Resumos
Nos últimos anos, no Brasil e em países do mundo desenvolvido, a obesidade se tornou um problema de saúde pública mais importante que a desnutrição. Com o aumento de prevalência de obesidade, identificou-se que, além do DM2 e da síndrome metabólica, outras entidades clínicas também estavam associadas à resistência à insulina. Nesta revisão, abordaremos algumas destas alterações, com destaque para a doença hepática gordurosa não alcoólica, mas incluindo também a SOP, a hiperuricemia, a doença renal crônica, a insuficiência cardíaca, alterações cognitivas e câncer.
Síndrome metabólica; Doença hepática gordurosa não alcoólica; Síndrome dos ovários policísticos; Hiperuricemia; Doença renal crônica; Insuficiência cardíaca; Alterações cognitivas; Câncer
In the past years, in Brazil and in developed countries, obesity has become a major public health problem. It was identified that besides DM2 and metabolic syndrome other clinical entities were associated with insulin resistance. In this review we describe some of these alterations emphasizing nonalcoholic fatty liver disease, but also including polycistic ovary disease, hyperuricemia, chronic renal failure, heart failure, cognitive decline and cancer.
Metabolic syndrome; Nonalcoholic fatty liver disease; Polycystic ovaries syndrome; Hyperuricemia; Chronic renal disease; Heart failure; Cognitive decline; Cancer
REVISÃO
Doenças associadas à resistência à insulina/hiperinsulinemia, não incluídas na síndrome metabólica
Insulin resistance/hyperinsulinemia associated diseases not included in the metabolic syndrome
José B.C. Carvalheira; Mario J.A. Saad
Departamento de Clínica Médica, FCM, Universidade Estadual de Campinas (UNICAMP), Campinas, SP
Endereço para correspondência Endereço para correspondência: Mario J.A. Saad Departamento de Clínica Médica, FCMUNICAMP Cidade Universitária Zeferino Vaz 13081-970 Campinas, SP Fax: (19) 3788-8950 E-mail: msaad@fcm.unicamp.br
RESUMO
Nos últimos anos, no Brasil e em países do mundo desenvolvido, a obesidade se tornou um problema de saúde pública mais importante que a desnutrição. Com o aumento de prevalência de obesidade, identificou-se que, além do DM2 e da síndrome metabólica, outras entidades clínicas também estavam associadas à resistência à insulina. Nesta revisão, abordaremos algumas destas alterações, com destaque para a doença hepática gordurosa não alcoólica, mas incluindo também a SOP, a hiperuricemia, a doença renal crônica, a insuficiência cardíaca, alterações cognitivas e câncer.
Descritores: Síndrome metabólica; Doença hepática gordurosa não alcoólica; Síndrome dos ovários policísticos; Hiperuricemia; Doença renal crônica; Insuficiência cardíaca; Alterações cognitivas; Câncer
ABSTRACT
In the past years, in Brazil and in developed countries, obesity has become a major public health problem. It was identified that besides DM2 and metabolic syndrome other clinical entities were associated with insulin resistance. In this review we describe some of these alterations emphasizing nonalcoholic fatty liver disease, but also including polycistic ovary disease, hyperuricemia, chronic renal failure, heart failure, cognitive decline and cancer.
Keywords: Metabolic syndrome; Nonalcoholic fatty liver disease; Polycystic ovaries syndrome; Hyperuricemia; Chronic renal disease; Heart failure; Cognitive decline; Cancer
A SÍNDROME METABÓLICA COMPREENDE um espectro de alterações que incluem resistência à insulina com ou sem diabete melito tipo 2 (DM2), hipertensão arterial, obesidade (especialmente central ou visceral) e dislipidemia. A anormalidade central associada à síndrome metabólica parece ser a resistência dos tecidos periféricos à insulina, a qual pode ser definida como um estado de resposta biológica subnormal aos níveis circulantes de insulina. Nos últimos anos, identificou-se que a resistência à insulina pode ser a base etiopatogênica ou fisiopatológica de outras entidades clínicas prevalentes na população. Nesta revisão, abordaremos algumas destas alterações, com destaque para a doença hepática gordurosa não alcoólica, mas incluindo também a SOP, a hiperuricemia, a doença renal crônica, a insuficiência cardíaca, alterações cognitivas e câncer.
