Abstracts
The calcium-sensing receptor (CASR) adjusts the extracellular calcium set point regulating PTH secretion and renal calcium excretion. The receptor is expressed in several tissues and is also involved in other cellular functions such as proliferation, differentiation and other hormonal secretion. High extracellular calcium levels activate the receptor resulting in modulation of several signaling pathways depending on the target tissues. Mutations in the CASR gene can result in gain or loss of receptor function. Gain of function mutations are associated to Autossomal dominant hypocalcemia and Bartter syndrome type V, while loss of function mutations are associated to Familial hypocalciuric hypercalcemia and Neonatal severe hyperparathyroidism. More than one hundred mutations were described in this gene. In addition to calcium, the receptor also interacts with several ions and polyamines. The CASR is a potential therapeutic target to treatment of diseases including hyperparathyroidism and osteoporosis, since its interaction with pharmacological compounds results in modulation of PTH secretion.
Familial hypocalciuric hypercalcemia; Autosomal dominant hypocalcemia; Mutations; CASR; Bartter syndrome type V; Neonatal severe hyperparathyroidism
O receptor sensor de cálcio (CASR) ajusta o set point do cálcio extracelular através da regulação da secreção de PTH e da excreção renal de cálcio. O receptor é expresso em diversos tecidos e também está envolvido em outras funções celulares como proliferação, diferenciação e secreção de outros hormônios. Concentrações altas de cálcio extracelular ativam o receptor resultando em modulação de inúmeras vias de sinais intracelulares dependendo do tecido-alvo. Mutações no gene do CASR podem resultar em ganho ou perda de função do receptor. Mutações com ganho de função são associadas à Hipocalcemia autossômica dominante e à Síndrome de Bartter tipo V, enquanto que mutações com perda de função são associadas à Hipercalcemia hipocalciúrica familiar e ao Hiperparatireoidismo neonatal grave. Mais de cem mutações foram descritas neste gene. Além do cálcio, o receptor também interage com inúmeros íons e poliaminas. CASR é um alvo terapêutico potencial para tratamento de doenças incluindo hiperparatireoidismo e osteoporose, pois a sua interação com compostos farmacológicos resulta em modulação da secreção de PTH.
Hipercalcemia hipocalciúrica familiar; Hipocalcemia autossômica dominante; Mutações; CASR; Síndrome de Bartter tipo V; Hiperparatireoidismo neonatal grave
REVIEW ARTICLE
The calcium-sensing receptor and related diseases
O receptor sensor de cálcio e doenças associadas
Lília D'Souza-Li
Pediatric Endocrinology Laboratory, Center for Investigation in Pediatrics and Department of Pediatrics, Faculty of Medical Science, State University of Campinas, São Paulo, Brazil
Address for correspondence Address for correspondence: Lília D'Souza-Li Center for Investigation in Pediatrics Rua Tessália Vieira de Camargo 126 Cidade Universitária "Zeferino Vaz" Caixa Postal 6111 13083-970 Campinas, SP, Brazil Fax: (55) (19) 3788-8972 E-mail: ldesouza@globo.com
ABSTRACT
The calcium-sensing receptor (CASR) adjusts the extracellular calcium set point regulating PTH secretion and renal calcium excretion. The receptor is expressed in several tissues and is also involved in other cellular functions such as proliferation, differentiation and other hormonal secretion. High extracellular calcium levels activate the receptor resulting in modulation of several signaling pathways depending on the target tissues. Mutations in the CASR gene can result in gain or loss of receptor function. Gain of function mutations are associated to Autossomal dominant hypocalcemia and Bartter syndrome type V, while loss of function mutations are associated to Familial hypocalciuric hypercalcemia and Neonatal severe hyperparathyroidism. More than one hundred mutations were described in this gene. In addition to calcium, the receptor also interacts with several ions and polyamines. The CASR is a potential therapeutic target to treatment of diseases including hyperparathyroidism and osteoporosis, since its interaction with pharmacological compounds results in modulation of PTH secretion.
