Abstracts
This review offers an overview of physiological agents, current therapeutics, as well as medications, which have been extensively used and those agents not currently available or non-classically considered anti-obesity drugs. As obesity - particularly that of central distribution - represents an important triggering factor for insulin resistance, its pharmacological treatment is relevant in the context of metabolic syndrome control. The authors present an extensive review on the criteria for anti-obesity management efficacy, on physiological mechanisms that regulate central and/or peripheral energy homeostasis (nutrients, monoamines, and peptides), on beta-phenethylamine pharmacological derivative agents (fenfluramine, dexfenfluramine, phentermine and sibutramine), tricyclic derivatives (mazindol), phenylpropanolamine derivatives (ephedrin, phenylpropanolamine), phenylpropanolamine oxytrifluorphenyl derivative (fluoxetine), a naftilamine derivative (sertraline) and a lipstatine derivative (orlistat). An analysis of all clinical trials - over ten-week long - is also presented for medications used in the management of obesity, as well as data about future medications, such as a the inverse cannabinoid agonist, rimonabant.
Obesity; Treatment; Anfepramone; Mazindol; Sibutramine; Orlistat; Rimonabant
Esta revisão faz um apanhado dos agentes fisiológicos e terapêutica atual, bem como de medicações que têm sido usadas extensivamente e de outros agentes ainda não disponíveis ou que são consideradas drogas anti-obesidade não clássicas. Como a obesidade - em especial aquela com distribuição central - representa um importante fator desencadeador de resistência à insulina, o seu tratamento farmacológico é relavente no contexto do controle da síndrome metabólica. Os autores apresentam uma revisão extensa dos critérios de eficácia do manuseio anti-obesidade, dos mecanismos fisiológicos que regulam a homeostase energética central e/ou periférica (nutrientes, monoaminas e peptídeos), dos agentes farmacologicamente derivados dos seguintes produtos: beta-fenetilamina (fenfluramina, dexfenfluramina, fentermina e sibutramina), tricíclicos (mazindol), fenilpropanolamina (efedrina, fenilpropanolamina), fenilpropanolamina oxitrifluorofenil (fluoxetina), naftilamina (sertralina) e lipstatina (orlistat). Também é apresentada uma análise de todos os ensaios clínicos com duração maior do que 10 semanas para medicações usadas no manuseio da obesidade, assim como dados sobre medicações futuras, como o agonista canabinóide inverso, rimonabant.
Obesidade; Tratamento; Anfepramona; Mazindol; Sibutramina; Orlistat; Rimonabant
REVISÃO
Pharmacological treatment of obesity
Tratamento farmacológico da obesidade
Marcio C. Mancini; Alfredo Halpern
Endocrinology and Metabology Division, Hospital das Clínicas, University of São Paulo Medical School, São Paulo, SP
Endereço para correspondência Endereço para correspondência: Marcio C. Mancini Rua Alves Guimarães 462/72 05410-000 São Paulo, SP Fax: (11) 3063-0063 E-mail: mmancini@usp.br
ABSTRACT
This review offers an overview of physiological agents, current therapeutics, as well as medications, which have been extensively used and those agents not currently available or non-classically considered anti-obesity drugs. As obesity particularly that of central distribution represents an important triggering factor for insulin resistance, its pharmacological treatment is relevant in the context of metabolic syndrome control. The authors present an extensive review on the criteria for anti-obesity management efficacy, on physiological mechanisms that regulate central and/or peripheral energy homeostasis (nutrients, monoamines, and peptides), on b-phenethylamine pharmacological derivative agents (fenfluramine, dexfenfluramine, phentermine and sibutramine), tricyclic derivatives (mazindol), phenylpropanolamine derivatives (ephedrin, phenylpropanolamine), phenylpropanolamine oxytrifluorphenyl derivative (fluoxetine), a naftilamine derivative (sertraline) and a lipstatine derivative (orlistat). An analysis of all clinical trials over ten-week long is also presented for medications used in the management of obesity, as well as data about future medications, such as a the inverse cannabinoid agonist, rimonabant.
Keywords: Obesity; Treatment; Anfepramone; Mazindol; Sibutramine; Orlistat; Rimonabant
RESUMO
Esta revisão faz um apanhado dos agentes fisiológicos e terapêutica atual, bem como de medicações que têm sido usadas extensivamente e de outros agentes ainda não disponíveis ou que são consideradas drogas anti-obesidade não clássicas. Como a obesidade em especial aquela com distribuição central representa um importante fator desencadeador de resistência à insulina, o seu tratamento farmacológico é relavente no contexto do controle da síndrome metabólica. Os autores apresentam uma revisão extensa dos critérios de eficácia do manuseio anti-obesidade, dos mecanismos fisiológicos que regulam a homeostase energética central e/ou periférica (nutrientes, monoaminas e peptídeos), dos agentes farmacologicamente derivados dos seguintes produtos: b-fenetilamina (fenfluramina, dexfenfluramina, fentermina e sibutramina), tricíclicos (mazindol), fenilpropanolamina (efedrina, fenilpropanolamina), fenilpropanolamina oxitrifluorofenil (fluoxetina), naftilamina (sertralina) e lipstatina (orlistat). Também é apresentada uma análise de todos os ensaios clínicos com duração maior do que 10 semanas para medicações usadas no manuseio da obesidade, assim como dados sobre medicações futuras, como o agonista canabinóide inverso, rimonabant.
Descritores: Obesidade; Tratamento; Anfepramona; Mazindol; Sibutramina; Orlistat; Rimonabant
THE PHARMACOLOGICAL MANAGEMENT of obesity has witnessed drastic changes and experienced the development of new products and treatment proposals. Information presented in this review offer an overview of physiological agents, current therapeutics, as well as medications widely used in the past and no longer available.
There is no specific strategy or medication to be recommended on a routine basis. The obese individual must be thoroughly examined regarding improper eating habits and exercising, depression symptoms, obesity-associated complications or conditions, and the possibility of developing side effects. The choice for anti-obesity medications is also based on patients' prior experience and previous therapies, although the failure of a previous treatment does not rule out an agent for later use.
The understanding of some key concepts is crucial in any discussion on the rationale of anti-obesity medications: 1) pharmacological treatment can only be justified when combined with diet and lifestyle changes. Efficacy of all agents depends on patients' compliance to nutritional and behavioral changes; 2) pharmacological treatment does not cure obesity when discontinued, weight gain is expected; 3) anti-obesity medications must be used under continuous medical supervision; 4) treatment and medication choice are patient-tailored. The risks associated to the use of a drug must be assessed considering the obesity persistence; 5) treatment should be maintained only when considered safe and effective for the patient.
Anti-obesity pharmacological treatment is indicated when body mass index (BMI) is over 30 kg/m2, or when morbidities are associated to overweight (BMI over 25 kg/m2) when dieting, physical activities and behavioral changes have proved unsuccessful (1).
Anti-obesity pharmacological agents are not recommended for children, since up to this point in time there is not enough evidence on their effects at this age group.
A useful medication for obesity treatment must: 1) be effective for body weight reduction and result in overweight-dependent conditions improvement; 2) have a long-term efficacy and safety; 3) be related to tolerable or transitory side effects; 4) not be addictive; 5) have known mechanism of action; 6) be reasonably affordable (2).
Obesity is a chronic and stigmatized disease (3) as it is the case of hypertension or hypercholesterolemia (4). Each of these chronic diseases is associated to a number of co-morbidities. Hypertension may cause heart failure and stroke while hypercholesterolemia commonly leads to atherosclerosis and coronary events. For obesity, its main consequences are numerous such as diabetes mellitus, systemic hypertension, dyslipidemia, cardiovascular diseases, certain types of cancer, sleep apnea, ostheoarthritis, among others.
Obesity is recognized as an epidemic condition that affects populations worldwide (1,5). Therefore, the need to improve the quality and efficacy of therapeutics has emerged. The core to current obesity management is based on specific behavioral therapies aiming to change eating habits and raise energy expenditure. Nutritional counseling to lower the intake of calories, particularly fat, associated with increased daily physical activities are highly necessary but compliance are very limited. Pharmacological management is seen as additional tool to this basic therapy.
As obesity particularly that of central distribution represents an important triggering factor for insulin resistance, its pharmacological treatment is relevant in the context of metabolic syndrome control.
Pharmacological treatment of obesity is subject to classification according to the mechanisms of action. Knowledge on body adiposity control and regulation had marked improvement in the last decades. One class of anti-obesity agents involves the control mechanism of energy intake. A second strategy against obesity relates to shift the normal nutrient metabolism and a third one to raise energy expenditure.
ANTI-OBESITY TREATMENT: CRITERIA FOR ASSESSING EFFICACY
A number of criteria have been proposed to assess the response to treatments for obesity. Nowadays, the most widely used criteria to assess the efficacy of anti-obesity therapies are those proposed by the Food and Drug Administration (FDA) in the United States, and by the Committee of the European Agency for the Evaluation of Medicinal Products (CPMP) in Europe. According to the FDA, a 5% weight loss significantly higher than placebo is consider response to treatment; CPMP suggests body weight loss over 10% as compared to placebo. In addition, the agencies suggest the inclusion of a run-in period, categoric analysis of the results (patients who have lost over 5% or 10% of their initial weight) and consider the improvement of obesity co-morbidities. Those and other secondary criteria are listed in a recent review (6). The basic difference between the criteria used by the American and the European agencies is the emphasis given to ancillary recommendations stronger on the part of the European agency including behavioral changes following the initial counseling in long-term studies, which raises body weight loss in the placebo group, thus allowing the detection of true effects of the active ingredient. If patients under study lose weight quickly under a behavioral change program or a very low calorie diet, it is harder to keep track of anti-obesity medications additional effects.
