Resumos
A disfunção endotelial está associada a diversas alterações vasculares, como a aterosclerose, hipertensão arterial, hiperlipidemia e diabetes mellitus, que têm em comum a resistência à insulina (RI). Citocinas são proteínas de baixo peso molecular, com diversas funções metabólicas e endócrinas, que participam da inflamação e resposta do sistema imune. Várias dessas citocinas são consideradas como fatores de risco independentes para doenças da artéria coronária e cerebrovascular. As principais fontes de citocinas (adipocinas) são os tecidos adiposos subcutâneo e visceral. Assim, aumento da massa de tecido adiposo está associado com alterações da produção de adipocina com aumento da expressão de fator de necrose tumoral alfa (TNF-alfa), interleucina 6 (IL-6), inibidor do fator ativador de plasminogênio 1 (PAI-1), e diminuição da expressão de adiponectina no tecido adiposo. A condição pró-inflamatória associada a essas alterações sugere ligação entre RI e disfunção endotelial no estágio inicial do processo de aterosclerose, em indivíduos obesos e em pacientes diabéticos tipo 2. A redução da massa de tecido adiposo, por redução de peso associada a exercício físico, reduz TNF-alfa, IL-6 e PAI-1, aumenta adiponectina, e melhora tanto a sensibilidade à insulina quanto a função endotelial. A interação entre adipocinas e insulina no controle da função endotelial será discutida, bem como o conceito de que a alteração da secreção de adiponectinas na RI e/ou obesidade piora a função endotelial, além de diminuir ainda mais a sensibilidade à insulina.
Resistência insulínica; Endotélio; Citocinas; Disfunção endotelial
Endothelial dysfunction is associated with several vascular conditions as atherosclerosis, hypertension, hyperlipidemia and diabetes mellitus. In all these conditions insulin resistance (IR) is present. Cytokines are low molecular weight proteins with several endocrine and metabolic functions that participate of inflammation and immune response. Several of these cytokines are independent risk factors for cerebrovascular and coronary artery disease. The major sources of cytokines (adipokines) are the visceral and subcutaneous adipose tissues. Thus, increased adipose tissue mass is associated with alteration in adipokine production as over expression of tumor necrosis factor alpha, interleukin 6, plasminogen activator inhibitor 1, and under expression of adiponectin in adipocite tissue. The pro-inflammatory status associated with these changes provides a potential link between IR and endothelial dysfunction, the early stage in the atherosclerotic process, in obese individuals, and type 2 diabetic patients. Reduction of adipose tissue mass through weight reduction in association with exercise reduces TNF-alpha, IL-6, and PAI-1, increases adiponectin, and is associated with improved insulin sensitivity and endothelial function. This review will focus on the evidence for regulation of endothelial function by insulin and the adypokines such as adyponectin, leptin, resistin, IL-6 and TNF-alpha. Interaction between insulin signaling and adypokines will be discussed, as well as the concept that aberrant adypokine secretion in IR and/or obesity impairs endothelial function and contributes further to reduce insulin sensitivity.
Insulin resistance; Endothelium; Cytokines; Endothelial dysfunction
REVISÃO
Citocinas, disfunção endotelial e resistência à insulina
Cytokines, endothelial dysfunction, and insulin resistance
Maria Helena C. de Carvalho; André Luiz Colaço; Zuleica Bruno Fortes
Laboratório de Hipertensão e Diabetes, Departamento de Farmacologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, SP
Endereço para correspondência Endereço para correspondência: Maria Helena Catelli de Carvalho Departamento de Farmacolgia Instituto de Ciências Biomédicas Universidade de São Paulo ICB/USP Av. Prof. Lineu Prestes 1524, 2º. andar sala 213 Prédio ICB1 Butantã 05508-000 São Paulo, SP E-mail: mhcarvalho@icb.usp.br
RESUMO
A disfunção endotelial está associada a diversas alterações vasculares, como a aterosclerose, hipertensão arterial, hiperlipidemia e diabetes mellitus, que têm em comum a resistência à insulina (RI). Citocinas são proteínas de baixo peso molecular, com diversas funções metabólicas e endócrinas, que participam da inflamação e resposta do sistema imune. Várias dessas citocinas são consideradas como fatores de risco independentes para doenças da artéria coronária e cerebrovascular. As principais fontes de citocinas (adipocinas) são os tecidos adiposos subcutâneo e visceral. Assim, aumento da massa de tecido adiposo está associado com alterações da produção de adipocina com aumento da expressão de fator de necrose tumoral a (TNF-a), interleucina 6 (IL-6), inibidor do fator ativador de plasminogênio 1 (PAI-1), e diminuição da expressão de adiponectina no tecido adiposo. A condição pró-inflamatória associada a essas alterações sugere ligação entre RI e disfunção endotelial no estágio inicial do processo de aterosclerose, em indivíduos obesos e em pacientes diabéticos tipo 2. A redução da massa de tecido adiposo, por redução de peso associada a exercício físico, reduz TNF-a, IL-6 e PAI-1, aumenta adiponectina, e melhora tanto a sensibilidade à insulina quanto a função endotelial. A interação entre adipocinas e insulina no controle da função endotelial será discutida, bem como o conceito de que a alteração da secreção de adiponectinas na RI e/ou obesidade piora a função endotelial, além de diminuir ainda mais a sensibilidade à insulina.