Keywords: Familial hypocalciuric hypercalcemia; Autosomal dominant hypocalcemia; Mutations; CASR; Bartter syndrome type V; Neonatal severe hyperparathyroidism
RESUMO
O receptor sensor de cálcio (CASR) ajusta o set point do cálcio extracelular através da regulação da secreção de PTH e da excreção renal de cálcio. O receptor é expresso em diversos tecidos e também está envolvido em outras funções celulares como proliferação, diferenciação e secreção de outros hormônios. Concentrações altas de cálcio extracelular ativam o receptor resultando em modulação de inúmeras vias de sinais intracelulares dependendo do tecido-alvo. Mutações no gene do CASR podem resultar em ganho ou perda de função do receptor. Mutações com ganho de função são associadas à Hipocalcemia autossômica dominante e à Síndrome de Bartter tipo V, enquanto que mutações com perda de função são associadas à Hipercalcemia hipocalciúrica familiar e ao Hiperparatireoidismo neonatal grave. Mais de cem mutações foram descritas neste gene. Além do cálcio, o receptor também interage com inúmeros íons e poliaminas. CASR é um alvo terapêutico potencial para tratamento de doenças incluindo hiperparatireoidismo e osteoporose, pois a sua interação com compostos farmacológicos resulta em modulação da secreção de PTH.
Descritores: Hipercalcemia hipocalciúrica familiar; Hipocalcemia autossômica dominante; Mutações; CASR; Síndrome de Bartter tipo V; Hiperparatireoidismo neonatal grave
ELECTROPHYSIOLOGICAL STUDIES SHOW that parathyroid cells possess a cell surface [Ca2+o] sensing mechanism that results in changes in phosphoinositide turnover and cytosolic calcium to regulate PTH secretion (1). Extracellular calcium regulates itself by serving as a first messenger and interacting with its receptor, the calcium-sensing receptor (CASR) on target tissues. The receptor was cloned in 1993 from bovine parathyroid (BoPCAR1) by expression cloning in Xenopus laevis oocytes and is a member of the G protein-coupled receptor super family (2). High calcium levels activate the CASR in the parathyroid cell surface to inhibit PTH secretion, and in the kidney to increase calcium excretion (3).
STRUCTURE OF THE CALCIUM-SENSING RECEPTOR
The human CASR gene is located on chromosome 3q13.3-21 (4,5) and spans over 50 kb of genomic DNA. It has a coding region of 3234 bp, which is contained within 6 exons (6). The human CASR is ~120 kDa protein, consisting of 1078 amino acid, with 612 amino acids in the extracellular domain (ECD), 250 amino acids of which comprise seven transmembrane spanning domains (TM), intracellular (ICL) and extracellular loops (ECL), and 216 amino acids of a long C-terminus cytoplasmic tail (ICD) (6). The CASR belongs to the metabotropic glutamate receptor subfamily, which comprises the metabotropic glutamate receptors (mGluR) (7), the GABAB receptor (8), the Vomero-nasal (pheromone) receptors (9), the taste receptors (10), the GPRC6A receptor (11) and five orphan receptors (12,13).
Studies, either with CASR cDNA transiently transfected in HEK293 cells (14) or expressed endogenously in rat inner medullary collecting duct endosomes (15), show that the CASR form homodimers via intermolecular disulfide linkages within the ECD. Dimers are the most abundant species present on the cell surface and intermolecular interactions within the dimeric CASR are important for receptor function (16,17). The ECD of the CASR contains nine potential N-linked glycosylation sites (6). The native CASR, as well as recombinant receptors transfected in HEK293 cells, present as three forms: 1) a 120 kDa band, which represents the non glycosylated species; 2) a 140 kDa band, which represents the immature glycosylated receptor; and 3) a 160 kDa band, which is the mature fully glycosylated receptor (18). Although the immature glycosylated receptor can reach the plasma membrane to a low extent, only the fully glycosylated receptor is functional (19,20).