DOENÇA HEPÁTICA GORDUROSA NÃO ALCOÓLICA (NAFLD)
A doença hepática gordurosa não alcoólica (NAFLD) é uma condição clínico-patológica comum, caracterizada por depósito de lipídeos no hepatócito do parênquima hepático (1-3). O quadro patológico lembra o da lesão hepática induzida pelo álcool, mas ocorre em indivíduos que não têm ingestão etílica significativa. O espectro de lesão hepática varia de esteatose macrovesicular simples para esteatoepatite, fibrose avançada e cirrose. A NAFLD é talvez a causa principal de morbidade e mortalidade ligadas a doenças do fígado, com potencial para progredir para insuficiência hepática. A progressão para fibrose ou cirrose parece ocorrer só em pacientes com evidência de esteatoepatite.
Epidemiologia
A prevalência mundial de NAFLD não foi ainda determinada, mas estima-se que seja de 1024% em várias populações (3). Embora possa haver erros nessas estimativas, a NAFLD é a doença hepática mais comum no mundo ocidental, e sua prevalência está aumentando. Afeta todos os grupos raciais e étnicos, sem predileção por sexo ou idade.
A NAFLD é a causa de elevação assintomática de aminotransferases em 4590% dos casos, excluídas outras causas bem estabelecidas. A prevalência de NAFLD aumenta significativamente em obesos, podendo chegar a 5075%. É provável que o aumento na prevalência de NAFLD seja paralelo ao aumento da prevalência de obesidade e diabetes em todos os grupos etários.
Etiopatogenia
Diferentes agentes e condições patológicas estão associados com NAFLD, como resistência à insulina adquirida, erros inatos do metabolismo, condições médicas ou cirúrgicas associadas à perda de peso, e algumas drogas e toxinas. Parece que a NAFLD, o DM2 e a dislipidemia compartilham mecanismos patogênicos. É provável que a esteatoepatite seja mediada pela resistência à insulina, um solo comum a estas condições.
Embora a patogênese exata da NAFLD permaneça desconhecida, a hipótese mais aceita é que diversas agressões estão envolvidas nesta condição. Em primeiro lugar, como resultado da resistência à insulina, há uma maior síntese e retenção de triglicérides no hepatócito, levando à esteatose macrovesicular. É provável que uma menor oxidação de ácidos graxos, por disfunção mitocondrial, possa contribuir para esta alteração. A segunda agressão é geralmente atribuída ao stress oxidativo, que causa peroxidação de lipídeos na membrana do hepatócito, produção de citocinas que são, em parte, responsáveis pela progressão de esteatose para esteatoepatite e cirrose (2,3). Toxinas bacterianas, hiperprodução de citocinas (especialmente TNFa), alteração dos estoques de ATP e da atividade da enzima citocromo P450 Cyp2E1 parecem ser gatilhos importantes para a progressão da doença e fibrogênese. Na obesidade, além da resistência à insulina, há também resistência à leptina (4), e os níveis deste hormônio estão elevados. O papel da leptina é ainda controverso, com estudos sugerindo que este hormônio promove esteatose hepática e esteatoepatite, e outros mostrando que os níveis de leptina se correlacionam com esteatose, mas não com inflamação e fibrose (5,6).
Diagnóstico
Sinais e sintomas
Como em outras doenças hepáticas, muitos pacientes com NAFLD (50100%) são assintomáticos. A doença hepática é muitas vezes descoberta acidentalmente, durante exames de rotina que revelam um aumento da AST. A NAFLD é a causa mais comum de aumentos não explicados de AST, desde que se exclua hepatite C ou outras doenças crônicas do fígado. Quando ocorrem os sintomas, em geral são pouco específicos. Dor vaga, no quadrante superior direito do abdômen, cansaço e fraqueza são os mais comuns. Ocasionalmente, prurido, anorexia e náusea se desenvolvem. Icterícia, ascite, sangramento gastrointestinal e manifestações de encefalopatia são indicativos de doença hepática avançada (cirrose descompensada), ocorrendo tardiamente na evolução.