Descritores: Resistência insulínica; Endotélio; Citocinas; Disfunção endotelial
ABSTRACT
Endothelial dysfunction is associated with several vascular conditions as atherosclerosis, hypertension, hyperlipidemia and diabetes mellitus. In all these conditions insulin resistance (IR) is present. Cytokines are low molecular weight proteins with several endocrine and metabolic functions that participate of inflammation and immune response. Several of these cytokines are independent risk factors for cerebrovascular and coronary artery disease. The major sources of cytokines (adipokines) are the visceral and subcutaneous adipose tissues. Thus, increased adipose tissue mass is associated with alteration in adipokine production as over expression of tumor necrosis factor a, interleukin 6, plasminogen activator inhibitor 1, and under expression of adiponectin in adipocite tissue. The pro-inflammatory status associated with these changes provides a potential link between IR and endothelial dysfunction, the early stage in the atherosclerotic process, in obese individuals, and type 2 diabetic patients. Reduction of adipose tissue mass through weight reduction in association with exercise reduces TNF-a, IL-6, and PAI-1, increases adiponectin, and is associated with improved insulin sensitivity and endothelial function. This review will focus on the evidence for regulation of endothelial function by insulin and the adypokines such as adyponectin, leptin, resistin, IL-6 and TNF-a. Interaction between insulin signaling and adypokines will be discussed, as well as the concept that aberrant adypokine secretion in IR and/or obesity impairs endothelial function and contributes further to reduce insulin sensitivity.
Keywords: Insulin resistance; Endothelium; Cytokines; Endothelial dysfunction
Resistência à insulina e disfunção endotelial
O TERMO "DISFUNÇÃO ENDOTELIAL" refere-se a um desequilíbrio na produção endotelial de mediadores que regulam o tônus vascular, agregação plaquetária, coagulação e fibrinólise, sendo o tônus vascular o aspecto mais estudado. A disfunção endotelial também é freqüentemente referida como piora no relaxamento dependente do endotélio, causado pela perda da biodisponibilidade do óxido nítrico (NO), muito embora a produção de outras substâncias vasoativas derivadas do endotélio, como a PGI2, EDHF, ET-1, Ang II, TXA2, também possa estar alterada (1).
A disfunção endotelial está presente em diversas doenças metabólicas e/ou cardiovasculares, como na obesidade, intolerância à glicose, hiperglicemia (diabetes mellitus), hipertensão arterial e dislipidemia. Em todas essas condições ocorre resistência insulínica, a qual se apresenta como um distúrbio metabólico que se manifesta por redução na utilização da glicose pelo músculo esquelético periférico (2), e tem sido fortemente associada à disfunção endotelial, que tem se mostrado ocorrer precocemente (3).
É importante ressaltar que em indivíduos relativamente jovens e obesos, a disfunção endotelial na artéria coronária é detectável juntamente com a resistência à insulina, antes mesmo do desenvolvimento de manifestações clínicas da síndrome metabólica. Demonstrou-se, também, reatividade anormal em irmãos e filhos não-diabéticos de pacientes com diabetes mellitus tipo 2 (DM2). Aqueles que eram resistentes à insulina apresentaram alteração da resposta vascular. Essa observação tem muitas implicações que relacionam a sinalização insulínica na vasculatura à disfunção endotelial como causa da resistência à insulina e a própria insulina contribuindo para acelerar o dano vascular (4).
A resistência à insulina tem componente genético ainda não completamente entendido, o qual é freqüentemente transmitido ao longo de gerações. Por outro lado, a obesidade que também tem importante componente genético, invariavelmente exacerba a resistência à insulina. Dessa forma, obesidade e resistência à insulina estão geralmente presentes por muitos anos antes do aparecimento de outras alterações como hipertensão arterial, dislipidemia, DM2 e doenças cardiovasculares (5). Entretanto, nem todos os indivíduos com resistência à insulina apresentam cada um dos componentes associados à síndrome da resistência à insulina ou síndrome X, a saber, DM2/intolerância à glicose, hipertensão arterial, dislipidemia, microalbuminúria, obesidade, hiperuricemia, sensibilidade ao sal, entre outros. Portanto, é provável que haja predisposição genética para desenvolvimento dessa síndrome.
Ressalta-se que a resistência à insulina pode estar presente por vários anos antes do aparecimento de alterações dos níveis plasmáticos de glicose. Dessa forma, indivíduos que irão desenvolver DM2 apresentam deterioração progressiva da tolerância à glicose. Eles geralmente progridem de normoglicêmicos a intolerantes à glicose e finalmente diabéticos.
O papel exercido pela resistência à insulina, que freqüentemente está associada ao hiperinsulinismo compensatório, não é ainda completamente compreendido. Uma possibilidade é que, na tentativa de sobrepujar a inibição da via de sinalização insulínica, a hiperinsulinemia possa continuar a estimular a via de sinalização mitogênica da insulina, exercendo seus efeitos indesejados (6). Deve-se ressaltar que a insulina apresenta ação vasodilatadora, a qual se deve à produção endotelial de NO. Assim, a resistência à insulina pode contribuir para a disfunção endotelial. Vários estudos também demonstraram que a vasodilatação mediada pelo óxido nítrico (NO) está diminuída em pacientes com DM2. A resposta da artéria braquial está alterada, seja a doadores endógenos ou exógenos de NO, sugerindo que há aumento da inativação do NO, possivelmente devido a aumento do seu metabolismo ou a resposta alterada do músculo liso devido a alterações da sinalização na via da guanilato ciclase.
Alteração da função endotelial também foi demonstrada em pacientes obesos sem DM2. Muitos desses pacientes apresentam tolerância diminuída à glicose e muitas das características da síndrome metabólica. Essas duas alterações de tolerância a carboidratos estão associadas a aumento da taxa de mortalidade por doença arterial coronariana, da ordem de 3 a 4 vezes no DM2, e de 2 a 3 vezes na tolerância diminuída à glicose.