Calcium binding sites
Due to the lack of a high-affinity ligand binding assay for the CASR, all the positions where calcium binds are unknown. Also, it is not known how many calcium ions bind to each receptor since there is the possibility of different affinity of each ligand binding domain for the ligand, cooperativity between the ligand binding sites and dimerization of the receptor (21). The pharmacology of the CASR is unusual for a receptor, as it only responds to the ligand in the millimolar ion concentration range, suggesting low affinity of the receptor for [Ca2+o] (22). However, this affinity range of the receptor is of physiological relevance as the free [Ca2+] range is 0.75 to 2.0 mM for extracellular fluids (22). The ECD of the CASR has homologous regions that align with the mGluRs and the related bacterial periplasmic amino acid binding protein, suggesting that the CASR might have evolved from an ancient family of cell-surface proteins binding essential extracellular solutes, and suggesting the existence of additional ion-sensing receptors (23). The Venus flytrap model is the proposed model of ligand receptor interaction for the metabotropic glutamate receptor family (24). In this model two ligand-bound forms have been observed: an open conformation with the ligand initially bound to one ligand pocket in the large ECD with low affinity, and a closed form in which the ligand binds to a second domain stabilizing a high-affinity closed conformation, enclosed within the cleft. The liganded N-terminal segment interacts with the membrane-associated domain to generate a signal (24). Alignment of the extracellular domain of the CASR with the metabotropic glutamate receptor and the related bacterial amino-acid binding protein suggested that Ser 147 and Ser 170 correspond to residues in the binding pockets in the CASR (25). Further mutation analysis associated to molecular modeling studies indicated that calcium interact with polar residues in the binding pockets in the ECD of the receptor, with residues Ser 170, Asp 190, Gln193, Ser 296 and Glu 297 directly involved in Ca2+ coordination and residues Tyr 218 and Phe 270 and Ser 147 contributing to complete the coordination (26).
Calcium-sensing receptor agonists
Although calcium is the endogenous ligand for the CASR, it also shows affinity for a variety of di-, tri-, and polyvalent cations in vitro such as Mg2+, Ba2+, Sr2+, Gd3+, La3+, neomycin, spermine, and protamine (6). The rank order of potency of some agonists is: Gd3+ > neomycin > Ca2+ > Mg2+, with a half-maximal response (EC50) of 20 µM, 70 µM, 3 mM and 10 mM, respectively (2). The physiological relevance of the interaction of CASR with ions other than Ca2+ is unknown. In addition to cations, studies suggest that the CASR also senses sodium and ionic strength in parathyroid cells (27,28).
CALCIUM-SENSING RECEPTOR SIGNALING
When BoPCaR1 is expressed in Xenopus laevis oocytes, agonists elicit an increase in inositol 1,4,5-trisphosphate, which is completely blocked by treatment with pertussis toxin, indicating that the response is mediated through Gai or Gao (2). However, in bovine parathyroid cells, high levels of [Ca2+o] activate phospholipase C (PLC) in a pertussis toxin-insensitive manner, suggesting that the CASR is coupled to PLC through a member of the Gq family (29). Interaction with Gaq is followed by activation of phospholipase Cb, breakdown of phosphatidylinositol 4,5-bisphosphate with formation of 1,2-sn-diacylglycerol and of inositol 1,4,5-trisphosphate (IP3). The accumulation of IP3 leads to the release of intracellular pools of calcium contributing to intracellular signaling and causing inhibition of PTH secretion through mechanisms that remain to be fully defined (30). The high [Ca2+o] also induces a sustained rise in [Ca2+i] in parathyroid and in CASR transfected HEK293 cells associated with activation of a Ca2+-permeable, nonselective cation channel (31). Activation of the receptor mediates different signal transduction pathways, depending on the cell line. In Chinese hamster ovary cells elicit phosphatidylinositol and arachidonic acid responses (32). In a mouse pituitary cell line (AtT-20) agonist-elicited increase in inositol phosphate is pertussis toxin-sensitive (33). In Madin-Darby canine kidney cell line, the CASR shows interactions of the receptor with Gai-2, Gai-3, and Gaq/11 (34). In the human astrocytoma cell line U87 (35), rat oligodendrocytes (36), rat microglia primary cultures (37), as well as in human lens-epithelial cells (38), the CASR activates an outwards K+ channel. In rat fibroblasts, CASR was shown to mediate cell proliferation through an increase in c-SRC and ERK1 tyrosine kinases activity (39). In the human colonic cell line Caco-2, low Ca2+ (via interaction with CASR) induces proliferation and c-myc proto-oncogene expression via PKC activation (40).
