Resumos
Além de influenciar o crescimento corpóreo, o hormônio do crescimento, ou somatotrófico, desempenha importante papel no metabolismo, composição corporal, perfil lipídico, estado cardiovascular e longevidade. Seu controle é multi-regulado por hormônios, metabólitos e peptídeos hipotalâmicos. Dados sobre a Deficiência Isolada de GH (DIGH) obtidos a partir da descrição da mutação IVS1+1G®A no gene do receptor do hormônio liberador do GH (GHRH-R) em indivíduos da cidade de Itabaianinha, SE, são revisados. São abordadas novas perspectivas sobre o modelo de resistência ao GHRH, a importância do GHRH no controle da secreção de GH, a freqüência das mutações do gene do GHRH-R, a relevância diagnóstica do IGF-I e os achados metabólicos, cardiovasculares e de qualidade de vida nestes indivíduos.
Hormônio do crescimento; Hormônio somatotrófico; Deficiência isolada do GH; Receptor do GHRH
In addition to stimulating body growth, growth or somatotrophic hormone plays an important role in metabolism, body composition, lipid profile, cardiovascular status and longevity. Its control is multiregulated by hormones, metabolites and hypothalamic peptides. Obtained data of the isolated growth hormone deficiency (IGHD) after the description of the IVS1+1G®A GHRH receptor gene mutation in individuals of Itabaianinha County are reviewed. New perspectives about the growth hormone resistance model, the importance of GHRH in the control of GH secretion, the frequency of GHRH-R gene mutations, the diagnostic relevance of IGF-I and the metabolic, cardiovascular and quality of life findings are approached.
Growth hormone; Somatotrophic hormone; Isolated growth hormone deficiency; GHRH receptor
PERSPECTIVAS
Hormônio do crescimento ou somatotrófico: novas perspectivas na deficiência isolada de GH a partir da descrição da mutação no gene do receptor do GHRH nos indivíduos da cidade de Itabaianinha, Brasil
Growth or somatotrophic hormone: new perspectives in isolated GH deficiency after description of the mutation in the GHRH receptor gene in individuals of Itabaianinha county, Brazil
Anita Hermínia O. SouzaI; Roberto SalvatoriII; Carlos E. Martinelli JrIII; Walter M.O. CarvalhoI; Carlos A. MenezesI; Elenilde S. de A. BarrettoI; José A.S. Barreto FilhoI; Marta R.S. de AlcântaraI; Carla R.P. OliveiraI; Paula R.S. de AlcântaraI; Roberto J.R. RamalhoI; Hélio A. OliveiraI; Ivana B. de LimaI; Jamille N. CarneiroI; Marcos M. SantosI; Matthew S. GillIV; Peter E. ClaytonIV; Manuel H. A.-OliveiraI
IServiço de Endocrinologia, Hospital Universitário da Universidade Federal de Sergipe, Aracaju, SE
IIThe Johns Hopkins University School of Medicine Baltimore, Maryland, USA
IIIServiço de Endocrinologia Pediátrica do Departamento de Puericultura e Pediatria Faculdade de Medicina de Ribeirão Preto USP, Ribeirão Preto, SP
IVEndocrine Sciences Research Group Department of Medicine University of Manchester, UK
Endereço para correspondência Endereço para correspondência Manuel Hermínio de Aguiar-Oliveira Departamento de Medicina, Hospital Universitário, UFS Rua Cláudio Batista s/n° 49060-100 Aracaju, SE Fax: (79) 214-2491 E-mail: herminio@infonet.com.br
RESUMO
Além de influenciar o crescimento corpóreo, o hormônio do crescimento, ou somatotrófico, desempenha importante papel no metabolismo, composição corporal, perfil lipídico, estado cardiovascular e longevidade. Seu controle é multi-regulado por hormônios, metabólitos e peptídeos hipotalâmicos. Dados sobre a Deficiência Isolada de GH (DIGH) obtidos a partir da descrição da mutação IVS1+1G®A no gene do receptor do hormônio liberador do GH (GHRH-R) em indivíduos da cidade de Itabaianinha, SE, são revisados. São abordadas novas perspectivas sobre o modelo de resistência ao GHRH, a importância do GHRH no controle da secreção de GH, a freqüência das mutações do gene do GHRH-R, a relevância diagnóstica do IGF-I e os achados metabólicos, cardiovasculares e de qualidade de vida nestes indivíduos.