The typical body weight history of overweight individuals is an approximate 0.25 kg gain per year (7). A very good objective, under a population point of view, would be the prevention of any kind of additional weight. For obese individuals, a sustained 5% loss may be considered the lowest criterion for success. A 5% to 10% sustained loss as compared to initial weight associated with partial or no improvement of risk factors would be from reasonable to good a response, whereas losses over 15% associated with normalization of risk factors and body weight reduction below 25 kg/m2 would be excellent and ideal, although hardly ever attainable in clinical practice.
Most studies usually report maximal body weight loss between 20 and 24 weeks of treatment. In a review of clinical trials, Bray calculated that weight loss in weeks 6, 12, and 18 in average corresponded to 44%, 72% and 89% of the 24-week loss (ref).
PHARMACOLOGICAL AGENTS MODULATING ENERGY HOMEOSTASIS
Pre-absortive agents
b-phenethylaminic and phenylpropanolaminic derivates
All central action anorectic medications except for mazindol are derived from b-phenethylamine. The b-phenethylaminic skeleton is also the structure of the neurotransmitters dopamine, norepinephrine (NE), and epinephrine (monoamines). Those neurotransmitters are tyrosine-synthetised at nervous terminations, stored in granules and released in the synaptic cleft to act on post-ganglionic receptors. After they act on those receptors, the monoamines may be deactivated through catechol-O-methyltransferase or be reuptaken by the nervous termination (8).
Chemical modifications in the structure of amphetamine (a-methyl-b-phenethylamine) resulted in the synthesis of a range of compounds, with different pharmacological actions and responses. On one end within the pharmacological spectrum, b-phenethylaminic derivates diethylpropion and phentermine influence noradrenergic and dopaminergic neurotransmission (stimulating release or blocking reuptake) which results in NE release from the nervous termination, raising the amount of NE interacting with post-synaptic receptors (17). On the other end, the substances affecting serotonin release and reuptake can be found, such as dexfenfluramine and its levorotatory isomer, l-fenfluramine or fenfluramine (9). Sibutramine can be found in the middle, blocking NE and serotonin reuptake (10).
Binding studies of [3H] marked phenethylaminic derivates have shown the presence of binding sites of variable affinity in the hypothalamus and in other cerebral regions (11). The binding affinity of the various phenethylaminic derivates is correlated to its anorectic potency, but not to its stimulative ability (20). The binding sites of those substances in the hypothalamus are regulated by glucose level through the action of ionic channels, stimulating sodium-potassium ATPase pump (12).
All b-phenethylaminic derivates have shown to be feeding-reducing in animal studies. Such action is the primary mechanism for weight-loss induction. When feeding is kept constant and paired with control group, weight loss is the same in the group receiving the active substance or placebo. These studies were carried out in animals using amphetamine, and in humans using fenfluramine (in metabolic units) (13,14). The effect is dose-related and takes place immediately after parenteral administrations of the substances (22).
Feeding pattern differs between compounds with primarily noradrenergic or serotoninergic action mechanisms. While amphetamine delays intake outset, fenfluramine does not, but rather anticipates food intake cessation (15). In animals, the administration of serotonin and fenfluramine primarily reduces fat intake (16), whereas NE injection at the paraventricular nucleus affects carbohydrate intake, and noradrenergic medications may have selective action on macronutrient choice (17).
Generally, b-phenethylaminic medications report thermogenic action in animal studies. Mazindol (in this paper, taken together with b-phenethylaminic medications despite a non-b-phenethylaminic derivate) stimulates oxygen consumption (just like diethylpropion) and raises NE stimulation in brown fat (just like amphetamine, fenfluramine, and dexfenfluramine) in rats. Sibutramine that blocks NE and serotonin reuptake reduces food intake and also stimulates thermogenesis in brown adipose tissue in animals (18).
Clinical pharmacokinetics
Noradrenergic anorectic medications are usually well absorbed in the gastrointestinal tract, reaching plasma peak levels in the first two hours (table 2). They are removed through metabolization or by hepatic conjugation, which produces active metabolites for some drugs (fenfluramine, dexfenfluramine, sibutramine) (19,29), but deactivates others (amphetamine, phentermine) (20). Half-life is short for most of those medications, and long for fenfluramine, dexfenfluramine and sibutramine metabolites (table 1) (29). Studies indicate that fenproporex is metabolically dealkylated leading to the production of amphetamine in animals (21,22), and its use results in amphetamine positive tests in humans (23). Even so, central nervous system stimulative effects with fenproporex are less notorious in clinical practice than with other agents such as diethylpropion and mazindol. Medical literature is hardly available regarding controlled clinical studies on that substance.
Human studies to assess food intake
Many anorectic medications have been studied to document human feeding reduction action in humans (24-26). It is interesting that d-amphetamine effects on appetite are attenuated by ondansetron, a 5-HT3 receptor antagonist, which suggests that serotoninergic pathways may be involved in the response to noradrenergic action as well (24).
Serotoninergic medications (fenfluramine and dexfenfluramine) reduce carbohydrate intake, although studies have been carried out with carbohydrate and fat-containing foods (25). Other studies have demonstrated that serotonin and fenfluramine led to protein and fat intake reduction, and that ingestion suppression under dexfenfluramine was more effective than under fenfluramine (24-26). Dexfenfluramine leads to meal size reduction and significant reduction in the habit of nibbling (39). Human studies have shown that dexfenfluramine selectively reduces fat intake (27).
Ritanserin, a 5-HT2C serotoninergic receptor antagonist, cancels dexfenfluramine-induced food intake reduction, as well as the increase or prolactin and temperature (28). m-chlorophenylpiperazine (mCPP), a 5-HT1B/2C agonist, was shown to reduce food intake in humans (29); similarly, sumatriptan, a selective 5-HT1B/D agonist, reduced food intake (especially fat intake) and raised plasma GH in a double-blind placebo-controlled study (30). Therefore, serotoninergic receptors 5-HT1B, 5-HT2C e 5-HTD are candidates for serotonine anorectic effects in humans.
Cardiovascular effects
Sympathomimetic vascular effects are predictable when using b-phenethylaminic substances, since their basic structure is the same of the monoamines NE, adrenaline and dopamine. After acute administration (except for fenfluramine and dexfenfluramine), a small stimulating effect occurs on heart rate and blood pressure (31). Treatment with sibutramine leads to a slight dose-proportional raise, 35 mmHg for diastolic blood pressure and 24 bpm for heart rate (32). Weight loss leads to blood pressure reduction in quite a number of patients (44) and long-term, clinically significant reductions may be obtained even with modest weight loss (5% reduction) (33). Mechanisms responsible for the hypotensive response to the weight loss are not fully understood, but they probably involve lower insulin level, followed by the reduction of sympathetic nervous system activity and natriuretic effect (34).
Dexfenfluramine and fenfluramine decrease blood pressure in obese patients who are normotensive (35), hypertensive (36), even in short-term studies (37). Those using blood pressure ambulatory monitoring have shown that the hypotensive effect occurred at daytime but not during the night (50). Dexfenfluramine resulted in plasma renin and noradrenaline reduction that was not dependent of weight loss (50).
Endocrine and metabolic effects
Weight reduction leads to the correction of a number of obesity-associated disturbances. Benefits occur even after modest weight loss (38), although improvement is accentuated as intentional weight loss progress (39).
Some studies have shown that fenfluramine and dexfenfluramine would have hypoglycemic action, which is not weight loss dependent (40,41). In addition, treatment with dexfenfluramine has been associated with visceral fat loss, which is correlated to insulin resistance improvement and intrahepatic fat reduction (55). Dexfenfluramine stimulates fatty acid oxidation and turnover (42). Fenfluramine, but especially dexfenfluramine, are powerful stimulanting factors of prolactin secretion; elevated prolactin levels are attenuated by naloxone (an opioid antagonist) in slim but not in obese women (43). Such elevation is lower in patients with endogenous depression, obsessive-compulsive and panic disorders; under depression therapy, the response this anti-obesity agent is better (44). Amphetamine, in particular, has no influence on prolactin secretion (56). The increase of ACTH and cortisol, detected in obese and non-obese women after naloxone administration, was attenuated by a seven-day treatment with dexfenfluramine (45). The same agent did not affect ACTH and cortisol responses to CRH (58); however, GH response to GHRH was shown to be increased in patients with android obesity, concomitantly with reduction in insulin levels (although the latter may be influenced by dietary habits) (46). Other authors have not detected increase in GH response to GHRH in obese women treated with dexfenfluramine (47).
Mazindol reduces insulin and GH responses to an oral glucose tolerance test, increases T4 but no change is observed in FSH, LH, testosterone, renin, angiotensin II and 17-ketosteroids levels and in baseline metabolic rate (48).
Weight loss due to sibutramine and energy restriction is associated to better metabolic control in type 2 diabetic obese patients (61,62).
Human studies to assess thermogenic effects
As previously mentioned, different animal studies have shown thermogenic action of various b-phenethylaminic derivates and mazindol (27-29). Their effects in human studies are not so clear, and contrasting results are frequently attributed to the heterogeneity of the obese patients studied.