ROLE OF THE CASR IN DIFFERENT TISSUES
Regulation of parathyroid function
The highest cell surface expression levels of CASR are found in parathyroid cells. CASR plays a crucial role in regulating PTH secretion and the parathyroid cells recognize remarkably small perturbations in the [Ca2+o], and respond by altering the secretion of PTH (22). [Ca2+o] has an inverse steep sigmoidal relationship with PTH secretion, and most of the sensing of [Ca2+o] in parathyroid cells occurs over changes in free [Ca2+] of approximately 0.25 mM (22). The set point of normal human parathyroid, defined as the calcium concentration at which PTH secretion is half-maximal, is ~1 mM, and it plays an important role in determining the level at which [Ca2+o] is set by the homeostatic system. Inactivating mutations in the CASR result in a mild increase in the set point for [Ca2+o]. In addition, [Ca2+o] exerts several other actions on parathyroid function including modulation of the intracellular degradation of PTH, cellular respiration and membrane voltage, but the role of the CASR in mediating these effects is not known (31). Bovine parathyroid cells maintained in culture for more than 24 hours reduce dramatically their responsiveness to [Ca2+o] (2). This is associated with a significant reduction in mRNA and protein levels of CASR (41).
Regulation of calcium excretion in kidney
Kidney is the major route for mineral ion excretion from the body and plays a key role in calcium homeostasis. In addition to PTH, the CASR plays an important role in regulating renal divalent mineral transport processes by both direct (by regulating calcium and water handling) and indirect (by modulating PTH secretion) mechanisms (42). [Ca2+o] modulates renal tubular divalent mineral and water transport processes by interacting with the CASR (42).
The CASR has been localized within several segments of the rat tubule, but it is expressed at highest levels in the cortical thick ascending limb (CTAL) (43). It is found mostly on the basolateral surface of tubular cells, but also to a lesser extent on the apical surface (31). Elevated peritubular [Ca2+o] and [Mg2+o] reduces the tubular reabsorption in isolated microperfused segments of CTAL in vitro (44). The reabsorption of Ca2+ and Mg2+ in CTAL occurs mainly through a paracellular pathway driven by a lumen-positive, transepithelial potential generated by the transport of Na+, K+, and Cl- by the apical Na-K-2Cl co-transporter combined with recycling of K+ into the lumen via an apical K+ channel (31). While PTH acts through its receptor in the kidney, stimulates cAMP accumulation, enhances the co-transport activity and results in an increase of Ca2+ and Mg2+ transport, [Ca2+o] inhibits the activity of the apical K+ channel resulting in a decrease in co-transporter activity and a reduction of Ca2+ and Mg2+ transport (31). High [Ca2+o] in the mouse CTAL decreases hormone-dependent cAMP accumulation as a result of a direct inhibition of adenylyl cyclase (AC) activity (45). An increase in Arginine vasopressin (AVP)-elicited osmotic water permeability in collecting ducts stimulates water reabsorption selectively via aquaporin-2 (AQP-2) water channels (46). CASR and AQP-2 were also found to co-express in rat kidney inner medullary collecting ducts (IMCD) suggesting a direct effect of CASR in inhibition of AVP-elicited osmotic water permeability and the consequent increase in diuresis (46). CASR and Ca2+-inhibitable AC were found to co-express and co-localize in the rat CTAL cells (47), and cAMP synthesis is inhibited by agents coupled to PLC or to Gai protein-mediated process suggesting that the CASR contributes to the effect observed for high [Ca2+o] (47). Additional evidence of the role of CASR in regulating Ca2+ and Mg2+ transport in CTAL is found in subjects with mutations in the CASR gene. In subjects with FHH due to an inactivating mutation in the CASR there is a PTH-independent increase in tubular Ca2+ reabsorption (48), while in ADH subjects there is increased urinary calcium excretion (31).