Descritores: Hormônio do crescimento; Hormônio somatotrófico; Deficiência isolada do GH; Receptor do GHRH
ABSTRACT
In addition to stimulating body growth, growth or somatotrophic hormone plays an important role in metabolism, body composition, lipid profile, cardiovascular status and longevity. Its control is multiregulated by hormones, metabolites and hypothalamic peptides. Obtained data of the isolated growth hormone deficiency (IGHD) after the description of the IVS1+1G®A GHRH receptor gene mutation in individuals of Itabaianinha County are reviewed. New perspectives about the growth hormone resistance model, the importance of GHRH in the control of GH secretion, the frequency of GHRH-R gene mutations, the diagnostic relevance of IGF-I and the metabolic, cardiovascular and quality of life findings are approached.
Keywords: Growth hormone; Somatotrophic hormone; Isolated growth hormone deficiency; GHRH receptor
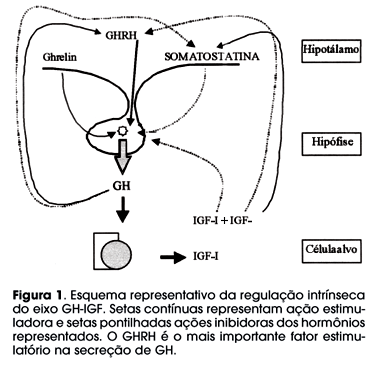
A EXISTÊNCIA DO HORMÔNIO DE CRESCIMENTO foi demonstrada por Evans e Long em 1921 (1), mas apenas em 1944, Li e Evans isolaram e sintetizaram o hormônio cristalino (2). Em 1973, Guilhermin e 1976 Schally isolaram a somatostatina, embora tenha sido Samuel McCann quem demonstrou sua existência (3). McCann, junto com Geoffrey Harris, estabeleceu a teoria dos fatores hipotalâmicos. McCann contribuiu com a formação de Ayrton Moreira e Antunes-Rodrigues da Faculdade de Medicina de Ribeirão Preto da USP, principais inspiradores do Serviço de Endocrinologia da Universidade Federal de Sergipe, onde foi produzida a maior parte dos dados a seguir apresentados sobre a importante mutação IVS1+1G®A no receptor do hormônio liberador de GH (GHRH-R), descrita em indivíduos da cidade de Itabaianinha, Sergipe, no Nordeste do Brasil. Dr. Michael Thorner, o descobridor do GHRH, 20 anos após o seu isolamento, confirma: "uma outra mutação no receptor do GHRH foi descrita em uma família brasileira com 110 indivíduos com acentuada baixa estatura severa. Estes experimentos da natureza demonstram que um déficit no GHRH-R impede o crescimento e enfatiza a vital importância do GHRH" (4). Apesar da descoberta recente do Ghrelin (5,6), um peptídeo produzido no estômago e no hipotálamo com capacidade estimuladora do GH através da ligação dos receptores GHS-R (Growth hormone secretagogues receptor), os dados oriundos de Itabaianinha demonstram a importância estimulatória do GHRH para a secreção de GH (figura 1). Neste artigo, revisaremos os principais dados publicados sobre esta mutação e as novas perspectivas que ela estabelece no estado da arte sobre a deficiência isolada de GH.
O Modelo da Resistência ao GHRH
O modelo de resistência hormonal, por problemas nos seus receptores, teve o seu primeiro exemplo definido no eixo GH-IGFs, quando Laron descreveu a Síndrome de Resistência ao GH caracterizada por mutações no receptor do GH, o GH-R (7). Neste modelo, há uma hipersecreção de GH com reduzidos níveis de IGF-I. Há mais de 200 indivíduos descritos com o Nanismo de Laron, e o maior agrupamento familiar com 70 indivíduos com resistência ao GH foi descrito no Equador (8).
A mutação IVS1+1GÆA no GHRH-R descrita em 105 indivíduos da cidade de Itabaianinha (9) foi a segunda mutação descrita no gene do GHRH-R, sendo precedido pela mutação E72X, análoga à do modelo experimental do little mouse, descrita em três famílias no sub-continente indiano, primeiro por Wajnrajch em duas crianças indianas primas (10), depois em 18 indivíduos da mesma família e geração no Paquistão, "os anões do Sindh" (11,12) e a seguir em duas irmãs do Srilanka (13). Os anões de Itabaianinha, a princípio confundidos com os portadores de resistência ao GH, na realidade são homozigóticos para uma mutação tipo splicing no início do intron 1 do gene do receptor do GHRH, com uma substituição de uma Guanina por Adenina (9). Esta mutação impede a formação do RNA mensageiro do GHRH-R, abolindo completamente sua expressão. O GHRH-R é um receptor acoplado à proteína G e possui um domínio N terminal extracelular, 7 hélices intramembrânicas e uma extremidade C intracelular. A ligação do GHRH ao GHRH-R na superfície dos somatotrófos leva ao aumento da atividade da adenilciclase, síntese do AMPc, ativação da via da proteína cinase A, causando proliferação celular e secreção de GH.
