Resumos
OBJETIVO: Apresentar a experiência relativa a pacientes com deficiência da enzima 5alfa-redutase tipo 2 provenientes de três serviços distintos no Brasil. CASUÍSTICA E MÉTODOS: Foram incluídos 25 pacientes com sinais clínicos e hormonais de deficiência da 5alfa-redutase 2 pertencentes a 23 famílias, 15 oriundas da Bahia, 7 de São Paulo e 1 de Minas Gerais. Foram avaliados dados clínicos, hormonais e moleculares. A análise molecular dos 5 éxons do gene SRD5A2 foi feita por meio da técnica de PCR, seguida de seqüenciamento automático ou manual. RESULTADOS: Em 10 famílias havia mutações no gene SRD5A2 em homozigose (5 com G183S, 2 com R246W, 1 com G196S, 1 com del642T, 1 com 217_218insC) e em 3 em heterozigose composta (1 com Q126R/IVS3+1G>A, 1 com Q126R/del418T e 1 com Q126R/G158R); em 3 casos os afetados eram heterozigotos, apresentando apenas uma mutação deletéria (1 com G196S, 1 com A207D e 1 com R246W). Em 7 casos não foram detectadas anormalidades ao seqüenciamento. Observou-se maior freqüência da G183S em pacientes miscigenados (Afro-Euro-Brasileiros) oriundos da Bahia. Os achados clínicos e hormonais não diferiram entre os casos com e sem mutação, à exceção da freqüência de consangüinidade e da maior gravidade da ambigüidade genital nos primeiros. CONCLUSÕES: Os resultados encontrados salientam a importância da investigação molecular para o diagnóstico dessa doença, ressaltando o achado de uma mutação bastante freqüente em nosso meio (G183S), especialmente em pacientes miscigenados oriundos da Bahia, e a descrição de mutações que até o momento só foram relatadas em pacientes brasileiros.
Dihidrotestosterona; Intersexo; Pseudo-hermafroditismo; Testosterona; 5alfa-redutase
OBJECTIVE: To report the experience regarding patients with steroid 5alpha-reductase type 2 deficiency from three different clinical services in Brazil. CASUISTIC AND METHODS: Twenty five patients with clinical and hormonal features of 5alpha-reductase deficiency from 23 families (15 from Bahia, 7 from São Paulo and 1 from Minas Gerais) were included in this study. Clinical, hormonal and molecular data were evaluated. The molecular analysis of the five exons of the SRD5A2 gene was done by automatic or manual sequencing of PCR products. RESULTS: In ten families, SRD5A2 mutations were found in homozygosis (5 with G183S, 2 with R246W, 1 with G196S, 1 with del642T, 1 with 217_218insC), in three in compound heterozygosis (1 with Q126R/IVS3+1G>A, 1 with Q126R/del418T, 1 with Q126R/G158R) while other three were heterozygous, with only one deleterious mutation (1 with G196S, 1 with A207D, and 1 with R246W). In seven cases, no sequencing abnormalities were detected. The G183S substitution was the most frequently found among miscigenated patients (Afro-Euro-Brazilians) from Bahia. Hormonal and clinical findings did not differ between patients with or without mutations, exception made to a higher frequency of consanguinity and greater severity of genital ambiguity in the first group. CONCLUSION: Our results reinforce the importance of molecular investigation for the diagnosis of this disease and point out to the finding of a very frequent mutation (G183S) in our series, especially in patients with mixed ethnic background from Bahia, and the description of mutations that have only been reported in Brazilian patients so far.
Dihydrotestosterone; Intersex; Pseudohermaphroditism; Testosterone; 5alpha-reductase deficiency
ARTIGO ORIGINAL
Deficiência de 5a-redutase tipo 2: experiências de Campinas (SP) e Salvador (BA)
5a-reductase type 2 deficiency: experiences from Campinas (SP) and Salvador (BA)
Christine HackelI,II,IV; Luiz Eduardo C. de OliveiraI; Maria Betania TorallesVI; Daniela Nunes-SilvaVI; Maria Manuela O. ToniniI; Lúcio Fábio Caldas FerrazI; Leandra SteinmetzVII; Durval DamianiVII; Laurione Cândido de OliveiraII; Andréa T. Maciel-GuerraIII,IV; Eliana Gabas Stuchi-PerezIII; Gil Guerra-JúniorIII,V
ICentro de Biologia Molecular e Engenharia Genética CBMEG
IILaboratório de Fisiologia do Departamento de Patologia Clínica
IIIGrupo Interdisciplinar de Estudos da Determinação e Diferenciação do Sexo GIEDDS
IVDepartamento de Genética Médica
VDepartamento de Pediatria da FCM da UNICAMP, Campinas, SP
VIDepartamento de Pediatria da FMUFBA, Salvador, BA
VIIInstituto da Criança da FMUSP, São Paulo, SP
Endereço para correspondência Endereço para correspondência Christine Hackel Centro de Biologia Molecular e Engenharia Genética (CBMEG) Laboratório de Genética Humana Universidade Estadual de Campinas UNICAMP 13083-970 Campinas, SP E-mail: hackel@unicamp.br
RESUMO
OBJETIVO: Apresentar a experiência relativa a pacientes com deficiência da enzima 5a-redutase tipo 2 provenientes de três serviços distintos no Brasil.
CASUÍSTICA E MÉTODOS: Foram incluídos 25 pacientes com sinais clínicos e hormonais de deficiência da 5a-redutase 2 pertencentes a 23 famílias, 15 oriundas da Bahia, 7 de São Paulo e 1 de Minas Gerais. Foram avaliados dados clínicos, hormonais e moleculares. A análise molecular dos 5 éxons do gene SRD5A2 foi feita por meio da técnica de PCR, seguida de seqüenciamento automático ou manual.