While some authors have shown higher resting metabolic rate, as well as higher response to feeding after dexfenfluramine administration (49,50), or attenuation of the usual decrease in resting metabolic rate during low-calorie diet in post-menopausal women in a three-month treatment period (51), others have found no difference in 24-hour energy expenditure after 1 week, 3 months (40), or even 13 months under dexfenfluramine or placebo (52).
Human data are also conflicting in sibutramine studies. In one study, no difference was found between baseline metabolic rate and three hours after the administration of sibutramine or placebo, neither after an 8-month treatment with sibutramine (53). However, when calorie expenditure was measured for a 5-hour period, there was an increase in thermogenesis both while fasting and after feeding in the last 3,5 hours after sibutramine administration. Such effect was not observed in the first study (54).
Phenylpropanolamine is an a1adrenergic agonist widely used in the United States for many years. In Brazil, experience with this agent is more limited. Only one study assessing the possible thermogenic effect of phenylpropanolamine did not report any gain in energy expenditure (55), which is consonant with a 12-week study carried out by our group; we evaluated 103 obese women under hypocaloric diet, with 3 daily administrations of capsules containing placebo, yohimbine (8 mg), T3 (25 µg), phenylpropanolamine (25 mg), or an ephedrine (25 mg) and aminophylline (100 mg) association (56). Only patients receiving phenylpropanolamine had a weight loss significantly higher than the placebo group, even though no difference was observed in resting metabolic rate measured by indirect calorimetry (69).
Ephedrine belongs to the phenylpropanolamines group and stimulates the release of noradrenaline. Structural changes result in increased peripheral action while reducing central action on adrenergic receptors. This made ephedrine prove to be potential treatment for asthma, and actually, for many years ephedrine either isolated or in combination with teophylline has been a first choice treatment to treat this disorder. Ephedrine causes a non-selective stimulation of sympathetic nervous system by acting on b-adrenoceptors (b3 included) and promoting thermogenesis (57).
Ephedrine has been studied in obese women, at 60 mg daily dose for 12 weeks, resulting in baseline metabolic rate increase. At higher doses 150 mg daily for 30 days it resulted in weight loss (58).
Ephedrine associated with methylxantins (like caffeine, teophyilline and aminophylline) or aspirin increases the duration of noradrenalin activity. Adenosin and prostaglandins, which decrease noradrenalin activity, are inhibited by caffeine and aspirin. Phosphodiasterase inhibition through caffeine seems to be the most important effect, since that enzyme is responsible for cyclic AMP metabolization, and its inhibition maximizes noradrenalin action (59,60).
Different combinations of caffeine and ephedrine have been analyzed in double-blind studies whose conclusion was that higher synergy occured at the dose of 200 mg caffeine and 20 mg of ephedrine (3 daily administrations) (61). In a randomized, double-blind study conducted by our group, 3 daily doses of a combination of ephedrine 22 mg, caffeine 20 mg and aminophylline 50 mg, resulted in a significantly higher weight loss, as compared to the group of patients not receiving the association, although baseline energy expenditure through calorimetry was not assessed (62). In a more recent study, 17 women with BMI of 34.5 kg/m2 and body weight of 87.2 kg, treated with 3 daily doses of aminophylline, 300 mg plus ephedrine, 75 mg, reached a weight loss of 5.6 kg (69).
Clinical trials and case reports in humans
Twenty-five years ago, an analysis including over 200 double-blind, controlled studies concerning appetite reduction medications (including amphetamine, phentetrazine, benzophetamine, fendimetrazine, phentermine, chlorphentermine, chlotermine, mazindol, fenfluramine and diethylpropion) was submitted to FDA to justify registrations of new drugs. Ninety percent of them showed a higher weight loss in the group of patients receiving active medication. The withdrawal rate was nearly 24% at first month and approximately 48% at the end of 3-to-8-week long treatments. A total of 4,543 patients were evaluated receiving placebo and 3,182 receiving active ingredients (8).
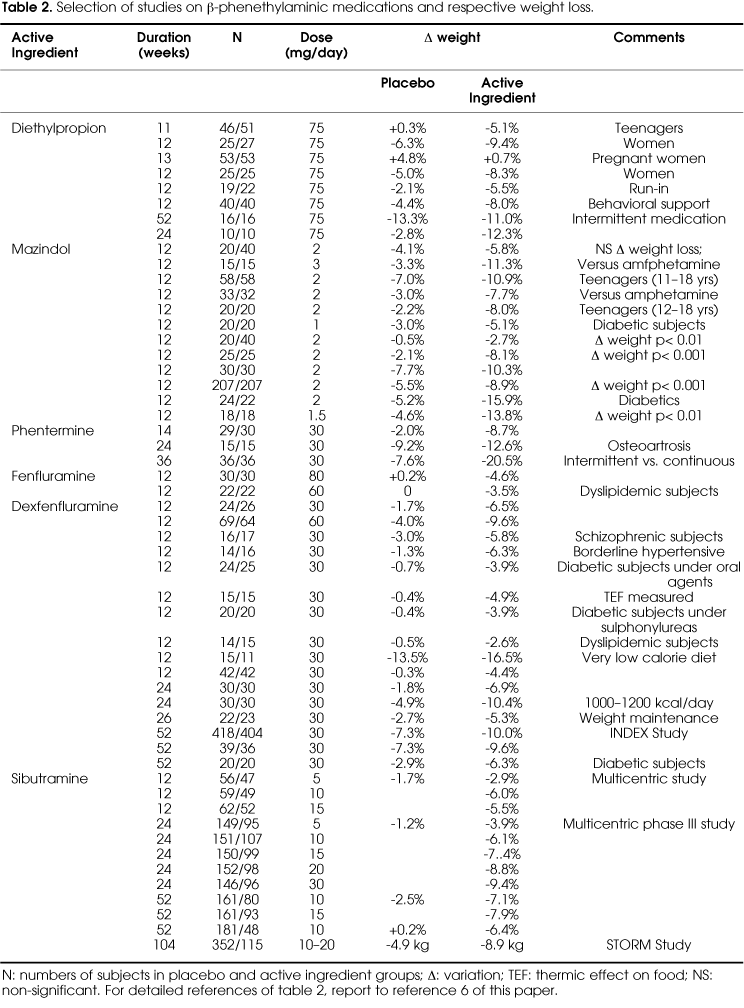
A review by Bray & Greenway (8) includes a detailed analysis of some of those studies. Table 2 shows a selection of 41 studies on phenethylaminics or tricyclics (diethylpropion, mazindol, phentermine, fenfluramine, dexfenfluramine), which lasted at least 10 weeks. Most of these studies are also described in another review (6).
Those medications fill the criteria currently used for anti-obesity medications, except for fenfluramine and dexfenfluramine. These 2 medications were withdrawn from world market in 1997 due to valvular abnormalities developed under combined therapy of phentermine plus fenfluramine (but not under phentermine monotherapy) (63,64), similar to carcinoid syndrome lesions. An echocardiographic study, including 76 obese women treated with dexfenfluramine for 6 months at the University of São Paulo Clinics Hospital Outpatient Unit, showed a prevalence of valvular injuries of 49%; from 37 women who presented echocardiographic abnormalities, 10 were re-examined 6 months after medication interruption and lesion regression was found in 5 of them (65). A larger prospective study with 1,072 participants did not detect increased risk of valvular injuries in patients using sustained-release dexfenfluramine for less than 3 months (66). A case-control study including 95 patients with pulmonary hypertension and 355 matched controls showed that fenfluramine use was associated with pulmonary hypertension (odds ratio [OR] 6.3; 95% CI 3.013.2). OR was higher in patients under fenfluramine for less than 12 months (OR 10.1; 3.429.9) or for a period longer than 3 months (OR 23.1; 6.977.7) (67).
Only one case of pulmonary hypertension, 12 months after interruption of mazindol therapy in a patient who had received for 10 weeks, was recently reported (68). Isolated cases of pulmonary hypertension (69) and psychosis (70) were associated to diethylpropion.
Although sibutramine is also a phenethylaminic derivate, it does present quite a different profile and much better tolerability. Table 3 summarizes sibutramine studies in which this agent was used for 10 weeks up to two years. References of these studies can be checked in another review published by the authors (6). The most common adverse effects were headache, dry mouth, constipation, insomnia, rhinitis and faringitis, reported by 1030% of patients under sibutramine. At 520 mg daily doses, diastolic and systolic blood pressure increases were in average 13 mmHg; heart rate increase was of 45 beats per minute (71). For the controlled hypertensive patients, the number who reported clinically significant increase in blood pressure (> 10 mmHg) in 3 successive visits was comparable to sibutramine and placebo groups. Nevertheless, hypertension was seen as its major adverse effect, resulting in discontinuation of patients in the study (72).
In Brazil, when phenylpropanolamine was used, controlled prescription was required. In contrast, for many years, in the US, this was an over-the-counter medication, more widely used than in our country. A case-control study (men and women, age range 1849 years), reported that when used as anti-obesity medication (opposedly to its use to fight influenza) phenylpropanolamine increased the risk of hemorrhagic stroke in the first 3 days of use (adjusted OR 15.9, p= 0.013) (73). Phenylpropanolamine was removed from the American and Brazilian markets in 2001.