Não há sinais específicos de NAFLD. A obesidade é a anormalidade mais comum no exame físico. Hepatomegalia é descrita em ~75% dos pacientes (por US pode chegar a 95%). Sinais de hipertensão portal são menos freqüentes, embora a esplenomegalia possa ser encontrada em 25% dos pacientes na época do diagnóstico. Dos sinais de insuficiência hepática, spider e eritema palmar são os mais comuns (3).
Achados laboratoriais
Elevações discretas ou moderadas nos níveis de AST e ALT são as alterações laboratoriais mais freqüentes. Não há correlação entre o grau de elevação destas enzimas com a gravidade histológica da inflamação ou fibrose. Diferente de pacientes com esteatoepatite induzida por álcool, que apresentam aumento maior de AST em relação à ALT, em pacientes com NAFLD a relação AST/ALT é menor que 1. Esta relação tende a aumentar com o desenvolvimento de cirrose, perdendo capacidade de discriminação diagnóstica. Os níveis de fosfatase alcalina podem ser discretamente elevados em 1/3 dos pacientes. Hiperbilirrubinemia, hipoalbuminemia e aumento do tempo de protrombina aparecem com menos freqüência, e geralmente são vistos quando há falência hepática.
Um pequeno percentual de pacientes com NAFLD pode apresentar baixos títulos (< 1/320) de anticorpos-antinucleares (ANA). O papel do ferro na patogênese da NAFLD é controverso. Alguns estudos mostram uma elevação da saturação de transferrina em ~10% e da ferritina em ~50% dos pacientes. Não há indicação para que se pesquise, rotineiramente, hemocromatose genética em pacientes com esteatoepatite não alcoólica.
É importante excluir outras causas secundárias de esteatose, para que se possa fazer um diagnóstico de NAFLD primária com segurança. A hepatite C (HCV) e a doença hepática pelo álcool são particularmente importantes, pela alta prevalência destes dois agentes hepatotóxicos. HCV pode induzir alterações histológicas que lembram a NAFLD, e testes sorológicos para excluir hepatites virais são pré-requisito para o diagnóstico de NAFLD. Pela definição, o diagnóstico de NAFLD não pode ser feito em pacientes com ingestão excessiva de álcool. Acredita-se que não se desenvolve esteatose com ingestões < 20g/dia para mulheres e < 30g/dia para homens.
Exames de imagem
Ultrassonografia (US), tomografia computadorizada (TC) e ressonância magnética (MRI) podem identificar a esteatose hepática. Destes, a US é a mais barata. Os achados sonográficos de alterações gordurosas difusas são ecotextura hiperecóica difusa e aumento de ecotextura comparado ao rim. Na TC, evidencia-se menor densidade do parênquima hepático. A comparação destes dois métodos mostra que o US é mais sensível na detecção de mudanças gordurosas difusas. Entretanto, quando as alterações na gordura são localizadas, a TC e a MRI são superiores à US. Deve ser destacado que nenhum dos métodos é capaz de distinguir esteatose de esteatoepatite, nem estimar a gravidade da alteração. Assim, a biópsia hepática é o melhor método diagnóstico para esteatoepatite.
Histologia hepática
O valor da biópsia hepática no diagnóstico de esteatoepatite (NASH) na prática clínica ainda é debatido. A ausência de tratamento médico adequado e os riscos associados à biópsia são argumentos propostos contra a obtenção de tecido hepático. Entretanto, a biópsia é o único método diagnóstico de esteatoepatite e a única maneira de se identificar a gravidade do dano hepático, bem como a prognóstico. Há dados clínico-laboratoriais que podem identificar pacientes com maior probabilidade de fibrose hepática, nos quais a biópsia pode ter maior valor prognóstico. Estes dados incluem: idade > 45 anos, presença de obesidade ou DM2 e relação AST/ALT > 1. A decisão de realizar a biópsia na prática clínica deve ser individualizada e compartilhada com o paciente. A figura 1 apresenta um algoritmo para a investigação diagnóstica de NAFLD.