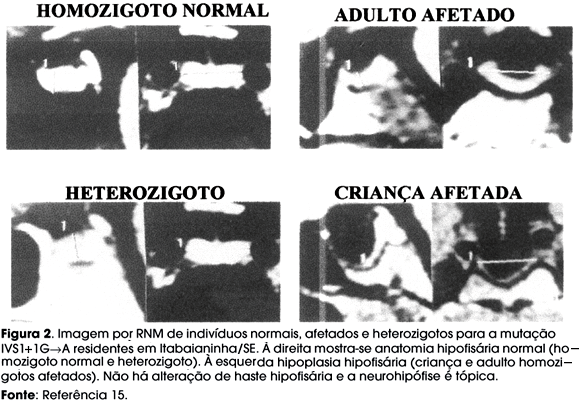
As mutações no GHRH-R definiram um modelo de resistência ao GHRH, causando uma deficiência genética isolada e grave do GH, cuja expressão fenotípica completa só pode ser estabelecida em Itabaianinha pelo grande número de indivíduos de várias gerações envolvidos (tabela 1). Como o GHRH é importante para o desenvolvimento dos somatotrófos hipofisários, os indivíduos homozigóticos para esta mutação apresentam uma hipoplasia da pituitária anterior pela acentuada redução dos mesmos, como sugerido em outras mutações bi-alélicas do GHRH-R, porém com menor número de pacientes afetados (13,14). Este achado foi demonstrado em estudo de morfologia pituitária utilizando Ressonância Nuclear Magnética (RNM) em 38 indivíduos de Itabaianinha com idade superior a 8 anos, homozigóticos afetados e normais e heterozigóticos. A altura da pituitária em mm não foi diferente em crianças (2,87 ± 0,79) e adultos (2,67 ± 0,87), porém, quando expressa em escores de desvio padrão, foi menor em crianças (-2,84 ± 0,79) que em adultos (-1,95 ± 0,58), sugerindo nos afetados uma ausente ou menos acentuada hiperplasia dos somatotrófos dos 10 aos 19 anos de idade, conforme acontece nos indivíduos normais. Não foram encontradas alterações da haste hipofisária e neuro-hipófise ectópica nos indivíduos afetados. Os indivíduos heterozigóticos apresentam volume hipofisário normal (figura 2) (15). Embora a hipoplasia da adeno-hipófise (RNM) não seja diagnóstica de mutações em um gene específico, sua ausência em um indivíduo com 8 ou mais anos de idade praticamente exclui uma mutação no gene do GHRH-R (15).
Este modelo homogêneo de deficiência genética isolada e grave do Hormônio de Crescimento permite estudar a ação do GH sobre o crescimento linear, suas ações anabólicas, como o aumento da massa muscular, e sua interação com vários reguladores metabólicos (glicose, aminoácidos, ácidos graxos livres) e hormônios como leptina (16) e Ghrelin (17), importantes para o estabelecimento adequado do tamanho (altura e peso) e composição corporal dos indivíduos (18). Estas importantes ações metabólicas que propiciaram a este hormônio o seu antigo nome, hormônio somatotrófico (17), serão analisadas neste artigo. A deficiência do hormônio de crescimento, ou somatotrófico, pode estar implicada com uma reduzida qualidade de vida e risco cardiovascular aumentado (17,18). No entanto, a maior parte dos trabalhos sobre deficiência do GH, seja no aspecto do crescimento, ou sobre os demais aspectos, são oriundas de casuísticas heterogêneas, onde a severidade da deficiência é variável e, em alguns casos, não há mesmo deficiência de GH.

Por outro lado, nas deficiências múltiplas, há sempre o problema da reposição de hormônio tireoideano, glicocorticóides e esteróides sexuais, cuja sub ou superdosagem podem apresentar morbidades inerentes, com repercussões sobre a qualidade de vida, aspectos cardiovasculares e longevidade. Desta forma, a comunidade de Itabaianinha talvez se constitua na única oportunidade de se averiguar as conseqüências da deficiência isolada e vitalícia do hormônio de crescimento ou somatotrófico.