Selective inhibitors of serotonin reuptake
Both fluoxetine and sertraline are selective inhibitors of serotonin reuptake despite diverse chemical structure. Fluoxetine is a phenylpropanolamin oxy-3-fluorphenyl derivate and sertraline is a naftilaminic one. Fluoxetine and sertraline inhibit serotonin reuptake at the pre-synaptic terminal; their main indication is to treat depression and bulimia, and they are not formally indicated to treat obesity. Both agents were found to reduce animal feeding experimentally (74). In humans, weight loss was a common finding during protocols for the approval of those medications as anti-depressants. Under sertraline which seems to be different from the drug action at muscarinic receptors weight loss was 0.450.91 kg in follow-ups of 8 to 16 weeks.
Human studies to assess food intake
Clinical trials to assess feeding reported the effect of those medications on patients' food intake size (75).
Endocrine and metabolic effects
A study including diabetic subjects reported that fluoxetine treatment was associated with a higher weight loss and their insulin requirements were reduced (76).
Human clinical trials
The key problem involving fluoxetine as anti-obesity agent is weight regain, as detected in long-term studies. In general, after the first 6 months of treatment, body weight is gradually recovered, although medication is maintained.
A study to assess weight loss under sertraline showed no difference when compared to the placebo groups (77). In another, sertraline increased the weight loss of patients under cognitive-behavioral treatment (78). Double-blind fluoxetine studies with at least 10-week duration are shown in table 3. References are detailed in another review published by the authors (6). Fluoxetine therapy for obesity management has been associated with gastrointestinal symptoms, sleep disorders, reduced libido, sweating, amnesia and thirst (79).
Selective inhibitors of serotonin reuptake are not, therefore, efficacious anti-obesity agents, although they may be useful for depressed obese patients, and for patients reporting other comorbidities for which those anti-depressants may be an appropriate treatment - for instance, sleep apnea - since fluoxetine leads to REM reduction, when most episodes of obstructive apnea occur.
Nutrients metabolism post-absorptive modifiers
Lipstatine analogues
Lipstatine is a compound from yeast - Streptomyces toxytricini. Orlistat is a stable lipstatine analogue, and partially hydrolized (tetra-hydrolipstatine).
Clinical pharmacokinetics
Orlistat is a powerful inhibitor of gastrointestinal (GI) lipases. Such enzymes catalize hydrolytic removal of triglycerides fatty acids and produce free fatty acids and monoglycerides. Orlistat binds irreversibly to lipase active sites through covalent binding. Approximately one-third of triglyceride intake does not undergo digestion, and is not absorbed by small intestines, crossing the GI tract and being eliminated. Orlistat has no systemic activity, and absorption by GI is minimal when administered up to 800 mg daily, with lipase inhibiting activity pharmacologically irrelevant (from 1,000 to 2,500 times lower than orlistat) (80).
Human studies to assess human intake
Orlistat has no direct effect on appetite regulating neuronal circuits. However, its pharmacological effect (reflected by the increased amount of fat in feces) stimulates long-term compliance to lower fat content food intake (81).
Cardiovascular effects
Weight loss resulting from orlistat is associated with significant reduction of systolic and diastolic blood pressure as compared to placebo (-4.9 vs -2.4 mmHg and -3.7 vs -1.8 mmHg, p< 0.05) (82). A meta-analysis of 5 studies demonstrated that patients reporting isolated systolic hypertension (systolic blood pressure > 140 mmHg) show higher reductions (-10.9 vs -5.1 mmHg, p< 0.05) (83).
Endocrine and metabolic effects
As previously mentioned, weight loss leads to the reversion of some obesity-associated disorders. This occurs even with modest weight loss but benefits are improved with intentional weight loss of greater magnitude.
In non-diabetic obese patients, the use of orlistat combined with calorie-fat restriction is associated with significant reductions in insulin (-5.05% vs +19.1%, vs. placebo, p= 0.001) and plasma glucose levels (-0.92% vs +2.33%, p< 0.05) (84). A one-year study, including controlled diabetic subjects under sulfonylureas, resulted in significant reduction of plasma glucose and glycated hemoglobin levels, as well as in the number of patients discontinuing oral anti-diabetic treatment (85). Those data have been confirmed by a Latin American multicenter 6-month-long trial (86). In our study, the use of orlistat was associated with higher weight loss and marked improvement in fasting (p= 0.036) and post-prandial glycemia (p= 0.05), and in glycated hemoglobin (p= 0.04). In addition, benefits in lipid profile - as seen by total cholesterol (p= 0.0001) and LDL-cholesterol level (p= 0.002) reductions - and in abdominal adiposity (p< 0.05) were observed (88).
Human clinical trials
The first orlistat clinical trials were 12-week long and multi-dosage, at 10 mg, up to 120 mg, 3 times a day (87,88). Another study, six-month long, was carried out at 30, 60, 120, and 240 mg of orlistat, 3 times a day (89). Significant difference in weight loss was reported from 60 mg doses (total daily dose= 180 mg), reaching a plateau at 120 mg (dose total daily dose= 360 mg). Higher doses did not increase the weight loss. Table 4 presents a selection of clinical trials concerning orlistat, also including diabetic subjects. The references of these studies can be checked in another review published by the authors (6). Trials under analysis reported no differences in the frequency of GI adverse effects comparing orlistat and placebo groups. GI effects are related to orlistat mechanism of action (oily stools, increased number of evacuation episodes, flatulence with or without fat discharge, fecal urgency); they are usually short-term and tend to decrease considerably after the first weeks of treatment. Such pattern seems to be related to the long-term patient compliance to low-fat foods.
TREATMENT PERSPECTIVES WITH PHARMACOLOGICAL AGENTS
Associations of two pharmacological agents
Although no randomized trial on the association of sibutramine and orlistat is available, in clinical practice this combination has been used for the management of obese patients, once mechanisms of action are distinctive. The authors evaluated the efficacy and tolerability of sibutramine combined to orlistat at regular doses up to 6 months in 214 patients (121 women and 93 men) (90). A reduction in body weight from baseline of 8% (1.5% to -24%) and -14.9% (-0.4 to -26.6%) was observerd after 3 months (n= 100) and 6 months (n= 36). In this study, the combination of sibutramine and orlistat for obesity management resulted in higher weight reduction when compared to randomized clinical trials, and tolerability was quite reasonable.
The use of pharmacological agents in childhood obesity management
Current clinical approach towards pediatric obesity mainly involves cognitive-behavioral therapies focusing eating and exercising pattern changes.
Foccus on the pathophysiology of obesity may lead to the development of the appropriate medications both for adults and children, possibly from substances regulating metabolic economy physiology. Orlistat already proved to be effective and its use is approved for teenagers. Development of trials in children and teenagers is critical, since one cannot assume that risks and benefits from the use of pharmacological agents in adults will be the same in children (90).
Use of anti-obesity agents in obese type 2 diabetic patients
Weight reduction has been shown to improve glycemic control and cardiovascular risk associated with insulin resistance in obese individuals with type 2 diabetes mellitus. Therapeutic options for these patients include promotion of weight loss (non-pharmacologic and pharmacologic treatments), which improves glycemic control, as well as treatment of commonly associated risk factors, such as hypertension and dyslipidemia. A recent review provides an overview of anti-obesity drugs used in the treatment of obese individuals with type 2 diabetes. The most widely investigated drugs, sibutramine and orlistat, resulted in modest, clinically worthwhile weight loss, but with marked improvement in several comorbidities, among them, type 2 diabetes. Studies involving these anti-obesity medications in cohorts of obese diabetic patients have been reviewed, as well as involving cathecolaminergic (diethylpropion [amfepramone], fenproporex, mazindol, ephedrine-caffeine combination), serotoninergic agents (fenfluramine, dexfenfluramine, fluoxetine), and others showing any benefit on weight loss (metformin, the anti-epileptic agent topiramate and zonisamide, and the antidepressive bupropion [amfebutamone]). These trials showed variable benefits in terms of effects on glucose metabolism (91). Orlistat was reported to prevent the development of type 2 diabetes in obese patients treated for 4 years (XenDOS study) (92).
Antagonists of endocannabinoid receptors
The ability of marijuana to increase hunger has been noticed for centuries, although research on its action started in the late 1960s. An endogenous neuromodulating system involved in feeding behaviour leads to the therapeutic use of a novel class of drugs, the selective cannabinoid type 1 receptor (CB1R) antagonists, for the treatment of obesity and eating disorders. The experience with the first agent from this class rimonabant was recently published. A 1-year blinded randomised clinical trial with doses of 5 mg or 20 mg of rimonabant was found to cause a pronounced reduction in bodyweight (respectively -3.4 kg and -6.6 kg), along with a decrease in waist circumference and a substantial amelioration of the metabolic profile and insulin resistance. Generally, it was well tolerated with mild and transient side effects (mainly nausea) (93).
REFERENCES
1. WHO Consultation on Obesity. Preventing and managing the global epidemic. Geneva: World Health Organization, 1998.
2. Guy-Grand B. Long-term pharmacoterapy in the management of obesity. In: Björntorp P, Rössner S, editors. From theory to practice: Obesity in Europe - 88. London: John Libbey, 1989. p.311-8.
3. Gortmaker SL, Must A, Perrin JM, Sobol AM, Dietz WH. Social and economic consequences of overweight in adolescence and young adulthood. N Engl J Med 1993;329:1008-12.
4. Bray GA. Obesity a time bomb to be refused. Lancet 1998;352:160-1.
5. Prentice AM, Jebb AS. Obesity in Britain: gluttony or sloth? Br Med J 1995;311:437-9.
6. Halpern A, Mancini MC. Treatment of obesity: an update on anti-obesity medications. Obes Rev 2003; 4:25-42.
7. Williamson DF. Dietary intake and physical activity as "predictors" of weight gain in observational, prospective studies. Nutr Rev 1996;54(suppl):S101-9.