RESULTADOS: Em 10 famílias havia mutações no gene SRD5A2 em homozigose (5 com G183S, 2 com R246W, 1 com G196S, 1 com del642T, 1 com 217_218insC) e em 3 em heterozigose composta (1 com Q126R/IVS3+1G>A, 1 com Q126R/del418T e 1 com Q126R/G158R); em 3 casos os afetados eram heterozigotos, apresentando apenas uma mutação deletéria (1 com G196S, 1 com A207D e 1 com R246W). Em 7 casos não foram detectadas anormalidades ao seqüenciamento. Observou-se maior freqüência da G183S em pacientes miscigenados (Afro-Euro-Brasileiros) oriundos da Bahia. Os achados clínicos e hormonais não diferiram entre os casos com e sem mutação, à exceção da freqüência de consangüinidade e da maior gravidade da ambigüidade genital nos primeiros.
CONCLUSÕES: Os resultados encontrados salientam a importância da investigação molecular para o diagnóstico dessa doença, ressaltando o achado de uma mutação bastante freqüente em nosso meio (G183S), especialmente em pacientes miscigenados oriundos da Bahia, e a descrição de mutações que até o momento só foram relatadas em pacientes brasileiros.
Descritores: Dihidrotestosterona; Intersexo; Pseudo-hermafroditismo; Testosterona; 5a-redutase
ABSTRACT
OBJECTIVE: To report the experience regarding patients with steroid 5a-reductase type 2 deficiency from three different clinical services in Brazil.
CASUISTIC AND METHODS: Twenty five patients with clinical and hormonal features of 5a-reductase deficiency from 23 families (15 from Bahia, 7 from São Paulo and 1 from Minas Gerais) were included in this study. Clinical, hormonal and molecular data were evaluated. The molecular analysis of the five exons of the SRD5A2 gene was done by automatic or manual sequencing of PCR products.
RESULTS: In ten families, SRD5A2 mutations were found in homozygosis (5 with G183S, 2 with R246W, 1 with G196S, 1 with del642T, 1 with 217_218insC), in three in compound heterozygosis (1 with Q126R/IVS3+1G>A, 1 with Q126R/del418T, 1 with Q126R/G158R) while other three were heterozygous, with only one deleterious mutation (1 with G196S, 1 with A207D, and 1 with R246W). In seven cases, no sequencing abnormalities were detected. The G183S substitution was the most frequently found among miscigenated patients (Afro-Euro-Brazilians) from Bahia. Hormonal and clinical findings did not differ between patients with or without mutations, exception made to a higher frequency of consanguinity and greater severity of genital ambiguity in the first group.
CONCLUSION: Our results reinforce the importance of molecular investigation for the diagnosis of this disease and point out to the finding of a very frequent mutation (G183S) in our series, especially in patients with mixed ethnic background from Bahia, and the description of mutations that have only been reported in Brazilian patients so far.
Keywords: Dihydrotestosterone; Intersex; Pseudohermaphroditism; Testosterone; 5a-reductase deficiency
A CONVERSÃO DA TESTOSTERONA (T) em 5a-dihidrotestosterona (DHT) é intermediada pela enzima 5a-redutase tipo 2. Uma das causas de pseudo-hermafroditismo masculino (PHM) é a deficiência desta enzima, de herança autossômica recessiva, na qual a conversão da T em DHT é nula ou defeituosa. A DHT induz a virilização da genitália externa para formar o pênis, a uretra, a próstata e a bolsa escrotal durante a vida fetal e atua no desenvolvimento dos caracteres sexuais secundários durante a puberdade. Os afetados são indivíduos com cariótipo 46,XY que, ao nascimento, apresentam freqüentemente ambigüidade genital. A variabilidade fenotípica é ampla, podendo haver desde uma genitália externa com estruturas quase femininas (hipospadia pseudovaginal períneo-escrotal) até um fenótipo masculino sem qualquer sinal clínico grave característico deste distúrbio (1).
A partir da análise de um banco de cDNA de próstata humana, foram isolados dois genes que codificam duas isoenzimas da 5a-redutase: o gene da 5a-redutase tipo 1 (SRD5A1) e o da 5a-redutase tipo 2 (SRD5A2), assim denominados de acordo com a ordem cronológica em que foram descobertos. Os genes SRD5A1 e SRD5A2 apresentam um mesmo arcabouço genético composto por 5 éxons e 4 íntrons e sítios de splicing idênticos. A homologia quanto à seqüência de bases é de aproximadamente 50% (2-4).
A isoenzima tipo 1 é uma proteína de 259 aminoácidos cujo gene SRD5A1 está localizado em 5p15, exibindo expressão preferencial na pele, fígado e em outros tecidos, com pouca importância fisiológica nos tecidos genitais (5). Já a 5a-redutase tipo 2 é uma proteína de 254 aminoácidos cujo gene SRD5A2 localiza-se em 2p23®22 (6), e se expressa principalmente nos tecidos genitais (próstata e rudimentos genitais externos), além de exibir alta afinidade por substratos esteróides. A ausência de mutações no gene SRD5A1 e a identificação de mutações (pontuais ou deleções) no gene SRD5A2 nos indivíduos afetados relacionou definitivamente o PHM por deficiência da 5a-redutase à isoenzima tipo 2 (3,7).
Apesar da aparente falta de importância da 5a-redutase tipo 1 no PHM por deficiência da 5a-redutase tipo 2, ela desempenha uma função fisiológica discreta. Uma das características da deficiência da 5a-redutase tipo 2 é a virilização da genitália externa durante a puberdade, e tal ocorrência tem merecido algumas tentativas de explicação. A primeira hipótese é a de que em algumas situações a enzima mutante ainda possuiria uma atividade residual suficiente para a formação da DHT, que, mesmo em concentrações mínimas, seria suficiente para a virilização na puberdade (8). Outra hipótese é a de que esta virilização seria fruto da própria ação de testosterona que, em concentrações séricas elevadas, poderia mimetizar os efeitos da DHT; finalmente, supõe-se que a 5a-redutase tipo 1 poderia estar promovendo esta virilização, uma vez que ocorre um aumento de sua expressão na pele durante a puberdade (5).