A Importância do GHRH no Controle da Secreção de GH
A regulação hipotalâmica da secreção de GH é única, pois envolve dois fatores estimulatórios (GHRH e Ghrelin) e outro inibitório (somatostatina). No modelo de resistência ao GHRH, provocado pela mutação IVS1+1G®A no GHRH-R, os indivíduos afetados não respondem ao estímulo agudo ou crônico com GHRH, nem aos testes com Hipoglicemia Insulínica, Clonidina (9,21), e ao estímulo com GRP-2, que atua à semelhança do Ghrelin sobre os receptores GHS-R, embora haja um pico de GH de 0,49 ± 0,41ng/ml, significativamente maior que os valores basais (22). Dados ainda não publicados demonstram, também, uma ausência de resposta do GH com exercício físico (teste que combina supressão do tônus somatostatinérgico com liberação de GHRH). Desta forma, a resistência ao GHRH reduz drasticamente a resposta do GH aos vários estímulos que agem via GHRH, somatostatina ou mesmo via outros receptores como o GRP-2 ou Ghrelin, sugerindo que a integridade da via do GHRH é condição básica para a resposta do GH aos diversos estímulos farmacológicos testados, neste modelo que combina a mutação no GHRH-R e redução importante da massa de somatotrófos.
Relevância Diagnóstica do IGF-I na Avaliação do Crescimento
O crescimento linear dos ossos longos se dá na cartilagem de conjugação. O principal eixo hormonal envolvido na regulação do crescimento é o denominado eixo somatotrófico ou, mais recentemente, eixo hormônio de crescimento (GH)sistema de fatores de crescimento insulina-símile (insulin-like growth factor, IGF) (eixo GH-IGF). Este eixo constitui a via final mediante a qual a maioria dos fatores que atuam no processo de crescimento exerce sua ação. O recrutamento das células a partir da zona de reserva da cartilagem de crescimento (diferenciação dos condrócitos) parece ser determinado pela ação direta do GH, enquanto que a hiperplasia e a hipertrofia das células recrutadas (expansão clonal) depende da ação dos IGFs (23). A primeira referência propondo que as ações do hormônio de crescimento na promoção do crescimento não seriam ações diretas, mas sim mediadas por algum outro fator cuja síntese encontrava-se sob seu controle, data de 1957 (24).O clássico conceito de que o crescimento era determinado pelas concentrações de IGF circulantes (Teoria da Somatomedina, 1980's) sofreu uma revisão inicial em 1999 quando demostra-se que a inativação parcial do gene do IGF-I em hepatócitos de ratos era compatível com o crescimento pós-natal normal destes animais, ressaltando a atividade parácrina/autócrina dos IGFs e praticamente relegando a ação dos IGFs circulantes ao campo metabólico e à regulação da secreção hipofisária de GH (Teoria da Somatomedina revisada) (25,26). Entretanto, em meados de 2002, ocorre a volta à origem, com a demonstração de que animais nos quais uma diminuição ainda maior no IGF-I circulante foi obtida apresentavam diminuição do crescimento (Teoria da Somatomedina resgatada), confirmando a participação tanto do IGF circulante quanto do local no processo de crescimento, mediante atuação endócrina, parácrina e autócrina (27). É interessante que os animais com depleção hepato-específica do gene do IGF-I (modelo que acopla redução importante do IGF-I e GH elevado) apresentam uma diminuição da acumulação de gordura dependente da idade em relação aos animais controles (28), sugerindo ação importante do GH no acumulação de gordura. Porém, não há mais dúvida da participação endócrina, sobretudo hepática, da IGF-I sobre o crescimento. Na clínica, a deficiência de GH deve ser considerada um dos tipos de deficiência de IGF-I, sendo que esta deve ser o referencial básico na avaliação das causas de baixa estatura, conforme explicado a seguir.
Nós verificamos que vários componentes do eixo GH-IGFs (IGF-I, IGF-II, IGFBP-3 e sub-unidade de Ácido Labil) são gravemente diminuídos na DIGH de Itabaianinha (21). Comprovar apenas esta grave redução seria menosprezar o modelo de grave e isolada DIGH, situação incomum na prática clínica. Mais importante seria estabelecer o poder diagnóstico destes parâmetros, para utilizar os mais adequados, frente às situações comuns de dúvida diagnóstica, como na avaliação de crianças com baixa estatura em geral. Desta forma, dividimos o menor valor dos indivíduos normais pelo maior valor dos indivíduos com DIGH em Itabaianinha, em diferentes idades (tabela 2), para definir este poder diagnóstico. A dosagem de IGF-I apresentou um elevado poder diagnóstico nas crianças, jovens e idosos. Em adultos com menos de 50 anos, ela foi suplantada pela dosagem da ALS livre. A dosagem do IGFBP-3 não se posiciona entre os três primeiros parâmetros em nenhum grupo etário. Desta forma, nós sugerimos excluir a dosagem do IGFBP-3 na avaliação inicial da baixa estatura. Além do mais, nós propomos um modelo de avaliação laboratorial da Baixa Estatura, testado em mais de 200 crianças com diferentes causas a partir da dosagem inicial de IGF-I, complementada, quando necessária, pelos testes de estímulo para o GH, dados de imagem e outros parâmetros (29).