Os achados histológicos da NAFLD são indistinguíveis dos decorrentes de doença hepática induzida por álcool. Há dois tipos de alterações associadas com NAFLD: 1) esteatose macrovesicular predominante, isoladamente, ou 2) esteatose macrovesicular predominante e graus variáveis de balonamento citológico e áreas de necrose, infiltrado inflamatório neutrofílico-linfocítico, corpúsculo hialino de Mallory e fibrose perisinusoidal. Nem todas as características da esteatoepatite estão presentes em cada caso. Entretanto, há padronizações descritas para a análise anátomo-patológica de NAFLD (7).
História natural
A história natural da NAFLD ainda não está bem estabelecida, mas parece ser determinada pela gravidade da lesão histológica. Estudos transversais de NAFLD indicam que a maioria dos indivíduos apresenta somente esteatose, e é rara a progressão para esteatoepatite ou fibrose na evolução (1-3). Em alguns estudos encontrou-se, no momento do diagnóstico, fibrose hepática avançada em 3040% dos casos, e cirrose bem estabelecida em 1015% dos pacientes. Pode haver progressão para carcinoma hepatocelular. A coexistência de esteatose com outras doenças hepáticas, como hepatite C, pode acelerar a progressão da doença hepática.
Nos pacientes com diagnóstico de esteatoepatite, estabelecido na biópsia, o seguimento demonstra que 1/3 tem progressão da fibrose, e 1/3 destes com progressão rápida para fibrose avançada. O único dado laboratorial que se correlacionou com a progressão histológica foi elevação dos níveis de AST.
Dados recentes sugerem que NASH pode ser a principal causa de cirrose "criptogênica". Há perda de infiltração gordurosa em pacientes com NASH associado à cirrose. Serão necessários grandes estudos prospectivos nessa área, para uma melhor definição da história natural da NAFLD.
Tratamento
Não há ainda tratamento efetivo que mude a história natural da NAFLD. Na ausência de terapêutica eficaz, o tratamento é direcionado à correção dos fatores de risco para NASH.
Perda de peso
Um programa apropriado de dieta e exercício é importante. Embora alguns estudos mostrem melhora bioquímica e histológica, com perda de peso gradual e moderada (~10%), não há estudos clínicos bem controlados sobre a perda de peso como tratamento da NAFLD. Tratamentos farmacológicos da obesidade, ou cirurgias bariátricas, não têm indicações precisas na NAFLD. Há o risco de piora da doença hepática quando a perda de peso é muito rápida (3).
Resistência à insulina
Não há estudos bem controlados sobre o uso de drogas que melhoram a sensibilidade à insulina em NASH. Há dados que mostram que a administração de rosiglitazona por 48 semanas induziu melhora da resistência à insulina e de marcadores histológicos de NASH (8). Também um estudo piloto com pioglitazona por um ano mostrou melhora da sensibilidade à insulina, dos níveis de AST e de dados histológicos (9). A administração de metformina por 4 meses também induziu melhora nos níveis de aminotransferases (10), mas não se obteve biópsia pós-tratamento, e o uso desta droga permanece experimental.
Não há evidências convincentes de que drogas que reduzem os níveis de triglicérides possam ter benefício no tratamento da NAFLD, e não há ainda estudos investigando os efeitos das estatinas nesta alteração hepática.