As concentrações séricas de testosterona e de DHT após o estímulo com gonadotrofina coriônica humana, o cálculo da razão T/DHT e a avaliação da atividade enzimática em fibroblastos de pele genital são os exames comumente realizados para diagnosticar a deficiência da 5a-redutase 2. Estes parâmetros permitem definir a etiologia nos casos mais gravemente afetados, onde a função enzimática se acha fortemente inibida, mas não são suficientes para diagnosticar formas mais discretas da deficiência (1). A avaliação molecular do gene SRD5A2 é necessária para a confirmação diagnóstica de todos os casos, particularmente em indivíduos gonadectomizados e em alguns pacientes pré-púberes, pois a dosagem hormonal depende da estimulação gonadal adequada, o que nem sempre é possível de se alcançar em pacientes que não atingiram a puberdade.
A virilização da genitália externa que ocorre na puberdade está associada, na maioria dos casos, ao surgimento de identificação masculina em pacientes criados como mulheres. Nesse contexto, a identificação de mutações no gene SRD5A2 tem contribuído não somente para confirmação da deficiência, mas também para o aconselhamento genético dos familiares dos afetados e para um melhor entendimento das bases moleculares da deficiência em 5a-redutase tipo 2.
O objetivo deste estudo foi o de apresentar as características clínicas, hormonais e moleculares de pacientes com ambigüidade genital, cariótipo 46,XY e quadro laboratorial sugestivo de deficiência da 5a-redutase 2 acompanhados em diferentes serviços de referência no Brasil.
CASUÍSTICA E MÉTODOS
A casuística foi composta de 25 pacientes com ambigüidade genital, gônadas palpáveis, história de consangüinidade entre os pais ou de casos semelhantes na família e relação T/DHT elevada. Destes, 14 eram acompanhados no Departamento de Pediatria da UFBA em Salvador (BA), 1 era acompanhado no Instituto da Criança da USP em São Paulo (SP) e os demais 10 eram acompanhados no Grupo Interdisciplinar de Estudos da Determinação e Diferenciação do Sexo (GIEDDS) da UNICAMP em Campinas (SP).
Todos os pacientes ou responsáveis assinaram o termo de consentimento pós-informação aprovado pelo Comitê de Ética em Pesquisa da FCM UNICAMP.
De todos os pacientes foram obtidos os dados de sexo social e idade no momento da avaliação, estado de origem dos pais, grupo racial, consangüinidade entre os pais, casos semelhantes na família, grau de ambigüidade genital segundo Prader (9), estadiamento puberal de pilificação de acordo com Marshall e Tanner (10), presença de próstata pela ultrassonografia pélvica, concentrações séricas de LH, FSH, T total (TT) e DHT. As dosagens hormonais foram realizadas no Laboratório de Fisiologia do Hospital de Clínicas da UNICAMP, sendo LH, FSH e TT pelo sistema modular Roche Elecsys® 2010 e a DHT por radioimunoensaio de fase sólida, por procedimento técnico de extração com n-hexano DSL®.
Foram colhidos cerca de 10 a 20mL de sangue total em frasco com EDTA e enviados ao Laboratório de Genética Humana do Centro de Biologia Molecular e Engenharia Genética (CBMEG) da UNICAMP, onde as amostras de DNA genômico foram extraídas a partir de leucócitos de sangue periférico conforme método descrito por Woodhead e cols. (11). Os cinco éxons e as regiões flanqueadoras foram amplificados por PCR (polymerase chain reaction) em reações independentes, utilizando-se cinco pares de primers (12), obtendo-se produtos de 171 a 369pb (pares de base). As reações foram realizadas em volumes de 50mL contendo 200 a 500ng de DNA genômico, 100mM de cada desoxinucleotídeo trifosfato (dATP, dCTP, dGTP e dTTP), 40pM de cada primer, 1 unidade de Taq DNA polimerase (Invitrogen, Califórnia, USA) em tampão Tris-HCl (pH 8,8 10mM) e MgCl2 (1,5mM). Após uma desnaturação inicial de 10 min a 95ºC, as amostras foram submetidas a 30 ciclos como se segue: desnaturação a 94ºC/1 min; anelamento a 60-68ºC/1 min e extensão a 72ºC/1 min.
Os produtos de amplificação foram submetidos a eletroforese em gel de agarose 2% em tampão Tris-Borato-EDTA (TBE) 1x. O gel foi corado com brometo de etídeo (0,2ng/mL) e os fragmentos amplificados foram visualizados sob iluminação ultravioleta a fim de se verificar a eficiência da reação.
Os produtos de PCR foram purificados utilizando-se o kit Wizard SV gel and PCR clean-up system (Promega). No início deste estudo, o seqüenciamento foi feito manualmente, empregando-se didesoxinucleotídeos 5-trifosfato (ddNTPs) marcados com a33-P e o Thermo SequenaseTM radiolabeled terminator cycle sequencing Kit (Amersham) de acordo com as indicações do fabricante. Subseqüentemente, as reações de seqüenciamento foram realizadas com o kit ABI Prism Terminator Big Dye Cycle Sequencing Ready Reaction Mix V. 3.1 (Perkin-Elmer), segundo recomendações do fabricante e analisadas no equipamento ABI Automated DNA Sequencer 377 (Perkin-Elmer). A fim de se excluir a possibilidade de que uma mutação de ponto tenha sido causada por um erro de incorporação da enzima Taq DNA polimerase, realizaram-se novas reações de PCR, seguidas por novas reações de seqüenciamento nos casos que apresentavam mutações.