Freqüência das Mutações no Gene do GHRH-R
A DIGH genética compreende quatro formas de acordo com grau de deficiência de GH e modo de herança:
Tipo IA: herança autossômica recessiva, com níveis séricos ausentes de GH. Os pacientes desenvolvem anticorpos anti-GH com o uso da terapia com GH. Deve-se principalmente a grandes deleções no gene GH1, localizado no cromossoma 17q23 (30,31).
Tipo IB: forma mais freqüente de DIGH, herança autossômica recessiva com níveis de GH severamente diminuídos. Não desenvolvem anticorpos anti-GH e respondem bem à terapia com GH. Deve-se a mutações no GH1 e no gene do GHRH-R localizado no cromossoma 7p14 (9-13,32-37).
Tipo II: herança autossômica dominante com níveis séricos de GH severamente diminuídos. Não desenvolvem anticorpos anti-GH e respondem bem à terapia com GH. A maioria ocorre por mutações que levam à perda do exon 3 do GH1 (38).
Tipo III: forma mais rara de DIGH, com herança ligada ao X com complexos achados clínicos e por vezes associadas à agamaglobulinemia.
Estudo de 151 indivíduos afetados (europeus do norte, mediterrâneos e asiáticos) em 83 famílias mostrou uma prevalência de 66,7% de GH1 mutações em afetados com DIGH tipo IA e apenas 1,7% nos pacientes com DIGH (39). Desta forma, a maioria dos casos de DIGH tipo IB não é causada por mutações no GH1. Mutações no GHRH-R são cada vez mais descritas como causas de DIGH, seja em homozigose ou heterozigose composta. Para determinar a prevalência de mutações no GHRH-R, analisamos 30 famílias com pelo menos 2 membros com DIGH IB, e a freqüência de mutações encontrada neste gene foi 10% (34). Atualmente, estas mutações incluem uma na região promotora (40) duas tipo splicing (37,41), uma mutação sem sentido (37), 6 de sentido trocado (32,34,40) e 2 microdeleções (37,42). Uma revisão atual dos aspectos moleculares destas mutações foi recentemente publicada (43).
Dentre estas, enfatizamos a mutação L144H, substituição de leucina por histidina no códon 144, em duas famílias, uma em Sergipe e outra na Espanha, ambas demonstradas com a mesma origem através de estudo de Linkage. Esta mesma mutação foi documentada em uma família da Pensilvânia, nos Estados Unidos, com um background genético diferente. A demonstração de uma mesma mutação em continentes diferentes com duas origens distintas atesta a importância e a dispersão destas mutações (33).
Modificações Metabólicas, Composição Corporal e Risco Cardiovascular
A deficiência do GH de Itabaianinha provoca importantes modificações metabólicas e na composição corporal. As crianças apresentam uma redução da massa magra, que persiste na puberdade e na fase adulta. O percentual da massa gorda é a imagem em espelho da redução da massa magra, sendo maior em todas as idades. Os níveis de Leptina são mais elevados nos pacientes que nos controles, seja em concentração ou quando estes foram divididos pela quantidade de massa gorda, indicando uma relação inversa entre a secreção de GH e Leptina nestes indivíduos (44). Os níveis de colesterol total e LDL são mais elevados que nos controles em crianças e adultos (45,46). Já em crianças, há comprovação de obesidade central que se acentua nos adultos (44,46). Nestes, a pressão sistólica é mais elevada que nos controles, porém sem sinais de remodelamento cardíaco à ecocardiografia (46). Estes dados sugerem a existência de fatores de risco cardiovascular nestes indivíduos, porém a ocorrência maior de doença vascular ateromatosa nesta população não foi ainda demonstrada até o momento. A inter-relação DIGH e doença vascular coronariana ou cerebral está sendo alvo de protocolos em curso. Estes dados demonstram que o GH, além do efeito sobre o crescimento linear, tem importantes ações metabólicas que sugerem conservar, ao menos, seu primeiro nome, hormônio somatotrófico. A longevidade dos indivíduos com DIGH e suas causas de morte não parecem ser distintas do restante dessa população rural, com acesso exato a alimentos naturais ou com pouca manipulação. Maior atividade física e menor consumo de alimentos industrializados podem exercer um efeito benéfico sobre aqueles aspectos na população estudada. Implicações importantes com a Síndrome Metabólica podem advir do modelo de DIGH por mutação no GHRH-R de Itabaianinha.