Terapia farmacológica que oferece proteção hepática
Diversos agentes terapêuticos, que potencialmente oferecem proteção hepática, foram utilizados na NAFLD. Para o uso do ácido ursodeoxicólico (UDCA) e dos anti-oxidantes vitamina E e betaina, há evidências científicas (3). Para outras drogas como lecitina, b-caroteno, selênio, N-acetilcisteína, não há dados controlados. Em alguns estudos, a suplementação com vitamina E (4001200 UI), usada por seu potencial de reduzir o stress oxidativo, mostrou efeitos na redução dos níveis de AST, mas os dados histológicos ou não foram disponíveis ou não mostraram alterações em comparação ao placebo. A eficácia da pioglitazona associada à vitamina E também foi avaliada, e se demonstrou que esta associação melhorava os dados histológicos e diminuía a AST, enquanto a vitamina E isoladamente só reduzia os níveis de AST (11). Um estudo piloto indica que o uso de betaina também pode induzir melhora nos dados bioquímicos e anátomo-patológicos de pacientes com NASH (12).
SÍNDROME DOS OVÁRIOS POLICÍSTICOS
A síndrome dos ovários policísticos (SOP) é a alteração endócrina mais comum em mulheres pré-menopausa, e hoje está bem estabelecido que a prevalência de resistência à insulina/hiperinsulinemia é significativamente maior nessas pacientes (13,14). A SOP é outro exemplo onde parece que a hiperinsulinemia compensatória (secundária à resistência à insulina em músculo e adiposo) agiria normalmente em outros tecidos, incluindo o ovário.
Há evidências de que, na SOP, há um aumento da secreção de testosterona pelo ovário, que parece ter, no mínimo, sensibilidade normal à insulina. Esta maior secreção de andrógenos parece ser induzida pela hiperinsulinemia, que tem ação sinérgica ao LH (15). Como os ovários policísticos compõem uma síndrome, é necessário enfatizar os seguintes pontos: nem todas as pacientes com resistência à insulina desenvolvem ovários policísticos, e nem todas as pacientes com SOP apresentam resistência à insulina. Entretanto, a importância da hiperinsulinemia na patogênese da SOP pode ser deduzida pela efetividade da metformina em pacientes com a síndrome, que é paralela à redução da hiperinsulinemia. A SOP é discutida em outra revisão deste fascículo.
HIPERURICEMIA
Elevações nas concentrações séricas de ácido úrico são comumente vistas em associação com intolerância à glicose, dislipidemia e hipertensão arterial, e há correlação significativa entre estas concentrações e resistência à insulina ou níveis de insulina no TOTG (16). Há evidências de que a insulina reduz o clearance urinário de ácido úrico (17). É interessante que indivíduos assintomáticos com hiperuricemia apresentam menor sensibilidade à insulina que indivíduos sem hiperuricemia. Assim, a hiperuricemia parece ser um componente das anormalidades da síndrome de resistência à insulina, explicando melhor a associação de elevação dos níveis de ácido úrico e doença cardiovascular.
DOENÇA RENAL CRÔNICA
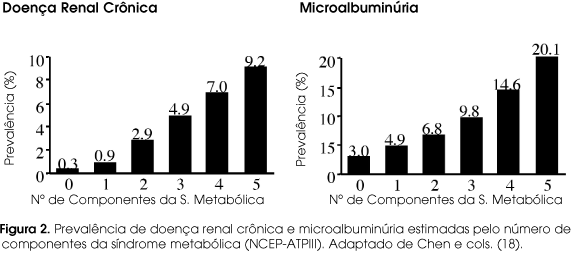
Recentemente, a relação entre indicadores de doença renal crônica e síndrome metabólica tem despertado atenção. A microalbuminúria é um dos critérios utilizados para o diagnóstico de síndrome metabólica pela definição da OMS. A freqüência de microalbuminúria aumenta quando se caminha de tolerância à glicose normal (510%) para síndrome metabólica (1220%), e para DM2 (2540%) (18-20). Há uma correlação significativa entre disfunção endotelial e microalbuminúria, sugerindo uma relação causal em que a microalbuminúria refletiria a disfunção endotelial expressa no glomérulo (21).