Os dados foram analisados de forma descritiva.
RESULTADOS
Os 25 casos avaliados pertenciam a 23 famílias distintas. Dois pacientes eram irmãos (casos 16a e 16b nas tabelas 1 e 2) e havia ainda duas primas em primeiro grau (casos 1a e 1b nas tabelas 1 e 2). Os 14 casos acompanhados no Departamento de Pediatria da UFBA pertenciam a 13 famílias oriundas de cidades do estado da Bahia (casos 1a, 1b, 2, 5, 8, 9,12,13,15, e 18 a 23 nas tabelas 1 e 2), e o caso acompanhado no Instituto da Criança da USP vinha de uma família oriunda da cidade de São Paulo (caso 11 nas tabelas 1 e 2). Os demais 10 casos (casos 3, 4, 6, 7, 10, 11, 16a, 16b e 17 nas tabelas 1 e 2), acompanhados no GIEDDS UNICAMP, pertenciam a 9 famílias, sendo 6 oriundas de cidades do estado de São Paulo, 2 da Bahia (casos 3 e 7 nas tabelas 1 e 2) e 1 de Minas Gerais (caso 6 nas tabelas 1 e 2).
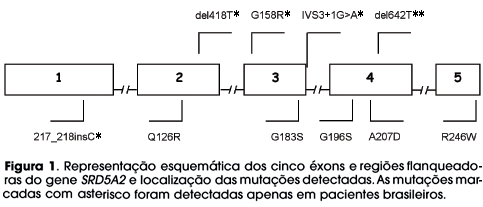
Em 10 das 23 famílias avaliadas foram encontradas mutações em homozigose, em 3 mutações em heterozigose composta e em 3 casos a mutação no gene SRD5A2 estava presente em heterozigose simples, totalizando 16 famílias (casos 1 a 16). Nas demais 7 famílias não foram identificadas mutações no gene SRD5A2 (casos 17 a 23) (tabela 1 e figura 1).
Das 10 famílias com mutações em homozigose, 8 apresentaram substituição de aminoácidos, sendo 5 com G183S (casos 1 a 5), 1 com G196S (caso 6) e 2 com R246W (casos 10 e 11); nas outras 2 havia alterações na fase de leitura, seja por perda de um nucleotídeo, del642T (caso 12) ou inserção de um nucleotídeo, 217_218insC (caso 13). Das três famílias com heterozigose composta, uma apresentou duas mutações que levam à substituição de aminoácidos (Q126R/ G158R) (caso 16), enquanto que nas outras duas foram observados genótipos compostos por uma mutação de troca de sentido em um dos alelos, associada à perda de um nucleotídeo no outro (Q126R/ del418T) (caso 14) ou a uma mutação em sítio doador de splicing (Q126R/IVS3+1G>A) (caso 15). Em três casos somente uma alteração molecular deletéria foi detectada, tratando-se, portanto, de heterozigotos para mutações no gene SRD5A2 que levam à substituição de aminoácidos, como G196S (caso 7), A207D (caso 8) e R246W (caso 11) (tabela 1 e figura 1).
Os dados referentes à origem das famílias e a classificação "racial" foram avaliados em relação às alterações moleculares observadas no gene SRD5A2 (tabela 1). Em relação às cinco famílias com a mutação G183S, quatro eram oriundas da Bahia e uma de São Paulo; todos os seis casos nestas famílias foram identificados como mulatos médios. Três famílias apresentaram a mutação R246W, sendo duas oriundas de São Paulo e uma da Bahia; duas com classificação racial de mulato claro e uma de branco. O mesmo ocorreu com a mutação Q126R, porém com 2 classificações raciais de branco e uma de mulato claro. Em relação às duas famílias com a mutação G196S, uma era oriunda de Minas Gerais e a outra de São Paulo, ambas classificadas como brancas.
As idades dos pacientes variaram de 21 dias a 27 anos (média= 10 anos, mediana= 8 anos e DP= 8,7 anos), sendo 10 acima de 14 anos, todos já com pubarca, entre os estádios 3 e 5 de Marshal e Tanner (10), e os demais abaixo de 9 anos (tabela 2).
Em sete das 16 famílias (43,7%) com mutação no gene da SRD5A2 havia consangüinidade entre os pais, e o mesmo dado somente foi observado em uma das sete (14,3%) famílias sem mutação (tabela 1). Em quatro das 16 famílias com mutação (25%) e em duas das 7 sem mutação (28,5%) houve história de casos semelhantes na família (tabela 1).
A ambigüidade genital variou entre os graus II e IV de Prader (9) entre os 18 casos com mutação (2 com grau II, 15 com grau III e 1 com grau IV), e entre os graus III e IV nos 7 casos sem mutação (1 com grau III e 6 com grau IV) (tabela 1). A próstata foi visualizada em apenas 3 dos 18 casos (16,7%) com mutação, e em 2 dos 7 casos (28,5%) sem mutação. Em 3 casos (12, 15 e 16b) já havia sido realizada a gonadectomia bilateral. O testículo foi palpado nos demais 22 casos, sendo em 17 deles na região inguinal e os demais 27 na região das saliências labioescrotais (tabela 1).
Na avaliação atual, seis indivíduos tinham sexo social e civil feminino, enquanto os demais 19, masculino. Havia história de mudança de sexo feminino para masculino em cinco casos, todos com mutação no gene SRD5A2 (tabela 1).
Na tabela 2, os casos são apresentados de acordo com a faixa etária e presença de pubarca para possibilitar uma melhor avaliação das dosagens hormonais. Três casos (12, 15 e 16b) já haviam realizado gonadectomia no momento desta avaliação e, portanto, não serão analisados em relação às dosagens hormonais. Portanto, restaram 22 casos, sendo 15 com mutação, dos quais 8 com pubarca, e 7 sem mutação, dos quais 2 com pubarca.