DIGH Pode Não Comprometer a Qualidade de Vida
Embora inúmeras citações referem uma deteriorização na qualidade de vida dos pacientes com DIGH, a heterogeneidade de pacientes estudados, muitos com pan-hipopituitarismo e com outras reposições (tireoideana, corticoterapia, esteróides sexuais), alguns com DIGH com origem na infância, outros na fase adulta, alguns submetidos à cirurgia hipofisária, outros à radioterapia, alguns com deficiência severa de GH, outros com deficiência moderada e alguns mesmo sem deficiência de GH, torna extremamente discutível o perfil da qualidade de vida nesses pacientes, que inclui diminuição do humor, tendência à depressão, menor capacidade laborativa e pior desempenho sexual. A tabela 3 apresenta dados de qualidade de vida através de questionário adaptado de Wallyamahmed (47) e World Health Organization Quality of Life (48) aplicado a 26 indivíduos com DIGH devido à mutação IVS1+1G®A no GHRH-R de Itabaianinha. Observa-se que o grau de satisfação foi superior ao grau de insatisfação nos itens vida familiar, sexo, diversão e sociabilidade, e não houve diferenças entre o percentual de respostas nos demais fatores inquiridos. Os mesmos são longevos, trabalham ativamente, são férteis, mesmo em idade avançada, não têm dificuldade para relacionamentos sociais ou afetivos. Estes dados não sugerem, nessa comunidade rural do Nordeste Brasileiro, uma piora da qualidade de vida dos pacientes com severa deficiência isolada de GH.
CONCLUSÕES
A mutação IVS1+1G®A, também encontrada em outras populações brasileiras, ao ser introduzida na comunidade de Itabaianinha, devido ao seu grande isolamento geográfico e alto índice de consangüinidade, propiciou pelo efeito fundador e deriva genética uma família singular que representa o mais valioso agrupamento até agora descrito para a caracterização da história natural da deficiência do hormônio do crescimento ou somatotrófico.
Os anões de Itabaianinha constituem um exemplo notável da interação genético-ambiental à época da codificação do genoma humano. Este experimento da natureza amplia os dados dos laboratórios de biologia molecular e de modelos animais de knock out genético para avaliação, seja da baixa estatura idiopática ou das conseqüências da deficiência vitalícia do hormônio de crescimento. De tema exótico, passa a ter repercussões sobre a atividade cotidiana do endocrinologista pediátrico ou geral. As principais características fenotípicas desta mutação foram descritas. Outros aspectos serão incorporados no futuro. Por enquanto, ela reincorpora à denominação atual (hormônio do crescimento), a antiga (hormônio somatotrófico) do GH.
Recebido em 19/11/03
Revisado em 04/03/04
Aceito em 14/03/04
- 1. Evans HM, Long JA. The effect of the anterior lobe administered intraperitoneal upon growth maturity an oestrus cycles of the rat. Anat Rec 1999;21:62-3.
- 2. Li CH, Evans HM. Chemistry of anterior pituitary hormones. In: Pincus G, Thieman KV, eds. The hormones. New York: New York Academic Press, 1948
- 3. Medvei VC. Present trends and outlook for the future Part II. In: The history of clinical endocrinology. London: Parthenon Publishing Group, 1993 p.337-64.
- 4. Thorner MO. The discovery of Growth Hormone Releasing Hormone. J Clin Endocrinol Metab 1999;84:4671-6.
- 5. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999;402:656-60.
- 6. Rosicka M, Krsek M, Jarkovska Z, Marek J, Schreiber V. Ghrelin a new endogenous growth hormone secretagogue. Physiol Res 2002;51:435-41.
- 7. Laron Z. Syndrome of familial dwarfism and high plasma immunoreactive growth hormone. Isr J Med Sci 1974;10:1247-53.
- 8. Berg MA, Guevara-Aguirre JG, Rosenbloom AL, Rosenfeld RG, Francke U. Mutation creating a new donor splice site in the growth hormone receptor genes of 37 Ecuadorian patients with Laron syndrome. Hum Mut 1999;1:24-34.
- 9. Salvatori R, Hayshida CY, Aguiar-Oliveira MH, Phillips III JA, Souza AHO, Gondo RG, et al. Familial Dwarfism due a Novel Mutation of the Growth Hormone Releasing Hormone Receptor gene. J Clin Endocrinol Metab 1999;84:917-23.
- 10. Wajnrajch MP, Gertner JM, Harbinson MD, Chua SC Jr, Leibel RL. Nonsense Mutation in the Human Growth Hormone-Releasing Hormone Receptor causes Growth Failure Analogous To the Little (lit) Mouse. Nat Genet 1996;12:88-90.
- 11. Baumann G, Maheshwari HG. The dwarfs of Sindh: severe growth hormone receptor (GH) deficiency caused by a mutation in the growth hormone releasing hormone receptor gene. Acta Pediatr Scand 1997;423:33-8.