A prevalência ou probabilidade de microalbuminúria e (ou) redução da filtração glomerular são progressivamente ampliadas pelo aumento do número de fatores de risco da síndrome metabólica (figura 2). Alterações patológicas específicas têm sido definidas como "glomerulopatia relacionada à obesidade". A alteração primária é uma glomerulomegalia (100% dos casos), glomerulosclerose focal e segmentar (80% dos casos) e aumento da celularidade da matriz mesangial (45% dos casos). Há achados que lembram também a glomerulopatia do diabetes e (ou) da hipertensão. A evolução clínica da glomerulopatia relacionada à obesidade parece ser progressiva. Após um seguimento de 27 meses, 14% dos pacientes dobram os níveis de creatinina ou atingem insuficiência renal terminal (22). Merece destaque que esta glomerulopatia relacionada à obesidade foi observada em uma criança com 3 anos de idade (23).
Os mecanismos moleculares responsáveis por esta glomerulopatia não são ainda esclarecidos, mas provavelmente refletem o processo inflamatório subclínico que ocorre na síndrome metabólica, com a ativação intracelular de vias inflamatórias como a do IKK/IkB/NFkB e a da JNK, que devem contribuir para a disfunção endotelial.
INSUFICIÊNCIA CARDÍACA
Diabetes e obesidade são fatores de risco para insuficiência cardíaca, e ambos são associados com resistência à insulina. Recentemente, uma investigação proveniente de um estudo longitudinal em Uppsala com homens adultos, demonstrou que a resistência à insulina é um fator de risco para insuficiência cardíaca, independente de outros fatores como diabetes (24). Assim, o mais provável é que a associação entre obesidade e desenvolvimento de insuficiência cardíaca é amplamente mediada pela resistência à insulina. A resistência à insulina também está associada à hipertrofia do miocárdio (25), e é possível que a angiotensina II tenha ações mitogênicas e de crescimento sinérgicas à insulina neste tecido (26,27).
ALTERAÇÕES COGNITIVAS
Estudo recente, avaliando uma população de idosos americanos, demonstrou que a presença de síndrome metabólica aumentava o risco de alterações cognitivas, independentemente de variações demográficas, de hábitos de vida e co-morbidades. Neste estudo, os critérios diagnósticos de síndrome metabólica usados foram os do NCEP-ATP III, e avaliaram-se também níveis de IL-6 e proteína C-reativa. O risco de redução cognitiva era ainda mais evidente em idosos com alterações inflamatórias mais acentuadas (28).
CÂNCER
Embora a obesidade seja reconhecida como importante causa de diabetes e doença cardiovascular, a associação entre obesidade e diferentes tipos de câncer tem recebido muito menos atenção. Apesar disso, os resultados de estudos epidemiológicos que começaram na década de 70 indicam que a obesidade contribui para o aumento da incidência e/ou mortalidade por câncer de cólon, mama (em mulheres na pós-menopausa), endométrio, rim, esôfago (adenocarcinoma), gástrico (cárdia), pâncreas, vesícula biliar e fígado (tabela 1). Com efeito, acredita-se que a obesidade atualmente seja a causa de 1520% de todos os cânceres nos Estados Unidos, constituindo-se, dessa maneira, no principal fator de risco para o desenvolvimento de câncer em indivíduos não fumantes (29).
Recentemente, a agência internacional para pesquisa em câncer (IARC) avaliou toda a literatura disponível sobre a associação entre obesidade e câncer, considerando tanto estudos epidemiológicos como ensaios clínicos e experimentais. Nessa revisão, concluiu-se que evitar o aumento de peso reduz o risco de desenvolver cânceres de cólon, mama (em mulheres na pós-menopausa), endométrio, rim e esôfago (adenocarcinoma). Essas conclusões foram baseadas em estudos epidemiológicos de indivíduos com sobrepeso e obesos comparados a indivíduos magros não em estudos de indivíduos que perderam peso. Infelizmente, poucos indivíduos conseguem manter perda de peso significativa após algum tempo, tornando extremamente difícil examinar a conseqüência da redução de peso em grandes populações que perderam peso. Conseqüentemente, o IARC concluiu que não existem evidências adequadas de que perder peso reduz o risco de desenvolver câncer (30).