Role of the calcium-sensing receptor in other tissues
The CASR is widely distributed and is also found in tissues that are not directly involved in calcium homeostasis. In these tissues it appears that high [Ca2+o], via interaction with CASR, regulates a series of cellular functions such as increased cell proliferation in fibroblasts (39), induction of cell differentiation in keratinocytes (49) and human colon epithelial cells (50), prevention of apoptosis in AT-3 prostate carcinoma cells (51), and cataract formation in lens epithelial cell (38). CASR was detected in a murine bone marrow-derived stromal cell line (ST2) (52), in osteoblast-like cell lines (53) and in rabbit mature osteoclasts (54), however its role in bone is still debatable (55,56). In the mouse pituitary cell line, AtT-20 cells, it was shown that the CASR was implicated in adrenocorticotropic hormone (ACTH) (57) and a-MSH release (58). The CASR was also demonstrated in human insulinoma primary cultures, causing released insulin upon [Ca2+o] stimulation (59), in hepatocytes stimulating bile flow (60) and in antral gastric cells stimulating gastrin secretion (61).
DISEASES ASSOCIATED WITH MUTATIONS IN THE CASR
Disorders due to loss of the calcium-sensing receptor function
Two autosomal disorders, Familial Hypocalciuric Hypercalcemia (FHH) and Neonatal Severe Primary Hyperparathyroidism (NSHPT), have been associated with loss of CASR function due to inactivating mutations.
Familial hypocalciuric hypercalcemia
FHH is characterized by moderate elevations of serum calcium concentration (hypercalcemia), lower urinary calcium excretion (hypocalciuria) and inappropriately normal parathyroid hormone (PTH) levels (62,63). This is not a life-threatening condition and most of the usual sequelae of hypercalcemia such as altered mental status, kidney stones, decreased urinary concentrating ability and hypertension are absent (64). Patients are usually asymptomatic or have nonspecific symptoms such as fatigue, weakness, painful joints and headache, with the diagnosis only suspected after a routine biochemical screening showing high blood calcium levels (63). Interestingly, some subjects with FHH present with an incomplete phenotype, lacking hypocalciuria. In some families a more severe phenotype suggestive of familiar isolated hyperparathyroidism is present (65,66). FHH is inherited as an autosomal dominant disorder, and all affected individuals with mutations in the CASR gene are heterozygous for the mutation (67). The dominant pattern of inheritance of this disease has been attributed to haploinsufficiency of the CASR gene, where protein receptor produced by a single normal allele cannot support normal function, although it may suffice for survival (68).
The gene responsible for FHH was linked to chromosome 3q 21-24 in four families (69) and later fluorescence in situ hybridization analysis identified the position of the gene as 3q 13.3-21 (4). Cloning of the CASR was followed by reports of inactivating mutations in this gene in FHH families (70,71). FHH is a heterogeneous disease, and the disease locus segregates with chromosome 3 in most of the families (FHH type 1); however, mutations in other genes may be responsible for similar phenotypes as the disease also segregates to chromosome 19p13.3 (72) in one family (FHH type 2) and 19q13 (73) in another family (FHH type 3) (74). In this last case, besides hypercalcemia and hypocalciuria, affected individuals present increase in PTH serum levels, hypophosphatemia and osteomalacia. In view of the lack of complications, medical treatment for lowering the calcium level is not indicated (75). Surgical exploration of the parathyroid glands is also not indicated, as parathyroidectomy does not cure the disorder (63).
Neonatal Several Hyperparathyroidism
NSHPT (76,77) represents the most severe expression of familial hypocalciuric hypercalcemia (68). In most patients in which mutations were found in the CASR, the two gene copies are mutated, with both parents having passed on a mutated copy and presenting with FHH. There are three reports of mutations being found de novo in individuals with NSHPT with only one copy mutated and no mutation found in the parents (78,79). Neonatal severe hyperparathyroidism causes a marked elevation in serum calcium and PTH levels. It appears very early, in the first days of life, and the baby presents with hypotonia, poor feeding, failure to thrive and respiratory distress associated with rib cage deformities (80). PTH concentrations are very high, associated with calcium levels that are life-threatening (80). In severe cases, surgical intervention is essential, with total parathyroidectomy still being the currently accepted method of treatment. However, there are reports of cases where symptoms are not life threatening and could be controlled using medical therapy to maintain calcium at levels compatible with normal life (81,82).