Defeitos na vasodilatação mediada pelo NO também podem contribuir para a resistência à insulina. Demonstrou-se que a infusão de inibidor do óxido nítrico sintase (NOS) não apenas diminui a vasodilatação dependente do endotélio, como também diminui a captação de glicose mediada pela insulina. Entretanto, há dados mostrando que a diminuição não ocorre em todos os órgãos e sistemas do organismo, apesar de ter ocorrido, com inibição da NOS, redução do fluxo do antebraço e aumento da pressão arterial. Apesar disso, a idéia de que a função endotelial pode regular a captação de glicose pela insulina é interessante e pode contribuir para explicar achado de dois ensaios clínicos em que a melhora da disfunção endotelial com agentes como os inibidores da enzima conversora da angiotensina e uma estatina não apenas retardaram a progressão da doença arterial coronariana e a morte, mas também preveniram o desenvolvimento do DM2 em pacientes de alto risco. Portanto, essas observações sugerem que a atenuação da doença cardiovascular é acompanhada de retardo na progressão do DM (4).
Nos tecidos-alvo, a insulina também estimula duas vias: a via do fosfatidilinositol 3-quinase e a via da proteína quinase ativada por mitógenos (MAPK). A ativação da fosfatidilinositol 3-quinase, que ocorre após a ligação da insulina com seu receptor, é crítica para a captação de glicose nos tecidos-alvo dependentes de insulina, como o músculo esquelético, coração e tecido adiposo. Demonstrou-se, também, que essa via regula a produção endotelial de NO mediada por insulina. Quando a via da fosfatidilinositol 3-quinase está disfuncional, ocorre piora da vasodilatação dependente do endotélio estimulada pela insulina. Dessa forma, disfunção sistêmica na via da fosfatidilinositol 3-quinase, que define resistência insulínica, provoca também defeito tanto na captação de glicose quanto na vasodilatação dependente do endotélio mediada por insulina.
A via da MAPK é importante para as ações proliferativas da insulina. Entretanto, na vasculatura, essa via medeia não somente o crescimento celular mas também a capacidade migratória das células endoteliais, do músculo liso vascular e dos monócitos. Além disso, ela parece mediar a expressão de um fator pró-trombótico, pró-fibrótico, o inibidor do ativador do plasminogênio 1 (PAI-1), em resposta a diversos estímulos. Dessa forma, por estimular o crescimento e a migração celular e as respostas pró-inflamatórias e pró-trombóticas, a via da MAPK pode ser aterogênica. Uma questão importante é saber se essa via também está atenuada em estados de resistência insulínica. Dados de vários estudos demonstram que a resistência insulínica e os defeitos associados com a síndrome metabólica são dependentes de defeito específico da via de sinalização da insulina, a via do fosfatidilinositol 3-quinase, enquanto as funções mediadas pela via da MAPK operam normalmente. De fato, há dados mostrando que diminuição da via do fosfatidilnositol 3-quinase pela insulina está associada a aumento da via da MAPK em células vasculares. Assim, na presença de insulina, é possível que a hiperinsulinemia possa ser aterogênica (4).
Papel dos ácidos graxos livres na disfunção endotelial
A obesidade central ou abdominal leva à resistência à insulina e à disfunção endotelial devido à formação de produtos metabólicos derivados de lipídeos, hormônios e citocinas. Por outro lado, a resistência à insulina pode levar à disfunção endotelial e alterações da via de sinalização da insulina, específicas ou compartilhadas, no músculo, tecido adiposo e células endoteliais, bem como novos fatores genéticos e não-tradicionais podem estar envolvidos. Estudos clínicos recentes demonstram que estratégias farmacológicas ou não-farmacológicas no combate à obesidade e/ou resistência à insulina podem melhorar a disfunção endotelial e a inflamação de baixa intensidade presentes nesses estados. Todos esses achados acrescentaram nova dimensão à associação obesidade, resistência à insulina e disfunção endotelial, o que pode se tornar alvo-chave na prevenção do DM2 e doenças cardiovasculares (5).
Nesse contexto, o aumento da massa gordurosa provoca aumento da lipólise e aumento das concentrações circulantes de ácidos graxos livres não esterificados, como observado em pacientes e cães obesos e hipertensos. Ativação simpática pode, por sua vez, reforçar esse aumento de ácidos graxos livres promovendo lipólise ou diminuindo a sua utilização ou re-esterificação. Estudos in vitro e in vivo demonstraram que os ácidos graxos livres podem alterar a função endotelial. A aplicação de ácidos graxos como o ácido oléico em cultura de células endoteliais inibiu a eNOS, bem como diminuiu a resposta vasodilatadora induzida por agonistas colinérgicos em artérias femorais isoladas de coelhos (acetilcolina) e na vasculatura da perna em humanos (metacolina). Além de inibir a eNOS, os ácidos graxos estimulam a geração de ânion superóxido por células endoteliais e vasculares via ativação da NADPH oxidase, o que contribui para a diminuição da biodisponibilidade do NO e dessa forma para disfunção endotelial.