- 12. Maheshwari HG, Silverman BL, Dupuis J, Baumann G. Phenotype and genetic analyses of a syndrome caused by an inactivating mutation in the growth hormone-releasing hormone receptor: dwarfism of Sindh. J Clin Endocrinol Metab 1998;83:4065-74.
- 13. Netchine I, Talon P, Dastot F, Vitaux F, Goossens M, Amselem S. Extensive phenotypic analysis of a family with growth hormone (GH) deficiency caused by a mutation in the GH-releasing hormone receptor gene. J Clin Endocrinol Metab 1998;83:432-6.
- 14. Murray RA, Maheshwari HG, Russel EJ, Baumann G. Pituitary hypoplasia in patients with a mutation in the growth hormone-releasing hormone receptor gene. Am J Neuroradiol 2000;21:685-9.
- 15. Oliveira HA, Salvatori R, Krauss MP, Oliveira CRP, Silva PRC, Aguiar-Oliveira MH. Magnetic resonance imaging study of pituitary morphology in subjects homozygous and heterozygous for a null mutation of the GHRH receptor gene. Eur J Endocrinol 2003;148:427-32.
- 16. Gill MS, Toogood AA, O'Neil PA, Adams JE, Thorner MO. Clayton PE, et al. Relationship between growth hormone (GH) status, serum leptin and body composition in healthy and GH deficient elderly subjects. Clin Endocrinol 1997;47:161-7.
- 17. Penalva A, Duran S, Otero XL, Popovic V, Spiliotis B, Dieguez C, et al. The role of growth hormone secretagogues in the regulation of the growth hormone-insulin-like growth factors-I axis. In: Lamberts SWJ, Ghigo, eds. The expanding role of octreotide II: Advances in endocrinology and eye diseases. Bristol. BioScientifica Ltd., 2002 p.195-207.
- 18. Dieguez C, Casnueva FF. Influence of metabolic substrates and obesity on growth hormone secretion. Trends Endocrinol Metab 1995;6:55-9.
- 19. Rosen T, Bengtsson BA. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet 1990;336:285-8.
- 20. Bates AS, Van't Hoff W, Jones PJ, Clayton RN. The effect of hypo-pituitarism on life expectancy. J Clin Endocrinol Metab 1996;81:1169-72.
- 21. Aguiar-Oliveira MH, Gill MS, Barreto ESA, Alcântara MRS, Miraki-Moud F, Menezes CA, et al. Effect of severe growth hormone (GH) deficiency due to a mutation in the GH-releasing hormone receptor on insulin-like growth factors (IGF-1), IGF-binding proteins, and ternary complex formation throughout life. J Clin Endocrinol Metab 1999;84:4118-25.
- 22. Gondo RG, Aguiar-Oiveira MH, Hayshida CY, Toledo SPA, Abelin N, Levine MA, et al. Growth hormone-releasing peptide-2 stimulates GH secretion in GH-deficient patients with mutated GH-releasing hormone receptor. J Clin Endocrinol Metab 2001;86:3279.
- 23. Clayton PE, Gill MS. Normal growth and its endocrine control. In: Brook CGD, Hindmarsh PC eds. Clinical paediatric endocrinology, 4th ed. Oxford: Blackwell Science, 2001 p.95-114.
- 24. Salmon WD Jr, Daughday WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med 1957;49:825-36.
- 25. Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Ohlsson C, et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci 1999;96:7088-92.
- 26. Butler AA, Le Roith D. Control of growth by the somatotropic axis: growth hormone and the insulin-like growth factors have related and independent roles. Annu Rev Physiol 2001;63:141-64.
- 27. Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CLL, Wu Y, Liu JL, et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest 2002;110:771-81.
- 28. Sjogren K, Jansson JO, Isakksson OG, Ohlsson C. A model for tissue-specific inducible insulin-like growth factor-I inactivation to determine the physiological role of liver derived IGF-I. Endocrine 2002;19:249-56.
- 29. Martinelli C, Oliveira CRP, Britto AVO, Costa FO, Silva PRC, Serpa MG, et al. Diagnóstico da deficiência do hormônio de crescimento, a rigor de IGF-I. Arq Bras Endocrinol Metab 2002;46:27-34.
- 30. Cogan JD, Phillips III JA. Growth disorders caused by genetics defects in the growth hormone pathway GH deficiency. In: Barness LA, Morron III G, Rudolph AM, eds. Advances in pediatrics. St. Louis: Mosby, 1998:45:337-61.
- 31. Phillips III JA, Hjelle BL, Seeburg PH. Molecular Basis for Familial Isolated Growth Hormone Deficiency. Proc Natl Acad Sci USA 1981;78:6372-5.