Entre os 15 casos com mutação, o LH variou de < 0,1 a 9,7UI/L, o FSH de 0,1 a 35,0UI/L, a TT de 1,5 a 15ng/mL, a DHT de 0,03 a 0,14ng/mL e a TT/DHT de 22 a 375. Entre os 7 casos sem mutação, o LH variou de < 0,1 a 11,8UI/L, o FSH de 0,5 a 35,6UI/L, a TT de 1,3 a 15ng/mL, a DHT de 0,03 a 0,14ng/mL e a TT/DHT de 43 a 160. As dosagens hormonais não diferenciaram os casos com mutação dos sem mutação (tabela 2).
DISCUSSÃO
A deficiência na atividade da 5a-redutase tipo 2 pode ser decorrente de formação de enzima não funcional, como resultado de deleções, mutações nonsense que resultam em códons de parada prematura, mutações nos sítios de splicing e mutações missense que levam à substituição de aminoácidos. Estas últimas também podem resultar na formação de enzima com estabilidade reduzida e/ou com afinidade diminuída por NADPH e/ou por T (13).
O estudo das mutações encontradas nesse gene permitiu a identificação de 2 domínios funcionais na enzima de 5a-redutase tipo 2: um domínio de ligação ao cofator NADPH e um domínio de ligação ao substrato esteróide (T). Mutações que afetam a ligação à T estão mapeadas nas extremidades do gene, nos éxons 1 e 5, enquanto que as mutações que afetam a ligação ao NADPH são mais numerosas e estão mapeadas principalmente nos éxons 3 e 4 (14).
Mais de 40 mutações diferentes já foram descritas, compreendendo em sua maioria mutações de ponto do tipo missense ou nonsense e, mais raramente, mutações que afetam o mecanismo de splicing, mutações frameshift (que alteram a fase de leitura) ou deleções completas do gene (8,13,15-20). Segundo Thigpen e cols. (15), aproximadamente 65% dos indivíduos afetados são homozigotos e 35% são heterozigotos compostos. A freqüência elevada de heterozigotos compostos sugere que a freqüência de portadores na população em geral pode ser alta, o que explicaria a recorrência de determinadas mutações em afetados não consangüíneos. Por outro lado, a recorrência de mutações idênticas em diferentes grupos étnicos e de regiões geográficas distintas tem sugerido a existência de regiões hot spots no gene; em outros casos, mutações similares dentro do mesmo grupo étnico seriam derivadas de mutações presentes em ancestrais comuns (8,13,21).
Além disso, variações polimórficas que levam à substituição de aminoácidos nos códons 49 (A49T) e 89 (V89L) vêm sendo relacionadas com deficiência de virilização e podem representar fatores genéticos de risco para a ocorrência de hipospadias (19,22).
Nesta série de pacientes, são apresentados os achados de mutações de ponto do tipo missense no gene SRD5A2 que já foram descritas em pacientes de outras populações (Q126R, G196S, G183S, A207D, R246W) ao lado de alterações moleculares que, até o momento, só foram encontradas em pacientes brasileiros. Estas últimas são representadas por uma mutação de ponto no éxon 4 que leva à substituição de aminoácido G158R, pelas deleções de uma timina nas posições 418 (del418T éxon 2) e 642 (del642T éxon 4), pela a inserção de uma citosina entre os nucleotídeos 217 e 218 no éxon 1 (217_218insC) e por uma mutação de ponto no sítio doador de splicing do íntron 3 (IVS3+1G>A) (20,23).
As seis substituições de aminoácidos (A207D, Q126R, G158R, G196S, G183S, R246W) causadas por mutações do tipo missense observadas nesta casuística ocorreram em regiões conservadas da enzima 5a-redutase tipo 2 tanto de origem humana quanto de rato e de macaco (24), sugerindo que essas posições são importantes para sua atividade. Com exceção da substituição G158R, as demais já foram anteriormente detectadas em pacientes com deficiência de 5 alfa redutase tipo 2 e constam da Human Gene Mutation Database do gene SRD5A2 (16).
Em relação às mutações missense já conhecidas, a recorrência da substituição G183S no presente trabalho foi observada em 4 famílias oriundas da Bahia e em uma do Estado de São Paulo. Esta mutação já havia sido observada anteriormente em dois pacientes brasileiros (13,15,25), também tendo sido detectada em 4 afetados de uma família da República Dominicana (26). Estudos in vitro revelaram que essa substituição diminui a afinidade da enzima pelo NADPH, com decréscimo de 90% da sua atividade enzimática (14). Embora possa se considerar o códon 183 como um hot spot no gene SRD5A2, é curioso notar que esta mutação foi identificada apenas em pacientes brasileiros miscigenados e em dominicanos, não havendo relato dessa mutação em outras partes do mundo, o que sugere a sua recorrência a partir de ancestrais comuns. Considerando-se a história da colonização do Brasil e da República Dominicana, constata-se que ambos os países apresentam um forte componente negróide na sua população, fruto do período da escravidão (27). Esses dados permitem supor que a substituição G183S surgiu em um grupo étnico ancestral (negros africanos, por exemplo) e distribuiu-se nas Américas à custa dos movimentos migratórios. O mesmo ocorreu neste estudo, sendo que todos os casos com esta mutação apresentaram classificação racial de mulato médio. Tendo em vista a elevada freqüência deste alelo mutante nesta série de pacientes brasileiros, pode-se sugerir que a triagem molecular para deficiência da 5a-redutase tipo 2 seja iniciada pela análise do éxon 3, especialmente entre pacientes Afro-descendentes oriundos da Bahia.