Os ácidos graxos também levam à resistência insulínica. O mecanismo pelo qual o aumento de ácidos graxos livres leva à diminuição da captação de glicose parece envolver o aumento da NADH e acetil CoA intramitocondriais. Como resultado, ocorre inibição da fosfofrutoquinase e hexoquinase II, levando ao aumento da concentração intracelular de glicose, bem como de sua captação. Entretanto, outros autores têm proposta diferente e sugerem que a inibição do transporte de glicose ou atividade de fosforilação precede a redução da síntese de glicogênio e oxidação de glicose induzida por ácidos graxos livres. Deve-se ressaltar que, aumentando-se os metabólitos intracelulares de ácidos graxos, pode haver ativação da cascata da quinase da serina/treonina, possivelmente via proteína quinase C delta, levando à fosforilação da serina/treonina do substrato para o receptor de insulina 1 (IRS-1). O IRS-1 fosforilado em serina não se liga nem ativa a PI-3 quinase, resultando em transporte diminuído de glicose e nas outras manifestações da resistência insulínica. Os ácidos graxos livres podem também interferir diretamente com a expressão, transcrição ou no recrutamento para a superfície celular do transportador GLUT-4. Além disso, os ácidos graxos livres reduzem o clearance hepático de insulina e aumentam a produção hepática de glicose, ambos potencializam os efeitos da resistência insulínica.
Inflamação e resistência insulínica
O desenvolvimento do conceito de que o DM2 é uma condição inflamatória é novo, interessante para a compreensão dessa condição, e tem implicações em termos de patogenia e complicações da doença. O conceito de inflamação em relação às condições metabólicas como obesidade e resistência insulínica remontam a 1993, quando se demonstrou que os adipócitos expressavam uma citocina pró-inflamatória, o TNF-a, que a expressão deste nos adipócitos de animais obesos estava aumentada, e que a neutralização do TNF-a levava à diminuição da resistência insulínica nesses animais. Estabeleceu-se, assim, a primeira conexão entre aumento da expressão e da concentração plasmática de citocina pró-inflamatória e resistência insulínica. Uma série de trabalhos posteriores na área de obesidade tem confirmado que a obesidade é um estado de inflamação crônica, pois há aumento da CRP, interleucina 6 (IL-6) e do inibidor do ativador do plasminogênio 1 (PAI-1). Alguns estudos também têm demonstrado que o DM2 é uma condição inflamatória caracterizada por concentrações elevadas de reagentes de fase aguda no plasma como o ácido siálico e IL-6.
O fato de a obesidade, o principal fator de risco para o DM2, e o próprio DM serem condições inflamatórias levou investigadores a pesquisar se os mediadores inflamatórios poderiam prever o desenvolvimento de DM2. Vários estudos confirmaram a presença de inflamação como preditores de desenvolvimento de DM2 (7).
Resistência à insulina e citocinas
Citocinas são proteínas de baixo peso molecular com diversas funções metabólicas e endócrinas que participam da inflamação e resposta do sistema imune. Várias dessas citocinas são fatores de risco independentes para doenças da artéria coronária e cerebrovascular. As principais fontes de citocinas (adipocinas/adipocitocinas) são os tecidos adiposos subcutâneo e visceral. Assim, aumentada massa de tecido adiposo está associada com alterações na produção de adipocina, como superexpressão de TNF-a, IL-6, PAI-1, e sub-expressão de adiponectina em tecido adiposo. A condição pró-inflamatória associada com essas alterações sugere ligação entre resistência à insulina e disfunção endotelial no estágio inicial do processo de aterosclerose, em indivíduos obesos e em pacientes com DM2.
Existem evidências para a regulação da função endotelial pela insulina e adipocinas como adiponectina, leptina, resistina, IL-6 e TNF-a, visto que, por exemplo, a redução da massa de tecido adiposo, através de redução de peso em associação com exercício, reduz TNF-a, IL-6 e PAI-1, aumenta adiponectina, e está associada com melhora na sensibilidade à insulina e função endotelial (8).
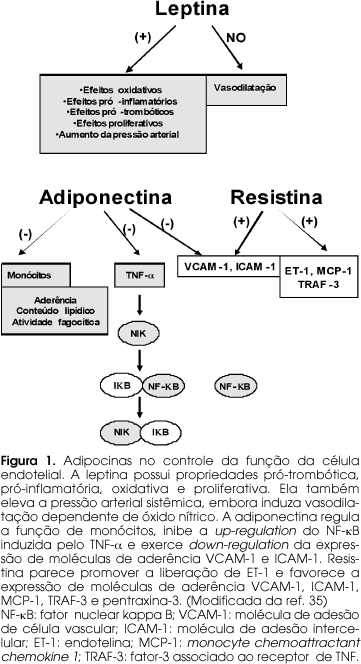
TNF-a (figura 1)
Entre os mediadores inflamatórios, o TNF-a é um possível candidato a induzir resistência insulínica. Ele é produzido por adipócitos e está aumentado no tecido adiposo de roedores obesos, bem como em humanos. Demonstrou-se que o tratamento com TNF-a leva à redução da autofosforilação do receptor da insulina estimulada pela própria insulina e inibição subseqüente da fosforilação de IRS-1. Além disso, o TNF-a induz modificação do IRS-1 por fosforilação em serina, o que torna essa molécula inibitória para a sinalização do receptor de insulina. Demonstrou-se também que a neutralização dos efeitos do TNF-a, em ratos obesos, provocou aumento da sensibilidade insulínica. Ressalta-se que camundongos deficientes em TNF-a permaneceram sensíveis à insulina quando colocados em dieta rica em gordura (9).