- 32. Carakushanski M, Whatmore AJ, Clayton PE, Shalet SM, Gleeson HK, Price DA, et al. A new missense mutation in the growth hormone releasing hormone receptor gene in familial isolated GH deficiency. Eur J Endocrinol 2003;148:25.
- 33. Salvatori R, Aguiar-Oliveira MH, Monte LBV, Hedgest L, Santos NL, Pereira RMC, et al. Detection of a recurring mutation in the human growth hormone releasing hormone receptor gene. Clin Endocrinol 2002;57.
- 34. Salvatori R, Fan X, Phillips III JA, Espigares-Martin R, Martin de Lara I, Freeman KL, et al. Three new mutations in the gene for the growth hormone (GH)-releasing hormone receptor in familial isolated GH deficiency type IB. J Clin Endocrinol Metab 2001;86:273-9.
- 35. Salvatori R, Fan X, Phillips III JA, Prince M, Levine MA. Isolated growth hormone (GH) deficiency due to compound heterozygosity for 2 new mutations in the GH-releasing hormone receptor gene. Clin Endocrinol 2001;54:681.
- 36. Salvatori R, Fan X, Mullis PE, Haile A, Levine MA. Decreased expression of the growth hormone-releasing hormone receptor gene due to a mutation in a Pit-1 binding site. Mol Endocrinol 2002;450.
- 37. Salvatori R, Fan X, Veldhuis J, Couch R. Serum GH response to pharmacological stimuli and physical exercise in two siblings with two new inactivating mutations in the GH-releasing hormone receptor gene. Eur J Endocrinol 2002;147:591.
- 38. Lee MS, Wajnrajch MP, Kim SS, Plotnick LP, Wang J, Gertner JM, et al. Autossomal dominant growth hormone (GH) deficiency type II: the Del 32-71-GH deletion mutant suppresses secretion of wild-type GH. Endocrinology 2000;141:8838-90.
- 39. Wagner JK, Eblé A, Hindmarsh PC, Mullis PE. Prevalence of human GH-1 alterations in patients with isolated growth hormone deficiency. Pediatr Res 1998;43:105-10.
- 40. Salvatori R, Fan X, Mullis PE, Haile A, Levine MA. Decreased expression of the GHRH receptor gene due to a mutation in a PIT-1 binding site. Mol Endocrinol 2002;16:450-8.
- 41. Roelfsema F, Biermasz NR, Veldman RG, Veldhuis JD, Frolich M, Stokvis-Brantsma WH, et al. Growth hormone (GH) secretion in patients with an inactivating defect of the GH-releasing hormone (GHRH) receptor is pulsatile: evidence for a role for non GHRH inputs in the generation of GH pulses. J Clin Endocrinol 2001;86:2459-64.
- 42. Horikawa R. Growth hormone-releasing hormone deficiency. Clin Pediatric Endocrinol 2000;9/14:35-9.
- 43. Alba M, Salvatori R. GHRH receptor mutations in familial GH deficiency. The Endocrinologist 2003;13:422-7.
- 44. Barretto ESA, Gill MS, Freitas MES, Magalhães MG, Souza AHO, Aguiar-Oliveira MH, et al. Serum leptin and body composition in children with familial GH deficiency (GHD) due to a mutation in the growth hormone-releasing hormone (GHRH) receptor. Clin Endocrinol 1999;51:559-64.
- 45. Gleeson HK, Souza AOH, Gill MS, Wieringa GE, Barretto ESA, Barretto-Fillho JAS, et al. Lipid profiles in untreated severe congenital isolated growth hormone deficiency through the lifespan. Clin Endocrinol 2002;57:89.
- 46. Barretto-Filho JA, Alcântara MR, Salvatori R. Familial isolated growth hormone deficiency is associated with increased systolic blood pressure, central obesity, and dyslipidemia. J Clin Endocrinol Metab 2002;87:2018.
- 47. Wallymahmed ME, Bakert GA, Humphris G, Dewey M, MacFarlane IA. The development, reliability and validity of a disease-specific quality of life model for adults with growth hormone deficiency. Cin Endocrinol (Oxf) 1996;44:403-11.
- 48. Katscnig. Instrumentos de avaliação de qualidade de vida. Organização Mundial de Saúde/World health Organization Quality of Life Assessment, 1998 Internet: www.hcpa.ufrgs.br/psiq
Endereço para correspondência
Datas de Publicação
-
Publicação nesta coleção
26 Ago 2004 -
Data do Fascículo
Jun 2004
Histórico
-
Aceito
14 Mar 2004 -
Revisado
04 Mar 2004 -
Recebido
19 Nov 2003