8. Samanin R, Garattini S. Neurochemical mechanism of action of anorectic drugs. Pharmacol Toxicol 1993;73:63-8.
9. Garattini S. Biological actions of drugs affecting serotonin and eating. Obes Res 1995;3:463-70.
10. Heal DJ, Frankland AT, Gosden J, Hutchins LJ, Prow MR, Luscombe GP, et al. A comparison of the effects of sibutramine hydrochloride, bupropion and methamphetamine on dopaminergic function: evidence that dopamine is not a pharmacological target for sibutramine. Psychopharmacology 1992;107:303-9.
11. Paul SM, Hulihan-Giblin B, Skolnick P. (+)-Amphetamine binding to rat hypothalamus: relation to anorexic potency of phenethylamines. Science 1982;218:487-90.
12. Angel I, Hauger RL, Luu MD, Giblin B, Skolnick P, Paul SM. Glucostatic regulation of (+)-[3H]amphetamine binding in the hypothalamus: correlation with Na+/K+-ATPase activity. Proc Natl Acad Sci USA 1985;82:6320-4.
13. Harris SC, Ivy AC, Searle LM. The mechanism of amphetamine-induced loss of weight. JAMA 1947;134:1468-75.
14. Petrie JC, Bewsher PD, Mowat JA, Stowers JM. Metabolic effects of fenfluramine a double-blind study. Postgrad Med J 1975;51:139-44.
15. Blundell JE, Hill AJ. Serotoninergic drug potentates the satiating capacity of food action of [scap]d-fenfluramine in obese subjects. Ann NY Acad Sci 1989;575:493-5.
16. Smith BK, York DA, Bray GA. Activation of hypothalamic serotonin receptors reduced intake of dietary fat and protein but not carbohydrate. Am J Physiol 1999;277:R802-11.
17. Foltin RW, Kelly TH, Fischman MW. Effect of amphetamine on human macronutrient intake. Physiol Behav 1995;58:899-907.
18. Stock MJ. Sibutramine: a review of the pharmacology of a novel anti-obesity agent. Int J Obes Relat Metab Disord 1997;21(suppl):S25-9.
19. Cheymol G, Weissenburger J, Poirier JM, Gellee C. The pharmacokinetics of dexfenfluramine in obese and non-obese subjects. Br J Clin Pharmacol 1995;39:684-7.
20. Hinsvark ON, Truant AP, Jenden DJ, Steinborn JA. The oral bioavailability and pharmacokinetics of soluble and resin-bound forms of amphetamine and phentermine in man. J Pharmacokinet Biopharm 1973;1:319-28.
21. Coutts RT, Nazarali AJ, Baker GB, Pasutto FM. Metabolism and disposition of N-(2-cyanoethyl) amphetamine (fenproporex) and amphetamine: study in the rat brain. Can J Physiol Pharmacol 1986;64:724-8.
22. Mattei R, Carlini EA. A comparative study of the anorectic and behavioral effects of fenproporex on male and female rats. Braz J Med Biol Res 1996;29:1025-30.
23. Musshoff E. Illegal or legitimate use? Precursor compounds to amphetamine and methamphetamine. Drug Metab Rev 2000;32:15-44.
24. Silverstone PH, Oldman D, Johnson B, Cowen PJ. Ondansetron, a 5-HT3 receptor antagonist, partially attenuates the effects of amphetamine: a pilot study in healthy volunteers. Int Clin Psychopharmacol 1992;7:37-43.
25. Wurtman JJ. Carbohydrate craving. Relationship between carbohydrate intake and disorders of mood. Drugs 1990;39:49-52.
26. Goodall EM, Feeney S, McGuirk J, Silverstone T. A comparison of the effects of d- and l-fenfluramine and d-amphetamine on energy and macronutrient intake in human subjects. Psychopharmacology 1992;106:221-7.
27. Lafreniere F, Lambert J, Rasio E, Serri O. Effects of dexfenfluramine treatment on body weight and postprandial thermogenesis in obese subjects. A double-blind placebo-controlled study. Int J Obes Relat Metab Disord 1993;17:25-30.
28. Goodall EM, Cowen PJ, Franklin M, Silverstone T. Ritanserin attenuates anorectic, endocrine and thermic responses to d-fenfluramine in human volunteers. Psychopharmacology (Berl) 1993;112:461-6.
29. Cowen PJ, Sargent PA, Williams C, Goodall EM, Orlikov AB. Hypophagic, endocrine and subjective responses to m-chlorophenylpiperazine in healthy men and women. Hum Psychopharmacol 1995;10:385-91.
30. Boeles S, Williams C, Campling GM, Goodall EM, Cowen PJ. Sumatriptan decreases food intake and increases plasma growth hormone in healthy women. Psychopharmacology (Berl) 1997;129:179-82.
31. Tuck ML, Sowers J, Dornfeld L, Kledzik G, Maxwell M. The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients. N Engl J Med 1981;304:930-3.
32. Bray GA, Blackburn GL, Ferguson JM, Greenway FL, Jain AK, Mendel CM, et al. Sibutramine produces dose-related weight loss. Obes Res 1999;7:189-98.
33. Stevens VJ, Obarzanek E, Cook NR, Lee I-M, et al; the Trials of Hypertension Prevention Research Group. Long-term weight loss and changes in blood pressure: results of the trials of hypertension prevention, phase II. Ann Intern Med 2001;134:1-11.
34. Mikhail N, Golub MS, Tuck ML. Obesity and hypertension. Prog Cardiovasc Dis 1999;42:39-58.
35. Noble RE. A six-month study of the effects of dexfenfluramine on partially successful dieters. Curr Ther Res 1990;47:612-9.
36. Kolanowski J, Younis LT, Vanbutsele R, Detry JM. Effect of dexfenfluramine treatment on body weight, blood pressure and noradrenergic activity in obese hypertensive patients. Eur J Clin Pharmacol 1992;42:599-606.
37. Fletchner Mors M, Ditschuneit HH, Yip I, Adler G. Blood pressure and plasma norepinephrine responses to dexfenfluramine in obese postmenopausal women. Am J Clin Nutr 1998;67:611-5.
38. Goldstein DJ. Beneficial health effects of modest weight-loss. Int J Obes Relat Metab Disord 1992;16:397-415.
39. Sjöström CD, Lissner L, Wedel H, Sjöström L. Reduction in incidence of diabetes, hypertension and lipid disturbances after intentional weight loss induced by bariatric surgery: the SOS intervention study. Obes Res 1999;7:477-84.
40. Turtle JR, Burgess JA. Hypoglycemic action of fenfluramine in diabetes mellitus. Diabetes 1973;22:858-67.
41. Andersen PH, Richelsen B, Bak J, Schmitz O, Sorensen NS, Lavielle R, et al. Influence of short-term dexfenfluramine therapy on glucose and lipid metabolism in obese non-diabetic patients. Acta Endocrinol (Copenh) 1993; 128:251-8.
42. Marks SJ, Moore NR, Clarck ML, Strauss BJ, Hockaday TD. Reduction of visceral adipose tissue and improvement of metabolic indices: effect of dexfenfluramine in NIDDM. Int J Obes Relat Metab Disord 1997;21:274-9.
43. Feeney S, Goodall E, Silverstone T. The prolactin response to d- and l-fenfluramine and to d-amphetamine in human subjects. Int Clin Psychopharmacol 1993;8:49-54.
44. Shapira B, Cohen J, Newman ME, Lerer B. Prolactin response to fenfluramine and placebo challenge following maintenance pharmacoterapy withdrawal in remitted depressed patients. Biol Psychiatry 1993;33:531-5.
45. Boushaki FZ, Rasio E, Serri O. Hypothalamic-pituitary-adrenal axis in abdominal obesity: effects of dexfenfluramine. Clin Endocrinol (Oxf) 1997;46:461-6.
46. Medeiros-Neto G, Lima N, Perozim L, Pedrinola F, Wajchenberg BL. The effect of hypocaloric diet with and without d-fenfluramine treatment on growth hormone release after growth hormone-releasing factor stimulation in patients with android obesity. Metabolism 1994;43:969-73.
47. Kars ME, Pijl H, Cohen AF, Frolich M, Schoemaker HC, Brandenburg HC, et al. Specific stimulation of brain serotonin mediated neurotransmission by dexfenfluramine does not restore growth hormone responsiveness in obese women. Clin Endocrinol (Oxf) 1996;44:541-6.
48. Jonderko K, Kucio C. Extra-anorectic actions of mazindol. Isr J Med Sci 1989;25:20-4.
49. Finer N, Bloom SR, Frost GS, Banks LM, Griffiths J. Sibutramine is effective for weight loss and diabetic control in obesity with type 2 diabetes: a randomised, double-blind, placebo-controlled study. Diabetes Obes Metab 2000;2:105-12.
50. Fujioka K, Seaton TB, Rowe E, Jelinek CA, Raskin P, Lebovitz HE, et al; Sibutramine/Diabetes Clinical Study Group. Weight loss with sibutramine improves glycaemic control and other metabolic parameters in obese patients with type 2 diabetes mellitus. Diabetes Obes Metab 2000;2:175-87.
51. Levitsky DA, Schuster JA, Stallone D, Strupp BJ. Modulation of the thermic effect of food by fenfluramine. Int J Obes 1986;10:169-73.