IL-6 (figura 1)
Dados recentes sugerem que, enquanto o TNF-a age de forma parácrina no adipócito, a IL-6 circula no plasma em concentrações relativamente altas, sendo portanto muito mais importante sistemicamente. De fato, ela é chamada de "citocina endócrina". Alguns dos efeitos metabólicos da IL-6 (uma citocina pleiotrópica) demonstram que, em estudos in vitro, essa citocina induziu inibição dependente de dose da liberação de insulina estimulada por glicose. In vivo, a IL-6 recombinante induziu alterações metabólicas usualmente encontradas em estados catabólicos, aumentando as concentrações plasmáticas de glicose sem alterar significativamente as concentrações plasmáticas de insulina ou peptídeo C. Apesar das células gordurosas contribuírem com 1/3 da concentração circulante de IL-6, existem outras fontes importantes de produção, como os monócitos. Esse mecanismo não parece ser importante em concentrações normais de glicose de jejum, mas pode ser importante no DM2. Nesse contexto, talvez a IL-6 represente um fator hormonal que causa resistência insulínica.
Outro fato importante é que pacientes com DM2 apresentam concentrações plasmáticas elevadas de IL-6, particularmente aqueles com características da síndrome de resistência insulínica. Uma interpretação para esse dado é que, no DM2 e na síndrome de resistência insulínica, há resposta de fase aguda devido ao aumento de IL-6 derivada de secreção imune ou do tecido adiposo, que não sofre restrição e que atua em adipócitos hipersensíveis. De fato, há dados mostrando que as concentrações plasmáticas de marcadores de fase aguda estão aumentadas nessas condições, como o CRP, proteína amiloide sérica A, glicoproteína acídica alfa 1, ácido siálico e cortisol. A recíproca também pode ser verdadeira, pois o aumento de IL-6 e dos marcadores de fase aguda pode estar contra-atacando a hiperglicemia e resistência insulínica. Quando a inflamação é mantida, crônica ou descontrolada e quando o estímulo se torna excessivo, não se consegue alcançar os efeitos desejados, e assim há piora da hiperglicemia e da resistência insulínica. Isso resultaria em inflamação somente quando a resistência insulínica fosse grave (10).
Leptina (figura 1)
A leptina (do grego leptos, que significa magro) é uma proteína de 167 aa secretada pelo tecido adiposo. Ela foi identificada em 1994 como uma molécula-chave na regulação do peso corpóreo e balanço de energia, visto que ela regula o apetite e o gasto de energia via sistema nervoso central (SNC).
A leptina é codificada pelo gene ob, o qual é predominantemente expresso pelos adipócitos, e seus níveis plasmáticos se correlacionam bem com a massa de gordura corporal. A expressão de leptina nos adipócitos é regulada transcripcionalmente, com a condição do armazenamento de energia no tecido adiposo branco e o tamanho do adipócito como principais determinantes. Além disso, a expressão de leptina e seus níveis séricos aumentam após a ingestão de comida. Em contraste, a expressão de leptina é rapidamente suprimida com a restrição de comida (11).
A leptina atua no seu receptor, denominado receptor para a leptina (LR). O LR é codificado pelo gene db. Das 5 isoformas do receptor, a forma longa é expressa por todo o corpo e tem sido localizada no hipotálamo, monócitos, células "natural killer", linfócitos CD4 e CD8, células b do pâncreas, enterócitos e células endoteliais (11).
Assim, alguns estudos têm demonstrado que a leptina não é restrita ao apetite e ingestão de comida, mas que é uma molécula pleiotrópica com uma ampla variedade de ações biológicas como função reprodutora, regulação do eixo hipotálamo-hipófise-adrenal (eixo HPA), metabolismo da insulina e glicose, lipólise, atividade do sistema nervoso simpático, resposta imune, hematopoiese e angiogênese. Nesse contexto, o papel da leptina na vasculatura é embasado em resultados experimentais mostrando que a reposição da leptina em camundongos obesos, deficientes em leptina ob/ob, reverte a disfunção endotelial.
Ressalta-se que a insulina pode interagir com a leptina para modular a função vascular. O mecanismo pelo qual a leptina induz a produção de óxido nítrico em alguns leitos vasculares é, em parte, relacionado à ativação da via de fosforilação AKt-eNOS. Assim, a insulina aumenta a vasodilatação dependente da leptina por aumentar a liberação da eNOS e por potencializar a AKt e a fosforilação da eNOS (12). Concomitantemente, a leptina aumenta a sensibilidade da insulina em ratos e pode melhorar a resposta vascular à insulina em estados de resistência à insulina. Assim, o cross-talk entre leptina e insulina poderia ter importantes implicações na fisiopatologia da disfunção vascular da síndrome metabólica, particularmente na obesidade relacionada à hipertensão arterial. Ressalta-se que, apesar de o TNF-a e da leptina estarem implicados na resistência insulínica associada ao DM2 e obesidade, existem outros fatores conectando o aumento da adiposidade e a resistência insulínica.
Adiponectina (figura 1)
A adiponectina é uma proteína plasmática de aproximadamente 30 KDa, relativamente abundante, que é secretada especificamente pelo tecido adiposo. Sua concentração plasmática em indivíduos sadios varia de 1,9 a 17 µg/mL (13) e sua meia-vida é de 2,5 h na corrente sanguínea (14). O gene da adiponectina humana localiza-se no cromossoma 3q27, um sítio associado com suscetibilidade a DM2 (15). Descreveu- se polimorfismo de um único nucleotídeo no gene da adiponectina e variações estruturais nas raças, o que contribui para o desenvolvimento de síndrome metabólica e diabetes mellitus tipo 2 (16,17).