Quanto à substituição R246W, os achados in vitro descritos na literatura revelam que a proteína mutante apresenta afinidade diminuída pelo NADPH, com menor atividade enzimática e vida média significativamente reduzida em relação à proteína normal (7). É possível que o códon 246 seja alvo de mutações recorrentes, uma vez que duas mutações afetando o mesmo códon (R246W e R246Q) ocorrem em um dinucleotídeo CG e já foram encontradas em sete grupos étnicos diferentes: afro-americanos, europeus, paquistaneses, dominicanos, brasileiros, egípcios (7) e chineses (19). Em pacientes brasileiros, a substituição R246W foi detectada pela primeira vez em homozigose por Thigpen e cols. em 1992 (15), em um indivíduo de origem caucasóide do Estado de São Paulo. Dos 3 pacientes aqui relatados, 2 também são oriundos do mesmo estado e outro da Bahia, e todos mulatos claros, ou seja, com componente caucasóide importante. No entanto, em um dos pacientes (caso 11) desta amostra, esta mutação foi detectada somente em um dos alelos do gene, dificultando a interpretação dos achados clínicos deste indivíduo. Situação semelhante foi observada em relação à substituição A207D (caso 8), encontrada em heterozigose com um alelo normal. Esta troca de aminoácido leva à inativação total da enzima mutante (7), tendo sido descrita pela primeira vez por Thigpen e cols. em 1992 (15) em um paciente heterozigoto composto (A207D/ R246Q) de origem européia, e posteriormente detectada em homozigose em dois irmãos mexicanos (28).
A mutação A>G que leva à substituição de aminoácido Q126R já foi descrita anteriormente em pacientes brasileiros, portugueses, creoles de ascendência francesa (13,25), franceses (29) e alemães (1). Esta substituição leva à completa inativação da enzima reduzindo drasticamente a sua meia-vida em células transfectadas in vitro (14). Nos pacientes aqui examinados, é curioso observar que esta mutação foi detectada somente nos heterozigotos compostos (famílias 14 a 16), sendo 2 famílias do Estado de São Paulo de origem caucasóide e em um mulato claro da Bahia. Isto pode sugerir que este alelo mutante seja relativamente freqüente na nossa população, em conseqüência do forte componente europeu no processo de formação do povo brasileiro.
Em relação às mutações associadas que foram encontradas nos heterozigotos compostos aqui relatados, cabe lembrar que nem sempre é possível estabelecer se as duas mutações ocorrem no mesmo alelo (situação em cis) ou em alelos diferentes (situação em trans), em função da metodologia utilizada (análise éxon a éxon). Em relação ao paciente identificado pelo número 14, foi possível estabelecer que ambos alelos são mutantes. Nesse caso, as duas mutações situam-se no mesmo éxon e o seqüenciamento dos produtos de PCR após clonagem permitiu revelar separadamente cada mutação (23). A segunda mutação, a deleção de uma timina na posição 418 do éxon 2, prediz a alteração da fase de leitura do RNA mensageiro a partir da deleção, levando à substituição dos aminoácidos dos códons 140 a 158 e introduzindo um códon de parada prematuro na posição 159 (23). Desse modo, mesmo que este RNA mensageiro venha a ser processado, a proteína resultante seria truncada na sua porção carboxi-terminal e, muito provavelmente, não funcional.
Quanto aos outros pacientes heterozigotos compostos para a substituição Q126R, não foi possível demonstrar a independência ou não das mutações, situadas em éxons diferentes. Contudo, além de ser menos provável a ocorrência de duas mutações distintas no mesmo alelo, as características fenotípicas exibidas pelos pacientes e a elevação significativa da relação T/DHT sugerem fortemente que se tratam de mutações em alelos distintos e que comprometem significativamente a atividade da 5a-redutase tipo 2. Assim, no paciente identificado pelo número 15, observou-se que a segunda mutação (IVS3+1G>A) ocorre no primeiro nucleotídeo do sítio doador de splicing do íntron 3. Nesse caso, pode-se prever uma alteração no processamento do RNA mensageiro, quer por retenção do íntron, quer pela utilização de outro eventual sítio de splicing, o que, de todo modo, não resultaria numa enzima normal. Com efeito, em relação ao gene SRD5A2, existe um relato semelhante descrito na literatura, de uma mutação de ponto (G>T) no sítio doador de splicing do íntron 4, em um indivíduo de origem paquistanesa com atividade de 5a-redutase praticamente nula em fibroblastos de pele genital (15).
Quanto aos dois irmãos com genótipo Q126R/G158R (família 16), ainda não existem estudos in vitro que demonstrem o efeito da substituição G158R. No entanto, o fato de se tratar da substituição de um aminoácido neutro (glicina) por um aminoácido positivamente carregado (arginina) em uma posição conservada da proteína sugere fortemente que a atividade enzimática seja prejudicada. De fato, até mesmo a substituição de aminoácidos de natureza semelhante em códons conservados pode comprometer sua atividade funcional de maneira significativa, tal como ocorre com a substituição do aminoácido glicina por serina na posição 196 (G196S). Estudos in vitro demonstraram que essa alteração leva a uma redução de aproximadamente 90% da atividade enzimática da 5a-redutase tipo 2 em decorrência de uma drástica diminuição da afinidade dessa enzima pelo NADPH (14).