52. Scalfi L, D'Arrigo E, Carandente V, Coltorti A, Contaldo F. The acute effect of fexfenfluramine on resting metabolic rate and postprandial thermogenesis in obese subjects: a double-blind placebo-controlled study. Int J Obes Relat Metab Disord 1993;17:91-6.
53. Van Gaal LF, Vansant GA, Steijaert MC, De Leeuw IH. Effects of dexfenfluramine on resting metabolic rate and thermogenesis in premenopausal obese women during therapeutic weight reduction. Metabolism 1995;44:42-5.
54. Breum L, Astrup A, Andersen T, Lambert O, Nielsen E, Garby L, et al. The effect of long-term dexfenfluramine treatment on 24-hour energy-expenditure in man a double-blind placebo controlled study. Int J Obes Relat Metab Disord 1990;14:613-21.
55. Seagle HM, Bessesen DH, Hill JO. Effects of sibutramine on resting metabolic rate and weight loss in overweight women. Obes Res 1998;6:115-21.
56. Hansen DL, Toubro S, Stock MJ, MacDonald IA, Astrup A. Thermogenic effects of sibutramine in humans. Am J Clin Nutr 1998;68:1180-6.
57. Alger S, Larson K, Boyce VL, Seagle H, Fontvieille AM, Ferraro RT, et al. Effect of phenylpropanolamine on energy expenditure and weight loss in overweight women. Am J Clin Nutr 1993;57:120-6.
58. Rascovski A, Millner TH, Batalha L, Reis C, Mancini MC, Halpern A. Eficácia e tolerabilidade das substâncias calorigênicas: ioimbina, triiodotironina, aminofilina combinada a efedrina e fenilpropanolamina no tratamento da obesidade a curto prazo. Arq Bras Endocrinol Metab 2000;44:95-102.
59. Liu YL, Toubro S, Astrup A, Stock MJ. Contribution of beta 3-adrenoceptor activation to ephedrine-induced thermogenesis in humans. Int J Obes Relat Metab Disord 1995;19:678-85.
60. Pasquali R, Cesari MP, Melchionda N, Stefanini C, Raitano A, Labo G. Does ephedrine promote weight loss in low-energy adapted obese women? Int J Obes 1987;11:163-8.
61. Halpern A, Mancini MC. Tratamento farmacológico da obesidade Drogas termogênicas. Arq Bras Endocrinol Metab 1996;40:224-7.
62. Dulloo AG, Seydoux J, Girardier L. Potentiation of the thermogenic antiobesity effects of ephedrine by dietary methylxantines: adenosine antagonism or phosphodiesterase inhibition. Metabolism 1992;41:1233-41.
63. Astrup A, Toubro S, Cannon S, Hein P, Madsen J. Thermogenic synergism between ephedrine and caffeine in healthy volunteers: a double-blind, placebo-controlled study. Metabolism 1991;40:323-9.
64. Mancini MC, Marsiaj HI, Hakoyama MM, Quantal IA, Correa NC, Halpern A. Ephedrine, caffeine and aminophilline preparation: na alternative in the treatment of obesity. Int J Obes 1990;14(suppl 2):141.
65. Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997;337:581-8.
66. Graham DJ, Green L. Further cases of valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997;337:635.
67. Leite CC, Mancini MC, Medeiros CCJ, Sbano JCN, Grinberg M, Halpern A. Echocardiographic evaluation of 70 patients using dexfenfluramine (abstracted). Int J Obes Relat Metab Disord 1998;22(suppl 3):S227.
68. Weissman NJ, Tighe JF, Gottdiener JS, Gwynne JT. An assessment of heart-valve abnormalities in obese patients taking dexfenfluramine, sustained-release dexfenfluramine, or placebo. N Engl J Med 1998; 339: 725-32.
69. Abenhaim L, Moride Y, Brenot F, et al. Appetite-supressant drugs and the risk of primary pulmonary hypertension. N Engl J Med 1996;335:609-16.
70. Hagiwara M, Tsuchida A, Hyakkoku M, et al. Delayed onset of pulmonary hypertension associated with an appetite suppressant, mazindol: a case report. Jpn Circ 2000;64:218-21.
71. Thomas SH, Butt AY, Corris PA, et al. Appetite supressants and primary pulmonary hypertension in the United Kingdom. Br Heart J 1995;74:660-3.
72. Little JD, Romans SE. Psychosis following readministration of diethylpropion: a possible role for kinding? Int Clin Psychopharmacol 1993;8:67-70.
73. Luque CA, Ray JA. Sibutramine: a serotonin-norepinephrine reuptake-inhibitor for the treatment of obesity. Ann Pharmacother 1999;33:968-78.
74. McMahon FG, Fujioka K, Singh BN, Mendel CM, et al. Efficacy and safety of sibutramine in obese white and African American patients with hypertension: a 1-year, double-blind, placebo-controlled multicenter trial. Arch Int Med 2000;160:2185-91.
75. Horwitz RI, Brass LM, Kernan WN, Viscoli CM. Phenylpropanolamine and risk of hemorrhagic stroke: final report of the hemorrhagic stroke project. http://fda.gov/ ohrms/dockets/ac/00/backgrd/3647b1_tab19.doc (accessed on March 2, 2001).
76. Neilsen JA, Chapin DS, Johnson Jr JL, Torgersen LK. Sertraline, a serotonin-uptake inhibitor, reduces food intake and body weight in lean rats and genetically obese mice. Am J Clin Nutr 1992;55(suppl):185S-9S.
77. McGuirk J, Silverstone T. The effect of the 5-HT re-uptake inhibitor fluoxetine on food intake and body weight in healthy male subjects. Int J Obes Relat Metab Disord 1990;14:361-72.
78. Gray DS, Fujioka K, Devine W, Bray GA. A randomized double-blind clinical trial of fluoxetine in obese diabetics. Int J Obes Relat Metab Disord 1992;16(suppl 4):S67-S72.
79. Wadden TA, Bartlett SJ, Foster GD, Greenstein RA, Wingate BJ, Stunkard AJ, et al. Sertraline and relapse prevention training following treatment by very-low-calorie diet: a controlled clinical trial. Obes Res 1995;3:549-57.
80. Ricca V, Mannucci E, Di Bernardo M, Rizzello SM, Cabras PL, Rotella CM. Sertraline enhances the effects of cognitive-behavioral treatment on weight reduction of obese patients. J Endocrinol Invest 1996;19:727-33.
81. Arterburn D, Noël PH. Extracts from "clinical evidence" Obesity. Br Med J 2001;322:1406-9.
82. Zhi J, Melia AT, Eggers H, et al. Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J Clin Pharmacol 1995;35:1103-8.
83. Aronne LJ. Modern medical management of obesity: the role for pharmacological intervention. J Am Diet Assoc 1998;98(suppl 2):S23-6.
84. Davidson MH, Hauptman J, DiGirolamo M, et al. Weight control and risk factor reduction in obese subjects treated for 2 years with orlistat. JAMA 1999;281:235-42.
85. Zavoral JH. Treatment with orlistat reduces cardiovascular risk in obese patients. J Hypertens 1998;16:2013-7.
86. Sjöstrom L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. Lancet 1998;352:167-72.
87. Hollander PA, Elbein SC, Hirsch IB, et al. Role of orlistat in the treatment of obese patients with type 2 diabetes. Diabetes Care 1998;21:1288-94.
88. Halpern A, Mancini MC, Suplicy H, Zanella MT, Repetto G, Gross J, et al. Latin-american trial of orlistat for weight loss and improvement in glycemic profile in obese diabetic patients. Diabetes Obes Metab 2003;5:180-8.
89. Drent ML, van der Veen EA. Lipase inhibition: a novel concept in the treatment of obesity. Int J Obes 1993;17:241-4.
90. Drent ML, Larsson I, William-Olsson T, Quaade F, et al. Orlistat, a lipase inhibitor, in the treatment of human obesity: a multiple dose study. Int J Obes 1995;19:221-6.
91. Van Gaal LF, Bloom JI, Enzi G, et al. Efficacy and tolerability of orlistat in the treatment of obesity: a 6-month-dose-ranging study. Eur J Pharmacol 1998;54:125-32.
92. Daniels S. Pharmacological treatment of obesity in paediatric patients. Paediatr Drugs 2001;3:405-10.
93. Halpern A, Mancini MC. Diabesity: are weight loss medications effective? Treat Endocrinol 2005;4:65-74.
94. Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004;27:155-61. Erratum in: Diabetes Care 2004;27:856.
95. van Gaal L, Rissanen AM, Scheen AJ, Ziegler O, Rössner S; the RIO-Europe Study Group. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet 2005;365:1389-97.
Recebido em 30/10/05
Aceito em 17/01/06
- 1. WHO Consultation on Obesity. Preventing and managing the global epidemic Geneva: World Health Organization, 1998
- 2. Guy-Grand B. Long-term pharmacoterapy in the management of obesity. In: Björntorp P, Rössner S, editors. From theory to practice: Obesity in Europe - 88 London: John Libbey, 1989 p.311-8.
- 3. Gortmaker SL, Must A, Perrin JM, Sobol AM, Dietz WH. Social and economic consequences of overweight in adolescence and young adulthood. N Engl J Med 1993;329:1008-12.
- 4. Bray GA. Obesity a time bomb to be refused. Lancet 1998;352:160-1.
- 5. Prentice AM, Jebb AS. Obesity in Britain: gluttony or sloth? Br Med J 1995;311:437-9.