Os níveis plasmáticos de RNA mensageiro de adiponectina estão reduzidos na obesidade e em estados de resistência à insulina. Eles se correlacionam negativamente com a porcentagem de gordura corpórea, distribuição de gordura central, insulina plasmática em jejum, tolerância oral de glicose, e com fatores de risco cardiovascular associados à obesidade, incluindo pressão arterial sistólica e diastólica, colesterol total, triglicérides, LDL-colesterol e ácido úrico. Entretanto, foi encontrada correlação positiva com a utilização de glicose durante clamp euglicêmico e com níveis HDL-colesterol (18).
Os níveis circulantes de adiponectina não variam substancialmente no período pós-prandial, entretanto durante o período noturno ocorre variação nos níveis plasmáticos com declínio à noite, atingindo nível mínimo no início da manhã (19).
Mulheres possuem nível de adiponectina aproximadamente duas vezes maior do que homens, o que reflete um efeito androgênico. Linsay e cols. (20) descreveram, na população indígena Pima, altas concentrações de adiponectina e detectaram menor probabilidade (37%) de essa população desenvolver DM2, refletindo uma associação de adiponectina com sensibilidade à insulina. Pacientes com doença arterial coronariana apresentam níveis de adiponectina menor do que indivíduos controles, ajustados tanto para idade quanto para o índice de massa corpórea. No estudo Health Professionals Follow-Up Study, com 18.225 pacientes, o nível plasmático de adiponectina foi fortemente correlacionado com risco diminuído de infarto do miocárdio (21).
Os efeitos metabólicos da adiponectina incluem aumento na sensibilidade à insulina no fígado, músculo e tecido adiposo, mediado por aumentada oxidação de gordura nos tecidos. Existe forte evidência de que a sinalização para adiponectina nesses tecidos seja através da via MAP quinase, a qual representa um sistema regulador metabólico que funciona em paralelo à sinalização para insulina (22). O acúmulo de lipídeos visceral e intravascular é uma marca da obesidade e da síndrome metabólica, e pode ser melhorado pela ativação da cascata da MAP quinase, a qual tem sido também demonstrada como um potencial mecanismo de ação das drogas antidiabetes como a metformina (23).
Na vasculatura, os níveis de adiponectina estão fortemente ligados à função endotelial (24). Adiponectina altera os efeitos vasculares adversos das citocinas, tais como TNF-a, suprime a geração de ânion superóxido na célula endotelial eliciado por agentes como a LDL oxidada, e aumenta a geração de óxido nítrico endotelial. Estudos em roedores, empregando lesão direta em vasos sanguíneos, mostram que a adiponectina pode atenuar a proliferação de músculo liso vascular e proteger contra estenose vascular. Interessantemente, Okamoto e cols. (25), utilizando a técnica de imuno-histoquímica, não conseguiram detectar adiponectina na parede de vaso sanguíneo de coelhos sadios, mas a detectaram em parede de vaso sanguíneo submetido à injúria por balão, sugerindo que o aumento dessa citocina na parede vascular lesada faz parte de uma resposta protetora contra processo celular inflamatório (25).
Embora não tenha sido demonstrado em todos os estudos, os níveis de adiponectina tendem a aumentar após perda de peso em indivíduos obesos (26,27). As alterações no hormônio da tireóide não afetam os níveis de adiponectina, mas uma variedade de outras condições têm sido identificadas e estão associadas com alterações nos níveis de adiponectina. Como era de se esperar, muitos desses efeitos estão também associados com alterações no risco cardiovascular.
Interessante ressaltar que as tiazolidinodionas usadas para o tratamento de DM2, aumentam os níveis circulantes de adiponectina, que poderiam aumentar a sensibilidade à insulina e a proteção vascular (28-30). Interessantemente, as mudanças iniciais nos níveis circulantes de adiponectina em resposta ao tratamento com tiazolidinodiona aumenta a proporção de complexos de alto peso molecular, sugerindo que eles possam ter influência na sensibilidade à insulina (31).
Resistina (figura 1)
A resistina é uma proteína pertencente à família de proteínas secretórias ricas em cisteína, denominadas "moléculas semelhantes à resistina" (RELM resistin-like molecules) ou "encontradas em locais de inflamação" (FIZZ found in the inflammatory zone), foi descoberta em 2001 e representa a mais nova das adipocinas. Ela foi assim denominada pela sua capacidade de promover resistência à insulina. Outros membros dessa família, que são semelhantes à resistina, foram detectados no trato gastrintestinal, originalmente nas células das criptas intestinais e induzida em células epiteliais brônquicas em resposta a estímulo inflamatório.
Os resultados dos estudos até agora existentes descrevem a resistina como um hormônio secretado pelo tecido adiposo branco, que ocorre no soro, e é induzido durante a adipogênese. Importante ressaltar que, em humanos, a fonte de resistina surpreendentemente não é o adipócito, mas os macrófagos, o que sugere um importante papel inflamatório (32).
Os níveis de resistina aumentam na obesidade genética ou induzida por dieta e, portanto, estão ligadas à resistência insulínica associada à obesidade. Descreveu-se a resistina como hormônio singular, cujos efeitos no metabolismo da glicose são antagônicos àqueles da insulina. A resistina regula, ainda, a diferenciação do adipócito por meio de mecanismo de retroalimentação negativa que limita a formação do tecido adiposo em resposta a aumento do consumo de energia (32).
Ao contrário da adiponectina, a resistina aumenta a expressão de moléculas de aderência VCAM-1 e ICAM-1, faz up-regulation da proteína quimiotáxica para monócitos (MCP-1) e promove ativação da célula endotelial via liberação de endotelina 1 (ET-1). Assim, embora muitos aspectos de sua função ainda devam ser elucidados, parece que a resistina irá adicionar conhecimento sobre a fisiopatologia da doença vascular e da síndrome metabólica.