Atualmente, os mecanismos que conectam a obesidade com o risco aumentado de desenvolver câncer envolvem os efeitos endócrinos e metabólicos da obesidade e as conseqüentes alterações que eles induzem na produção de peptídeos e hormônios esteróides. Nesse contexto, destacam-se a hiperinsulinemia crônica, alteração da secreção de hormônios esteróides sexuais e esteatoepatite não alcoólica (29).
A hiperinsulinemia crônica está associada com a patogênese do câncer de cólon e com os cânceres de mama, pâncreas e endométrio. Esses efeitos podem ser mediados diretamente pela presença de receptores de insulina nas células (pré) neoplásicas, estimulando o crescimento, ou ter a sua gênese mediada por mecanismos comuns que ocasionam a resistência à insulina como, por exemplo, a inflamação crônica subclínica com o aumento do TNFa, que agiria como agente promotor do crescimento tumoral.
O aumento da adiposidade influencia a síntese e viabilidade dos hormônios esteróides sexuais, através de pelo menos três mecanismos. Primeiro, o tecido adiposo aumenta a conversão de andrógenos a estrógenos. Segundo, a obesidade ocasiona a redução da síntese da globulina carreadora de hormônios esteroidais (SHBG) aumentando a biodisponibilidade dos estrógenos. Finalmente, a hiperinsulinemia per se pode levar a aumento da síntese de andrógenos ovarianos, como pode ser observado em mulheres com síndrome dos ovários policísticos. Estudos epidemiológicos correlacionam essas alterações com o aumento da incidência de câncer de mama e endométrio.
Como visto anteriormente, uma das conseqüências da resistência à insulina é o desenvolvimento de doença gordurosa do fígado (NAFLD). A NAFLD é caracterizada por um espectro de alterações do tecido hepático que varia do acúmulo de gordura no fígado a esteatoepatite não alcoólica (NASH), cirrose e hepatocarcinoma. Assim, a síndrome metabólica contribui para o risco de hepatocarcinoma através de NAFLD e NASH.
Recebido em 17/01/06
Aceito em 17/01/06
- 1. Sheth SG, Gordon FD, Chopra S. Nonalcoholic steatohepatitis. Ann Intern Med 1997;126:137-45.
- 2. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002;346:1221-31.
- 3. Sass DA, Chang P, Chopra KB. Nonalcoholic fatty liver disease: a clinical review. Dig Dis Sci 2005;50:171-80.
- 4. Carvalheira JB, Ribeiro EB, Folli F, Velloso LA, Saad MJ. Interaction between leptin and insulin signaling pathways differentially affects JAK-STAT and PI 3-kinase-mediated signaling in rat liver. Biol Chem 2003;384:151-9.
- 5. Uygun A, Kadayifci A, Yesilova Z, Erdil A, Yaman H, Saka M, et al. Serum leptin levels in patients with nonalcoholic steatohepatitis. Am J Gastroenterol 2000;95:3584-9.
- 6. Chitturi S, Farrell G, Frost L, Kriketos A, Lin R, Fung C, et al. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology 2002;36:403-9.
- 7. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313-21.
- 8. Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPARg ligand rosiglitazone. Hepatology 2003;38:1008-17.
- 9. Promrat K, Lutchman G, Uwaifo GI, Freedman RJ, Soza A, Heller T, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 2004;39: 188-96.
- 10. Marchesini G, Brizi M, Bianchi G, Tomassetti S, Zoli M, Melchionda N. Metformin in non-alcoholic steatohepatitis. Lancet 2001;358:893-4.
- 11. Sanyal AJ, Mofrad PS, Contos MJ, Sargeant C, Luketic VA, Sterling RK, et al. A pilot study of vitamin E versus vitamin E and pioglitazone for the treatment of nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 2004;2:1107-15.