Nesta série de pacientes brasileiros, a mutação de ponto que leva a essa substituição foi identificada em 2 famílias distintas (casos 6 e 7). O caso 6, com 16 anos, filho de consangüíneos e criado no sexo feminino, vinha apresentando sinais de virilização e ausência de desenvolvimento mamário, acne ou pêlos faciais (30) e procurou o serviço médico na puberdade para mudança de registro civil; o caso 7, ainda pré-pubere e sem antecedentes familiares dignos de nota. No caso 6, homozigoto para a mutação, observou-se uma relação T/DHT significativamente elevada, o que não ocorreu no caso 7, onde a mutação foi detectada em heterozigose. Na literatura, a substituição G196S foi identificada em homozigose em três pacientes pré-puberes criados no sexo masculino, sendo um de origem grega (15) e os outros dois, não aparentados e filhos de consangüíneos, de origem turca (1). Subseqüentemente, foi encontrada em heterozigose com outra mutação missense (G196S/ H231R) em uma irmandade de origem sueca com três indivíduos afetados com hipospadias, micropênis, bolsa testicular bem desenvolvida e criptorquidia (31). Nessa irmandade, após as correções cirúrgicas, dois dos irmãos afetados já tiveram filhos (com paternidade comprovada) provavelmente porque ambas as substituições de aminoácidos levam a enzimas parcialmente funcionais.
A deleção de uma timina (del642T), detectada em homozigose no paciente identificado pelo número 12, provoca uma alteração no quadro de leitura do RNA mensageiro a partir do códon 215, levando ainda à incorporação de 22 aminoácidos adicionais na proteína correspondente, em conseqüência do deslocamento do códon de parada. Assim, é provável que esta alteração leve ao comprometimento da atividade enzimática, tendo em vista o quadro clínico apresentado pelo paciente. No entanto, a avaliação hormonal no presente caso foi prejudicada pelo fato de que este paciente foi submetido à gonadectomia cerca de um ano antes de sua inclusão no estudo.
Em relação à inserção 217_218insC encontrada no paciente identificado pelo número 13, como já comentado anteriormente, a alteração do quadro de leitura leva à criação de um códon de parada prematuro na posição 135. Assim, mesmo que esta proteína truncada seja sintetizada, haveria perda dos domínios de ligação ao NADPH (éxons 3 e 4) e à testosterona (éxon 5).
A ausência de alterações moleculares deletérias no gene SRD5A2 nos pacientes 17 a 23, bem como o achado de apenas um alelo mutante nos pacientes 7, 8 e 11, permite supor a existência de mutações em seqüências intrônicas ou em região promotora, que não foram estudadas no presente trabalho. Fenômenos epigenéticos (tais como metilação da região promotora) que bloqueiem a transcrição do alelo normal também devem ser considerados. Contudo, a região promotora do gene SRD5A2 ainda não foi devidamente caracterizada quanto às seqüências reguladoras que poderiam ser importantes no controle de sua expressão, restando esta questão em aberto para futuras investigações.
Do ponto de vista clínico, deve-se ressaltar que neste estudo, entre os casos com mutação do gene SRD5A2, observou-se uma maior freqüência de casamentos consangüíneos entre os pais, maior gravidade da ambigüidade genital, menor freqüência de casos com visualização da próstata e presença de casos com mudança de registro civil de feminino para masculino.
No entanto, ainda devido à variabilidade fenotípica e hormonal exibida pelos pacientes com deficiência de 5a-redutase tipo 2, os resultados encontrados salientam a importância da investigação molecular para o diagnóstico dessa doença. Deve-se ressaltar os achados de uma mutação (G183S) bastante freqüente em nosso meio, especialmente em pacientes com componente negróide oriundos da Bahia, a recorrência de mutações já descritas em outros grupos étnicos e a descrição de alterações moleculares que até o momento só foram relatadas em pacientes brasileiros.
AGRADECIMENTOS
Agradecemos ao CNPq pelo auxílio à pesquisa (GGJr, 472638/2003-3), à FAPESP e ao CNPq pelas bolsas de iniciação científica (LECO, FAPESP-02/13237-4 e MMOT, PIBIC 1997-1998), à CAPES pelas bolsas de mestrado (LFCF, 1997-1999 e EGSP, 1998-1999) e ao FAEP-UNICAMP pelo apoio inicial a esta pesquisa (CH, 566/1997).
Recebido em 05/10/04
Aceito em 04/11/04
- 1. Sinnecker GHG, Hiort O, Dibbelt L, Albers N, Dorr HG, Hauss H, et al. Phenotypic classification of male pseudo-hermaphroditism due to steroid 5a-reductase 2 deficiency. Am J Med Genet 1996;63:223-30.
- 2. Andersson S, Russell DW. Structural and biochemical properties of cloned and expressed human and rat steroid 5a-reductases. Proc Natl Acad Sci USA 1990;87:3640-4.
- 3. Jenkins EP, Andersson S, Imperato-McGinley J, Wilson JD, Russell DW. Genetic and pharmacological evidence for more than one human steroid 5a-reductase. J Clin Invest 1992;89:293-300.
- 4. Labrie F, Sugimoto Y, Luu-The V, Simard J, Lachance Y, Bachvarov D, et al. Structure of human type II 5a-reductase gene. Endocrinology 1992;131:1571-3.
- 5. Thigpen AE, Silver RI, Guileyardo JM, Casey ML, McConnell JD, Russell DW. Tissue distribution and ontogeny of steroid 5a-reductase isozyme expression. J Clin Invest 1993;92:903-10.
- 6. Morissette J, Durocher F, Leblanc J-F, Normand T, Labrie F, Simard J. Genetic linkage mapping of the human steroid 5a-reductase type 2 gene (SRD5A2) close to D2S352 on chromosome region 2p23Æp22. Cytogen Cell Genet 1996;73:304-7.
- 7. Russell DW, Wilson JD. Steroids 5a-reductases: two genes/two enzymes. Annu Rev Biochem 1994;63:25-61.
- 8. Forti G, Falchetti A, Santoro S, Davis DL, Wilson JD, Russell DW. Steroid 5a-reductase 2 deficiency: virilization in early infancy may be due to partial function of mutant enzyme. Clin Endocrinol 1996;44:477-82.
- 9. Prader A. Der genital befund bein pseudohermaphroditismus femininus des kongenitalen adrenogenitalen syndrome. Helv Pediatr Acta 1954;9:231-48.