Resistência à insulina e hipertensão arterial
A hipertensão arterial está associada a hiperinsulinismo em pacientes diabéticos obesos, indivíduos obesos não-diabéticos e em pacientes com hipertensão arterial essencial. Parentes de primeiro grau normotensos desses pacientes apresentam resistência à insulina e dislipidemia. Ambas foram também encontradas em animais com hipertensão genética. Esses achados sugerem predisposição genética comum para a hipertensão essencial e resistência à insulina. Por outro lado, hiperinsulinismo sem resistência à insulina e concentrações aumentadas de insulina dentro da faixa normal não predispõem à hipertensão arterial (33).
A patogenia da hipertensão arterial associada à obesidade é complexa e certamente não deve estar relacionada a um único gene. Muitos fatores atuam em conjunto para promover vasoconstrição e retenção de sódio. A principal hipótese sugere que a leptina, os ácidos graxos livres e a insulina, cujas concentrações estão aumentadas na obesidade, atuem sinergisticamente estimulando a atividade simpática e a vasoconstrição. Além disso, a resistência à insulina e a disfunção endotelial atuam como amplificadores da resposta vasoconstritora. Finalmente, o aumento da reabsorção renal de sódio pode também ocorrer, causado por aumento da atividade simpática renal, por efeito direto da insulina, hiperatividade do sistema renina angiotensina e possivelmente por alteração de forças físicas intrarrenais (9).
Assim, a resistência à insulina e/ou hiperinsulinemia tem sido sugerida como responsável pelo aumento da pressão arterial em alguns pacientes com hipertensão arterial. Esta característica é agora amplamente reconhecida como parte da síndrome X, ou síndrome metabólica marcada também por obesidade central, dislipidemia (especialmente triglicérides aumentado) e hipertensão. Apesar de estar claramente demonstrado que uma fração substancial da população de hipertensos apresenta resistência à insulina e hiperinsulinemia, a existência de associação ainda não é tão clara.
Como dito anteriormente, a resistência à insulina é comum em pacientes com DM2 e em obesos, que são condições mais comuns em pacientes hipertensos do que em indivíduos normotensos. Entretanto, vários estudos têm demonstrado que hiperinsulinemia e resistência à insulina estão presentes em pacientes magros sem DM2, sugerindo que esta relação é mais do que uma coincidência.
Para entendermos a contribuição da resistência à insulina na gênese da hipertensão arterial temos que avaliar os efeitos da resistência à insulina e hiperinsulinemia sobre os fatores que contribuem para a elevação da pressão arterial. Assim, a hiperinsulinemia pode aumentar a pressão arterial por um ou mais mecanismos. Uma afirmação subjacente em cada caso é que alguns, mas nem todos, tecidos-alvos da insulina são resistentes a seus efeitos. Especificamente, os tecidos envolvidos na homeostase da glicose são resistentes, enquanto os tecidos envolvidos no processo hipertensivo não o são, produzindo, portanto, hiperinsulinemia.
Primeiro, altos níveis circulantes de insulina causam retenção de sódio e outros efeitos vasculares, tais como proliferação e expansão de matriz celular (34). Na presença de hiperinsulinemia, fatores neuro-humorais tais como angiotensina II (Ang II), endotelina (ET) e vasopressina (VP) também potenciam a proliferação de células do músculo liso vascular e endotelial. Outro mecanismo é a hipertrofia do músculo liso vascular, secundária à ação mitogênica da insulina. Terceiro, a insulina modifica o transporte iônico através da membrana celular, aumentando, desse modo, os níveis de cálcio citosólico dos tecidos renal e vascular sensíveis à insulina. Além disso, o efeito da insulina sobre diversos fatores de crescimento contribui para o desenvolvimento da lesão vascular através de sua potencialização do processo aterosclerótico. Em pessoas geneticamente predispostas à hipertensão arterial e/ou nefropatia, esse fatores podem potencializar a lesão da vasculatura e órgãos-alvo.
Finalmente, a resistência à insulina pode ser um marcador para outros processos patológicos, isto é, a não-modulação, o qual poderia ser o mecanismo principal para aumentar a pressão arterial. É importante ressaltar que o papel da insulina no controle da pressão arterial é pouco conhecido e sua participação como fator patogênico na hipertensão permanece indefinida.
Recebido em 07/12/05
Aceito em 17/01/06
- 1. Mombouli J, Vanhoute PM. Endothelial dysfunction: from physiology to therapy. J Mol Cell Cardiol 1999;31:61-74.
- 2. Ferrannini E, Natali A, Capaldo B, Lehtovirta M, Jacob S, Yki-Järvinen H. Insulin resistance, hyperinsulinemia, and blood pressure: role of age and obesity. Hypertension 1997;30:1144-9.
- 3. Hsueh WA, Lyon CJ, Quiñones MJ. Insulin resistance and the endothelium. Am J Med 2004;117:109-17.
- 4. Hsueh WA, Quiñones MJ. Role of endothelial dysfunction in insulin resistance. Am J Cardiol 2003;92:10J-7J.
- 5. Caballero AE. Endothelial dysfunction in obesity and insulin resistance: a road to diabetes and heart disease. Obesity Res 2003;11:1278-89.
- 6. Wang CCL, Goalstone ML, Draznin B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes 2004;53:2735-40.
- 7. Dandona P, Aljada A, Chaudhuri A, Mohanty P. Endothelial dysfunction, inflammation and diabetes. Rev Endocr Metab Dis 2004;5:189-97.