- 6. Halpern A, Mancini MC. Treatment of obesity: an update on anti-obesity medications. Obes Rev 2003; 4:25-42.
- 7. Williamson DF. Dietary intake and physical activity as "predictors" of weight gain in observational, prospective studies. Nutr Rev 1996;54(suppl):S101-9.
- 8. Samanin R, Garattini S. Neurochemical mechanism of action of anorectic drugs. Pharmacol Toxicol 1993;73:63-8.
- 9. Garattini S. Biological actions of drugs affecting serotonin and eating. Obes Res 1995;3:463-70.
- 10. Heal DJ, Frankland AT, Gosden J, Hutchins LJ, Prow MR, Luscombe GP, et al. A comparison of the effects of sibutramine hydrochloride, bupropion and methamphetamine on dopaminergic function: evidence that dopamine is not a pharmacological target for sibutramine. Psychopharmacology 1992;107:303-9.
- 11. Paul SM, Hulihan-Giblin B, Skolnick P. (+)-Amphetamine binding to rat hypothalamus: relation to anorexic potency of phenethylamines. Science 1982;218:487-90.
- 12. Angel I, Hauger RL, Luu MD, Giblin B, Skolnick P, Paul SM. Glucostatic regulation of (+)-[3H]amphetamine binding in the hypothalamus: correlation with Na+/K+-ATPase activity. Proc Natl Acad Sci USA 1985;82:6320-4.
- 13. Harris SC, Ivy AC, Searle LM. The mechanism of amphetamine-induced loss of weight. JAMA 1947;134:1468-75.
- 14. Petrie JC, Bewsher PD, Mowat JA, Stowers JM. Metabolic effects of fenfluramine a double-blind study. Postgrad Med J 1975;51:139-44.
- 15. Blundell JE, Hill AJ. Serotoninergic drug potentates the satiating capacity of food action of [scap]d-fenfluramine in obese subjects. Ann NY Acad Sci 1989;575:493-5.
- 16. Smith BK, York DA, Bray GA. Activation of hypothalamic serotonin receptors reduced intake of dietary fat and protein but not carbohydrate. Am J Physiol 1999;277:R802-11.
- 17. Foltin RW, Kelly TH, Fischman MW. Effect of amphetamine on human macronutrient intake. Physiol Behav 1995;58:899-907.
- 18. Stock MJ. Sibutramine: a review of the pharmacology of a novel anti-obesity agent. Int J Obes Relat Metab Disord 1997;21(suppl):S25-9.
- 19. Cheymol G, Weissenburger J, Poirier JM, Gellee C. The pharmacokinetics of dexfenfluramine in obese and non-obese subjects. Br J Clin Pharmacol 1995;39:684-7.
- 20. Hinsvark ON, Truant AP, Jenden DJ, Steinborn JA. The oral bioavailability and pharmacokinetics of soluble and resin-bound forms of amphetamine and phentermine in man. J Pharmacokinet Biopharm 1973;1:319-28.
- 21. Coutts RT, Nazarali AJ, Baker GB, Pasutto FM. Metabolism and disposition of N-(2-cyanoethyl) amphetamine (fenproporex) and amphetamine: study in the rat brain. Can J Physiol Pharmacol 1986;64:724-8.
- 22. Mattei R, Carlini EA. A comparative study of the anorectic and behavioral effects of fenproporex on male and female rats. Braz J Med Biol Res 1996;29:1025-30.
- 23. Musshoff E. Illegal or legitimate use? Precursor compounds to amphetamine and methamphetamine. Drug Metab Rev 2000;32:15-44.
- 24. Silverstone PH, Oldman D, Johnson B, Cowen PJ. Ondansetron, a 5-HT3 receptor antagonist, partially attenuates the effects of amphetamine: a pilot study in healthy volunteers. Int Clin Psychopharmacol 1992;7:37-43.
- 25. Wurtman JJ. Carbohydrate craving. Relationship between carbohydrate intake and disorders of mood. Drugs 1990;39:49-52.
- 26. Goodall EM, Feeney S, McGuirk J, Silverstone T. A comparison of the effects of d- and l-fenfluramine and d-amphetamine on energy and macronutrient intake in human subjects. Psychopharmacology 1992;106:221-7.
- 27. Lafreniere F, Lambert J, Rasio E, Serri O. Effects of dexfenfluramine treatment on body weight and postprandial thermogenesis in obese subjects. A double-blind placebo-controlled study. Int J Obes Relat Metab Disord 1993;17:25-30.
- 28. Goodall EM, Cowen PJ, Franklin M, Silverstone T. Ritanserin attenuates anorectic, endocrine and thermic responses to d-fenfluramine in human volunteers. Psychopharmacology (Berl) 1993;112:461-6.
- 29. Cowen PJ, Sargent PA, Williams C, Goodall EM, Orlikov AB. Hypophagic, endocrine and subjective responses to m-chlorophenylpiperazine in healthy men and women. Hum Psychopharmacol 1995;10:385-91.
- 30. Boeles S, Williams C, Campling GM, Goodall EM, Cowen PJ. Sumatriptan decreases food intake and increases plasma growth hormone in healthy women. Psychopharmacology (Berl) 1997;129:179-82.
- 31. Tuck ML, Sowers J, Dornfeld L, Kledzik G, Maxwell M. The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients. N Engl J Med 1981;304:930-3.
- 32. Bray GA, Blackburn GL, Ferguson JM, Greenway FL, Jain AK, Mendel CM, et al. Sibutramine produces dose-related weight loss. Obes Res 1999;7:189-98.
- 33. Stevens VJ, Obarzanek E, Cook NR, Lee I-M, et al; the Trials of Hypertension Prevention Research Group. Long-term weight loss and changes in blood pressure: results of the trials of hypertension prevention, phase II. Ann Intern Med 2001;134:1-11.
- 34. Mikhail N, Golub MS, Tuck ML. Obesity and hypertension. Prog Cardiovasc Dis 1999;42:39-58.
- 35. Noble RE. A six-month study of the effects of dexfenfluramine on partially successful dieters. Curr Ther Res 1990;47:612-9.
- 36. Kolanowski J, Younis LT, Vanbutsele R, Detry JM. Effect of dexfenfluramine treatment on body weight, blood pressure and noradrenergic activity in obese hypertensive patients. Eur J Clin Pharmacol 1992;42:599-606.
- 37. Fletchner Mors M, Ditschuneit HH, Yip I, Adler G. Blood pressure and plasma norepinephrine responses to dexfenfluramine in obese postmenopausal women. Am J Clin Nutr 1998;67:611-5.
- 38. Goldstein DJ. Beneficial health effects of modest weight-loss. Int J Obes Relat Metab Disord 1992;16:397-415.
- 39. Sjöström CD, Lissner L, Wedel H, Sjöström L. Reduction in incidence of diabetes, hypertension and lipid disturbances after intentional weight loss induced by bariatric surgery: the SOS intervention study. Obes Res 1999;7:477-84.
- 40. Turtle JR, Burgess JA. Hypoglycemic action of fenfluramine in diabetes mellitus. Diabetes 1973;22:858-67.
- 41. Andersen PH, Richelsen B, Bak J, Schmitz O, Sorensen NS, Lavielle R, et al. Influence of short-term dexfenfluramine therapy on glucose and lipid metabolism in obese non-diabetic patients. Acta Endocrinol (Copenh) 1993; 128:251-8.
- 42. Marks SJ, Moore NR, Clarck ML, Strauss BJ, Hockaday TD. Reduction of visceral adipose tissue and improvement of metabolic indices: effect of dexfenfluramine in NIDDM. Int J Obes Relat Metab Disord 1997;21:274-9.
- 43. Feeney S, Goodall E, Silverstone T. The prolactin response to d- and l-fenfluramine and to d-amphetamine in human subjects. Int Clin Psychopharmacol 1993;8:49-54.
- 44. Shapira B, Cohen J, Newman ME, Lerer B. Prolactin response to fenfluramine and placebo challenge following maintenance pharmacoterapy withdrawal in remitted depressed patients. Biol Psychiatry 1993;33:531-5.
- 45. Boushaki FZ, Rasio E, Serri O. Hypothalamic-pituitary-adrenal axis in abdominal obesity: effects of dexfenfluramine. Clin Endocrinol (Oxf) 1997;46:461-6.
- 46. Medeiros-Neto G, Lima N, Perozim L, Pedrinola F, Wajchenberg BL. The effect of hypocaloric diet with and without d-fenfluramine treatment on growth hormone release after growth hormone-releasing factor stimulation in patients with android obesity. Metabolism 1994;43:969-73.
- 47. Kars ME, Pijl H, Cohen AF, Frolich M, Schoemaker HC, Brandenburg HC, et al. Specific stimulation of brain serotonin mediated neurotransmission by dexfenfluramine does not restore growth hormone responsiveness in obese women. Clin Endocrinol (Oxf) 1996;44:541-6.
- 48. Jonderko K, Kucio C. Extra-anorectic actions of mazindol. Isr J Med Sci 1989;25:20-4.
- 49. Finer N, Bloom SR, Frost GS, Banks LM, Griffiths J. Sibutramine is effective for weight loss and diabetic control in obesity with type 2 diabetes: a randomised, double-blind, placebo-controlled study. Diabetes Obes Metab 2000;2:105-12.
