobulin deficiency. Proc Natl Acad Sci USA 1978;75:74-8.
- 94. van Voorthuizen WF, de Vijlder JJ, van Dijk JE, Tegelaers WH. Euthyroidism via iodide supplementation in hereditary congenital goiter with thyroglobulin deficiency. Endocrinology 1978;103:2105-11.
- 95. Veenboer GJ, de Vijlder JJ. Molecular basis of the thyroglobulin synthesis defect in Dutch goats. Endocrinology 1993;132:377-81.
- 96. Sterk A, van Dijk JE, Veenboer GJ, Moorman AF, de Vijlder JJ. Normal-sized thyroglobulin messenger ribonucleic acid in Dutch goats with a thyroglobulin synthesis defect is translated into a 35,000 molecular weight N-terminal fragment. Endocrinology 1989;124:477-83.
- 97. Beamer WG, Maltais LJ, DeBaets MH, Eicher EM. Inherited congenital goiter in mice. Endocrinology 1987; 120:838-40.
- 98. Basche M, Beamer WG, Schneider AB. Abnormal properties of thyroglobulin in mice with inherited congenital goiter (cog/cog). Endocrinology 1989;124:1822-9.
- 99. Kim PS, Hossain SA, Park YN, Lee I, Yoo SE, Arvan P. A single amino acid change in the acetylcholinesterase-like domain of thyroglobulin causes congenital goiter with hypothyroidism in the cog/cog mouse: a model of human endoplasmic reticulum storage diseases. Proc Natl Acad Sci USA 1998;95:9909-13.
- 100.Umezu M, Kagabu S, Jiang J, Sato E. Evaluation and characterization of congenital hypothyroidism in rdw dwarf rats. Lab Anim Sci 1998;48:496-501.
- 101.Sakai Y, Yamashina S, Furudate SI. Missing secretory granules, dilated endoplasmic reticulum, and nuclear dislocation in the thyroid gland of rdw rats with hereditary dwarfism. Anat Rec 2000;259:60-6.
- 102.Oh-Ishi M, Omori A, Kwon JY, Agui T, Maeda T, Furudate SI. Detection and identification of proteins related to the hereditary dwarfism of the rdw rat. Endocrinology 1998;139:1288-99.
- 103.Hishinuma A, Furudate S, Oh-Ishi M, Nagakubo N, Namatame T, Ieiri T. A novel missense mutation (G2320R) in thyroglobulin causes hypothyroidism in rdw rats. Endocrinology 2000;141:4050-5.
- 104.Kim PS, Ding M, Menon S, Jung CG, Cheng JM, Miyamoto T, et al. A missense mutation G2320R in the thyroglobulin gene causes non-goitrous congenital primary hypothyroidism in the WIC-rdw rat. Mol Endocrinol 2000;14:1944-53.
- 105.De La Vieja A, Dohan O, Levy O, Carrasco N. Molecular analysis of the sodium/iodide symporter: impact on thyroid and extrathyroid pathophysiology. Physiol Rev 2000;80:1083-105.
- 106.Tazebay UH, Wapnir IL, Levy O, Dohan O, Zuckier LS, Zhao QH, et al. The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat Med 2000;6:871-8.
- 107.Mandell RB, Mandell LZ, Link CJ. Radioisotope concentrator gene therapy using the sodium/iodide symporter gene. Cancer Res 1999;59:661-8.
- 108.Spitzweg C, O'Connor MK, Bergert ER, Tindall DJ, Young CY, Morris JC. Treatment of prostate cancer by radioiodine therapy after tissue-specific expression of the sodium iodide symporter. Cancer Res 2000;60:6526-30.
Endereço para correspondência
Publication Dates
-
Publication in this collection
28 May 2004 -
Date of issue
Feb 2004
History
-
Accepted
15 Dec 2003 -
Received
30 Nov 2003