- 8. Aldhahi W, Hamdy O. Adipokines, inflammation, and endothelium in diabetes. Curr Diab Rep 2003;3:293-8.
- 9. Montani JP, Antic V, Yang Z, Dulloo A. Pathways from obesity to hypertension: from the perspective of a vicious triagle. Int J Obesity 2002;26:S28-S38.
- 10. Fernández-Real JM, Ricart W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr Rev 2003;24:278-301.
- 11. Peelman F, Waelput W, Iserentant H, Lavens D, Eyckerman S, Zabeau L, et al. Leptin: linking adipocyte metabolism with cardiovascular and autoimmune diseases. Prog Lipid Res 2004;43:283-301.
- 12. Vecchione C, Aretini A, Maffei A, Marino G, Selvetella G, Poulet R, et al. Cooperation between insulin and leptin in the modulation of vascular tone. Hypertension 2003;42:166-70.
- 13. Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab 2002;13:84-9.
- 14. Hoffstedt J, Arvidsson E, Sjolin E, Wahlen K, Arner P. Adipose tissue adiponectin production and adiponectin serum concentration in human obesity and insulin resistance. J Clin Endocrinol Metab 2004;89:1391-6.
- 15. Kissebah AH, Sonnenberg GE, Myklebust J, Goldstein M, Broman K, James RG, et al. Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc Natl Acad Sci USA 2000;97:14478-83.
- 16. Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, et al. Impaired multimerization of human adiponectin mutants associated with diabetes: molecular structure and multimer formation of adiponectin. J Biol Chem 2003;278:40352-63.
- 17. Kishida K, Nagaretani H, Kondo H, Kobayashi H, Tanaka S, Maeda N, et al. Disturbed secretion of mutant adiponectin associated with the metabolic syndrome. Biochem Biophys Res Comm 2003;306:286-92.
- 18. Matsuzawa Y, Funahashi T, Kihara S, Shimomura I. Adiponectin and metabolic syndrome. Arterioscler Thromb Vasc Biol 2004;24:29-33.
- 19. Gavrila A, Chan JL, Yiannakouris N, Kontogianni M, Miller LC, Orlova C, et al. Serum adiponectin levels are inversely associated with overall and central fat distribution but are not directly regulated by acute fasting or leptin administration in humans: cross-sectional and interventional studies. J Clin Endocrinol Metab 2003;88:4823-31.
- 20. Lindsay RS, Funahashi T, Hanson RL, Matsuzawa Y, Tanaka S, Tataranni PA, et al. Adiponectin and development of type 2 diabetes in the Pima Indian population. Lancet 2002;360:57-8.
- 21. Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 2004;291:1730-7.
- 22. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metabolism 2005;1:15-25.
- 23. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001;108:1167-74.
- 24. Goldstein BJ, Scalia R. Adiponectin: a novel adipokine linking adipocytes and vascular function. J Clin Endocrinol Metab 2004;89:2563-8.
- 25. Okamoto Y, Arita Y, Nishida M, Muraguchi M, Ouchi N, Takahashi M, et al. An adipocyte-derived plasma protein, adiponectin, adheres to injured vascular walls. Horm Metab Res 2000;32:47-50.
- 26. Esposito K, Marfella R, Ciotola M, Di Palo C, Giugliano F, Giugliano G, et al. Effect of a Mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA 2004;292:1440-6.
- 27. Abbasi F, Lamendola C, McLaughlin T, Hayden J, Reaven GM, Reaven PD. Plasma adiponectin concentrations do not increase in association with moderate weight loss in insulin-resistant, obese women. Metabolism Clin Exper 2004;53:280-3.
- 28. Yu JG, Javorschi S, Hevener AL, Kruszynska YT, Norman RA, Sinha M, et al. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes 2002;51:2968-74.
- 29. Combs TP, Wagner JA, Berger J, Doebber T, Wang WJ, Zhang BB, et al. Induction of adipocyte complement-related protein of 30 kilodaltons by PPARg agonists: A potential mechanism of insulin sensitization. Endocrinology 2002;143:998-1007.
- 30. Phillips SA, Ciaraldi TP, Kong AP, Bandukwala R, Aroda V, Carter L, et al. Modulation of circulating and adipose tissue adiponectin levels by antidiabetic therapy. Diabetes 2003;52:667-74.
- 31. Tonelli J, Li W, Kishore P, Pajvani UB, Kwon E, Weaver C, et al. Mechanisms of early insulin-sensitizing effects of thiazolidinediones in type 2 diabetes. Diabetes 2004;53:1621-9.
- 32. Wolf G. Insulin resistance and obesity: resistin, a hormone secreted by adipose tissue. Nutr Rev 2004;62:389-94.
- 33. Stas SN, El-Atat FA, Sowers R. Pathogenesis of hypertension in diabetes. Rev Endocr Metab Dis 2004;5:221-5.
- 34. Stehouwer CDA, Lambert J, Donker AJ, van Hinsbergh VW. Endothelial dysfunction and pathogenesis of diabetic angiopathy. Cardiovasc Res 1997;34:55-68.
- 35. Kougias P, Chai H, Lin PH, Yao Q, Lumsden AB, Chen C. Effects of adipocyte-derived cytokines on endothelial functions: implication of vascular disease. J Surg Res 2005;126:121-9.
Datas de Publicação
-
Publicação nesta coleção
23 Maio 2006 -
Data do Fascículo
Abr 2006
Histórico
-
Aceito
17 Jan 2006 -
Recebido
07 Dez 2005