- 50. Fujioka K, Seaton TB, Rowe E, Jelinek CA, Raskin P, Lebovitz HE, et al; Sibutramine/Diabetes Clinical Study Group. Weight loss with sibutramine improves glycaemic control and other metabolic parameters in obese patients with type 2 diabetes mellitus. Diabetes Obes Metab 2000;2:175-87.
- 51. Levitsky DA, Schuster JA, Stallone D, Strupp BJ. Modulation of the thermic effect of food by fenfluramine. Int J Obes 1986;10:169-73.
- 52. Scalfi L, D'Arrigo E, Carandente V, Coltorti A, Contaldo F. The acute effect of fexfenfluramine on resting metabolic rate and postprandial thermogenesis in obese subjects: a double-blind placebo-controlled study. Int J Obes Relat Metab Disord 1993;17:91-6.
- 53. Van Gaal LF, Vansant GA, Steijaert MC, De Leeuw IH. Effects of dexfenfluramine on resting metabolic rate and thermogenesis in premenopausal obese women during therapeutic weight reduction. Metabolism 1995;44:42-5.
- 54. Breum L, Astrup A, Andersen T, Lambert O, Nielsen E, Garby L, et al. The effect of long-term dexfenfluramine treatment on 24-hour energy-expenditure in man a double-blind placebo controlled study. Int J Obes Relat Metab Disord 1990;14:613-21.
- 55. Seagle HM, Bessesen DH, Hill JO. Effects of sibutramine on resting metabolic rate and weight loss in overweight women. Obes Res 1998;6:115-21.
- 56. Hansen DL, Toubro S, Stock MJ, MacDonald IA, Astrup A. Thermogenic effects of sibutramine in humans. Am J Clin Nutr 1998;68:1180-6.
- 57. Alger S, Larson K, Boyce VL, Seagle H, Fontvieille AM, Ferraro RT, et al. Effect of phenylpropanolamine on energy expenditure and weight loss in overweight women. Am J Clin Nutr 1993;57:120-6.
- 58. Rascovski A, Millner TH, Batalha L, Reis C, Mancini MC, Halpern A. Eficácia e tolerabilidade das substâncias calorigênicas: ioimbina, triiodotironina, aminofilina combinada a efedrina e fenilpropanolamina no tratamento da obesidade a curto prazo. Arq Bras Endocrinol Metab 2000;44:95-102.
- 59. Liu YL, Toubro S, Astrup A, Stock MJ. Contribution of beta 3-adrenoceptor activation to ephedrine-induced thermogenesis in humans. Int J Obes Relat Metab Disord 1995;19:678-85.
- 60. Pasquali R, Cesari MP, Melchionda N, Stefanini C, Raitano A, Labo G. Does ephedrine promote weight loss in low-energy adapted obese women? Int J Obes 1987;11:163-8.
- 61. Halpern A, Mancini MC. Tratamento farmacológico da obesidade Drogas termogênicas. Arq Bras Endocrinol Metab 1996;40:224-7.
- 62. Dulloo AG, Seydoux J, Girardier L. Potentiation of the thermogenic antiobesity effects of ephedrine by dietary methylxantines: adenosine antagonism or phosphodiesterase inhibition. Metabolism 1992;41:1233-41.
- 63. Astrup A, Toubro S, Cannon S, Hein P, Madsen J. Thermogenic synergism between ephedrine and caffeine in healthy volunteers: a double-blind, placebo-controlled study. Metabolism 1991;40:323-9.
- 64. Mancini MC, Marsiaj HI, Hakoyama MM, Quantal IA, Correa NC, Halpern A. Ephedrine, caffeine and aminophilline preparation: na alternative in the treatment of obesity. Int J Obes 1990;14(suppl 2):141.
- 65. Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997;337:581-8.
- 66. Graham DJ, Green L. Further cases of valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997;337:635.
- 67. Leite CC, Mancini MC, Medeiros CCJ, Sbano JCN, Grinberg M, Halpern A. Echocardiographic evaluation of 70 patients using dexfenfluramine (abstracted). Int J Obes Relat Metab Disord 1998;22(suppl 3):S227.
- 68. Weissman NJ, Tighe JF, Gottdiener JS, Gwynne JT. An assessment of heart-valve abnormalities in obese patients taking dexfenfluramine, sustained-release dexfenfluramine, or placebo. N Engl J Med 1998; 339: 725-32.
- 69. Abenhaim L, Moride Y, Brenot F, et al. Appetite-supressant drugs and the risk of primary pulmonary hypertension. N Engl J Med 1996;335:609-16.
- 70. Hagiwara M, Tsuchida A, Hyakkoku M, et al. Delayed onset of pulmonary hypertension associated with an appetite suppressant, mazindol: a case report. Jpn Circ 2000;64:218-21.
- 71. Thomas SH, Butt AY, Corris PA, et al. Appetite supressants and primary pulmonary hypertension in the United Kingdom. Br Heart J 1995;74:660-3.
- 72. Little JD, Romans SE. Psychosis following readministration of diethylpropion: a possible role for kinding? Int Clin Psychopharmacol 1993;8:67-70.
- 73. Luque CA, Ray JA. Sibutramine: a serotonin-norepinephrine reuptake-inhibitor for the treatment of obesity. Ann Pharmacother 1999;33:968-78.
- 74. McMahon FG, Fujioka K, Singh BN, Mendel CM, et al. Efficacy and safety of sibutramine in obese white and African American patients with hypertension: a 1-year, double-blind, placebo-controlled multicenter trial. Arch Int Med 2000;160:2185-91.
- 75. Horwitz RI, Brass LM, Kernan WN, Viscoli CM. Phenylpropanolamine and risk of hemorrhagic stroke: final report of the hemorrhagic stroke project. http://fda.gov/ ohrms/dockets/ac/00/backgrd/3647b1_tab19.doc (accessed on March 2, 2001).
- 76. Neilsen JA, Chapin DS, Johnson Jr JL, Torgersen LK. Sertraline, a serotonin-uptake inhibitor, reduces food intake and body weight in lean rats and genetically obese mice. Am J Clin Nutr 1992;55(suppl):185S-9S.
- 77. McGuirk J, Silverstone T. The effect of the 5-HT re-uptake inhibitor fluoxetine on food intake and body weight in healthy male subjects. Int J Obes Relat Metab Disord 1990;14:361-72.
- 78. Gray DS, Fujioka K, Devine W, Bray GA. A randomized double-blind clinical trial of fluoxetine in obese diabetics. Int J Obes Relat Metab Disord 1992;16(suppl 4):S67-S72.
- 79. Wadden TA, Bartlett SJ, Foster GD, Greenstein RA, Wingate BJ, Stunkard AJ, et al. Sertraline and relapse prevention training following treatment by very-low-calorie diet: a controlled clinical trial. Obes Res 1995;3:549-57.
- 80. Ricca V, Mannucci E, Di Bernardo M, Rizzello SM, Cabras PL, Rotella CM. Sertraline enhances the effects of cognitive-behavioral treatment on weight reduction of obese patients. J Endocrinol Invest 1996;19:727-33.
- 81. Arterburn D, Noël PH. Extracts from "clinical evidence" Obesity. Br Med J 2001;322:1406-9.
- 82. Zhi J, Melia AT, Eggers H, et al. Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J Clin Pharmacol 1995;35:1103-8.
- 83. Aronne LJ. Modern medical management of obesity: the role for pharmacological intervention. J Am Diet Assoc 1998;98(suppl 2):S23-6.
- 84. Davidson MH, Hauptman J, DiGirolamo M, et al. Weight control and risk factor reduction in obese subjects treated for 2 years with orlistat. JAMA 1999;281:235-42.
- 85. Zavoral JH. Treatment with orlistat reduces cardiovascular risk in obese patients. J Hypertens 1998;16:2013-7.
- 86. Sjöstrom L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. Lancet 1998;352:167-72.
- 87. Hollander PA, Elbein SC, Hirsch IB, et al. Role of orlistat in the treatment of obese patients with type 2 diabetes. Diabetes Care 1998;21:1288-94.
- 88. Halpern A, Mancini MC, Suplicy H, Zanella MT, Repetto G, Gross J, et al. Latin-american trial of orlistat for weight loss and improvement in glycemic profile in obese diabetic patients. Diabetes Obes Metab 2003;5:180-8.
- 89. Drent ML, van der Veen EA. Lipase inhibition: a novel concept in the treatment of obesity. Int J Obes 1993;17:241-4.
- 90. Drent ML, Larsson I, William-Olsson T, Quaade F, et al. Orlistat, a lipase inhibitor, in the treatment of human obesity: a multiple dose study. Int J Obes 1995;19:221-6.
- 91. Van Gaal LF, Bloom JI, Enzi G, et al. Efficacy and tolerability of orlistat in the treatment of obesity: a 6-month-dose-ranging study. Eur J Pharmacol 1998;54:125-32.
- 92. Daniels S. Pharmacological treatment of obesity in paediatric patients. Paediatr Drugs 2001;3:405-10.
- 93. Halpern A, Mancini MC. Diabesity: are weight loss medications effective? Treat Endocrinol 2005;4:65-74.
- 94. Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004;27:155-61.
- Erratum in: Diabetes Care 2004;27:856.
- 95. van Gaal L, Rissanen AM, Scheen AJ, Ziegler O, Rössner S; the RIO-Europe Study Group. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet 2005;365:1389-97.
Publication Dates
-
Publication in this collection
23 May 2006 -
Date of issue
Apr 2006
History
-
Received
30 Oct 2005 -
Accepted
17 Jan 2006