- 10. Marshall WA, Tanner JM. Variation in pattern of pubertal changes in boys. Arch Dis Child 1969;44:201-3.
- 11. Woodhead JL, Fallon R, Figueredo H, Langdale J, Malcom ADB. Alternative methodology of gene diagnosis. In: Davies KE, editor. Human genetics diseases A practical approach Oxford:IRL Press Limited; 1986p.51-64.
- 12. Katz MD, Cai LQ, Zhu Y, Herrera C, DeFillo-Ricart M, Shackleton HL, et al. The biochemical and phenotypic characterization of females homozygous for 5a-reductase deficiency. J Clin Endocrinol Metab 1995;80:3160-7.
- 13. Wilson JD, Griffin JE, Russell DW. Steroid 5a-reductase 2 deficiency. Endocr Rev 1993;14:577-93.
- 14. Wigley WC, Prihoda JS, Mowszowicz I, Mendonça BB, New MI, Wilson JD, et al. Natural mutagenesis study of the human steroid 5a-reductase 2 isozyme. Biochemistry 1994;33:1265-70.
- 15 Thigpen AE, Davis D, Milatovich A, Mendonça BB, Imperato-McGinley J, Griffin JE, et al. Molecular genetics of 5a-reductase deficiency. J Clin Invest 1992;90:799-809.
-
1616. Human Gene Mutation Database SRD5A2 gene (http://uwcmml1s.uwcm.ac.uk/uwcm/mg/search/127343.html). Acessado em 28/09/2004.
- 17. Ocal G, Adiyaman P, Berberoglu M, Cetinkaya E, Akar N, Uysal A, et al. Mutations of the 5alpha-steroid reductase type 2 gene in six Turkish patients from unrelated families and a large pedigree of an isolated Turkish village. J Pediatr Endocrinol Metab 2002;15:411-21.
- 18. Fernandez-Cancio M, Nistal M, Garcia R, Molina MA, Tovar JA, Esteban C, et al. Compound heterozygous mutations in the SRD5A2 gene exon 4 in a male pseudohermaphrodite patient of Chinese origin. J Androl 2004;25:412-6.
- 19. Wang Y, Qiang L, Xu J, Liu Q, Wang W, Lin Y, et al. Mutation analysis of five candidate genes in Chinese patients with hypospadias. Eur J Hum Genet 2004;12:706-12.
- 20. Hackel C, Oliveira LEC, Ferraz LFC, Tonini MMO, Silva DN, Toralles MB, et al. New mutations, hotspots and founder effects in Brazilian patients with steroid 5a-reductase deficiency type 2. J Mol Med 2004; in press.
- 21. Mazen I, Gad YZ, Hafez M, Sultan C, Lumbroso S. Molecular analysis of 5alpha-reductase type 2 gene in eight unrelated Egyptian children with suspected 5alpha-reductase deficiency: prevalence of the G34R mutation. Clin Endocrinol 2003;58:627-31.
- 22. Silver RI, Russell DW. 5a-reductase type 2 mutations are present in some boys with hypospadias. J Urology 1999;162:1142-5.
- 23. Ferraz LFC, Baptista MTM, Maciel-Guerra AT, Guerra-Junior G, Hackel C. New frameshift mutation in the 5alpha-reductase type 2 gene in a Brazilian patient with 5alpha-reductase deficiency. Am J Med Genet 1999;87:221-5.
- 24. Levy MA, Brandt M, Sheedy KM, Holt DA, Heaslip JI, Trill JJ, et al. Cloning, expression and functional characterization of type 1 and type 2 steroid 5a-reductases from Cynomolgus monkey: comparisons with human and rat isoenzymes. J Steroid Biochem Molec Biol 1995;52:307-19.
- 25. Mendonça BB, Inácio M, Costa EM, Arnhold IJ, Silva FA, Nicolau W, et al. Male pseudohermaphroditism due to steroid 5alpha-reductase 2 deficiency. Diagnosis, psychological evaluation, and management. Medicine (Baltimore) 1996;75:64-76.
- 26. Cai L-Q, Zhu Y-S, Katz MD, Herrera C, Baéz J, DeFillo-Ricart M, et al. 5a-reductase-2 gene mutations in the Dominican Republic. J Clin Endocrinol Metab 1996;81:1730-5.
- 27. Weil TE, Black JK, Blutstein HI, Johnston KT, McMorris DS, Munson FP. Social system. In: Area Handbook for the Dominican Republic Superintendent of Documents:US Government Printing Office; 1973p.49-55.
- 28. Canto P, Vilchis F, Chávez B, Mutchinick O, Imperato-McGinley J, Pérez-Palacios G, et al. Mutations of the 5a-reductase type 2 gene in eight Mexican patients from six different pedigrees with 5a-reductase-2 deficiency. Clin Endocrinol 1997;46:155-60.
- 29. Boudon C, Lumbroso S, Lobaccaro JM, Szarras-Czapnik M, Romer TE, Garandeau P, et al. Molecular study of the 5a-Reductase type 2 gene in three European families with 5a-Reductase deficiency. J Clin Endocrinol Metab 1995;80:2149-53.
- 30. Ferraz LFC, Guerra-Junior G, Baptista MTM, Maciel-Guerra AT, Hackel C. Detection of Gly-196-Ser mutation in 5a-reductase type II gene in Brazilian patient with female assignment and behavior. J Pediatr Endocrinol Metab 1998;11:465-6.
- 31. Nordenskold A, Ivarsson S-A. Molecular characterization of 5a-reductase type 2 deficiency and fertility in a Swedish family. J Clin Endocrinol Metabol 1998;83:3236-8.
Datas de Publicação
-
Publicação nesta coleção
09 Maio 2005 -
Data do Fascículo
Fev 2005
Histórico
-
Aceito
04 Nov 2004 -
Recebido
05 Out 2004