- 12. Abdelmalek MF, Angulo P, Jorgensen RA, Sylvestre PB, Lindor KD. Betaine, a promising new agent for patients with nonalcoholic steatohepatitis: results of a pilot study. Am J Gastroenterol 2001;96:2711-7.
- 13. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev 1997;18:774-800.
- 14. Nestler JE, Jakubowicz DJ, Evans WS, Pasquali R. Effects of metformin on spontaneous and clomiphene-induced ovulation in the polycystic ovary syndrome. N Engl J Med 1998;338:1876-80.
- 15. Carvalho CR, Carvalheira JB, Lima MH, Zimmerman SF, Caperuto LC, Amanso A, et al. Novel signal transduction pathway for luteinizing hormone and its interaction with insulin: activation of Janus kinase/signal transducer and activator of transcription and phosphoinositol 3-kinase/Akt pathways. Endocrinology 2003;144:638-47.
- 16. Wyngaarden JB, Kelley WN. Gout. Metabolic basis of inherited disease 5th ed. New York: McGraw-Hill; 1983 p.1043.
- 17. Zavaroni I, Mazza S, Fantuzzi M, Dall'Aglio E, Bonora E, Delsignore R, et al. Changes in insulin and lipid metabolism in males with asymptomatic hyperuricaemia. J Intern Med 1993;234:25-30.
- 18. Chen J, Muntner P, Hamm LL, Jones DW, Batuman V, Fonseca V, et al. The metabolic syndrome and chronic kidney disease in US adults. Ann Intern Med 2004;140:167-74.
- 19. Jones CA, Francis ME, Eberhardt MS, Chavers B, Coresh J, Engelgau M, et al. Microalbuminuria in the US population: third National Health and Nutrition Examination Survey. Am J Kidney Dis 2002;39:445-59.
- 20. Palaniappan L, Carnethon M, Fortmann SP. Association between microalbuminuria and the metabolic syndrome: NHANES III. Am J Hypertens 2003;16:952-8.
- 21. Clausen P, Jensen JS, Jensen G, Borch-Johnsen K, Feldt-Rasmussen B. Elevated urinary albumin excretion is associated with impaired arterial dilatory capacity in clinically healthy subjects. Circulation 2001;103:1869-74.
- 22. Kambham N, Markowitz GS, Valeri AM, Lin J, D'Agati VD. Obesity-related glomerulopathy: an emerging epidemic. Kidney Int 2001;59:1498-509.
- 23. Cohen AH. Massive obesity and the kidney. A morphologic and statistical study. Am J Pathol 1975;81:117-30.
- 24. Ingelsson E, Sundstrom J, Arnlov J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA 2005;294:334-41.
- 25. Burchfiel CM, Skelton TN, Andrew ME, Garrison RJ, Arnett DK, Jones DW, et al. Metabolic syndrome and echocardiographic left ventricular mass in blacks: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation 2005;112:819-27.
- 26. Carvalheira JB, Calegari VC, Zecchin HG, Nadruz W Jr., Guimarães RB, Ribeiro EB, et al. The cross-talk between angiotensin and insulin differentially affects phosphatidylinositol 3-kinase- and mitogen-activated protein kinase-mediated signaling in rat heart: implications for insulin resistance. Endocrinology 2003;144:5604-14.
- 27. Carvalho CR, Thirone AC, Gontijo JA, Velloso LA, Saad MJ. Effect of captopril, losartan, and bradykinin on early steps of insulin action. Diabetes 1997;46:1950-7.
- 28. Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, et al. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA 2004;292:2237-42.
- 29. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 2004;4:579-91.
-
30International Agency for Research on Cancer. IARC handbooks of cancer prevention, weight control and physical activity Lyon: International Agency for Research on Cancer, 2002
Datas de Publicação
-
Publicação nesta coleção
23 Maio 2006 -
Data do Fascículo
Abr 2006
Histórico
-
Aceito
17 Jan 2006 -
Recebido
17 Jan 2006