Disorders due to gain of calcium-sensing receptor function
An autosomal dominant hypocalcemia (ADH) and Bartter syndrome type V have been associated with gain of CASR function due to activating mutations in the receptor.
Autosomal Dominant Hypocalcemia
ADH presents with a wide clinical spectrum, from severe hypocalcemia in the neonatal period to an incidental finding in adulthood (83). Associated problems include seizures, mental deficiency, orodental problems, basal ganglia calcification, kidney stones and renal failure (84). Individuals present with hypocalcemia, hyperphosphatemia, low serum PTH levels and hypercalciuria (84). Autosomal dominant hypocalcemia was initially classified as familial isolated hypoparathyroidism, a heterogeneous group of disorders characterized by PTH deficiency, hypocalcemia and hyperphosphatemia. Within this group, different modes of inheritance were identified with transmission patterns consistent with autosomal dominant, autosomal recessive, and X-linked forms. Finegold et al. linked one form of autosomal dominant hypoparathyroidism to chromosome 3q13 (85) and Pollak et al. (86) described the first activating mutation in the CASR in a family with ADH, and the terminology was recommended to be changed to autosomal dominant hypocalcemia, as a direct contrast to the hypercalcemia in FHH (87). In most individuals where mutations have been found, familial inheritance is clear, with one parent being affected with the same mutation (67). However, de novo mutations found in individuals where no mutation was found in the parents have also been described (67). A careful treatment for this condition is required, as attempts to normalize blood calcium levels with regular doses of vitamin D tend to exacerbate urinary calcium levels and increase the risk of kidney stone and renal impairment (88). Treatment should be limited to symptomatic patients. Hydrochlorothiazide has been used to control hypercalciuria in these patients (88). Recombinant human PTH to improve hypocalcemia symptoms has been described, however longer follow-up studies are required (89).
Bartter Syndrome type V
Bartter syndrome is a heterogeneous rare disease due to deficiency in sodium and chloride absorption. Biochemical profile is renal salt wasting, hypokalemic metabolic alkalosis, elevated renin and aldosterone levels with low blood pressure. In some individuals hypercalciuria is also present. Gain of function mutations in the CASR has been described in some patients with Batter syndrome associated to hypocalcemia and hypercalciuria (90,91). Functional studies showed that these mutations (L125P, C131W and A843E) result in a more severe receptor activation when compared to other activating mutations described (90,91). Of interest, the mutation A843E is the only constitutive mutation described in the CASR, presenting a high basal activity in the absence of [Ca2+o] (92). Clinical data in the literature may be biased towards the most severely affected individuals in both ADH and FHH/NSHPT and may not reflect the whole spectrum of the disease.
Calcium-sensing abnormalities in other disorders
Autoimmune hypoparathyroidism
Autoimmune hypoparathyroidism (AH) manifests biochemically by hypocalcemia and hyperphosphatemia caused by a deficiency of PTH. It represents an integral part of type I autoimmune polyglandular syndrome, a rare disorder characterized by the presence of AH, Addison's disease, and mucocutaneous candidiasis and can be associated with female primary hypogonadism, keratopathy, alopecia, vitiligo, parietal cell atrophy, insulin-dependent diabetes mellitus, autoimmune hepatitis and hypothyroidism (93). In one study, an epitope within the ECD of the CASR was specifically recognized in 14 of 25 individuals (56%) with AH, suggesting that the CASR is a key antigen in directing the immune response against parathyroid tissue in this condition (94). The mechanism of the hypoparathyroidism is destruction of the parathyroid gland due to the inflammatory reaction and complement fixation.
Autoimmune hypocalciuric hypercalcemia
The ECD of the CASR is also antigen for antibodies that instead of inducing parathyroid cell destruction, interferes with the normal activation of the receptor resulting in increase PTH levels (95). Patients may manifest clinically with hypercalcemia, not suppressed PTH levels and hypocalciuria similar of FHH patients. However, it is not associated to mutation in the CASR gene (95). In addition to the hypercalcemia, patients may present other autoimmune disease such as thyroiditis, celiac sprue, psoriasis, hypophysitis, uveitis and rheumatoid arthritis (95).
Mutations in the calcium-sensing receptor
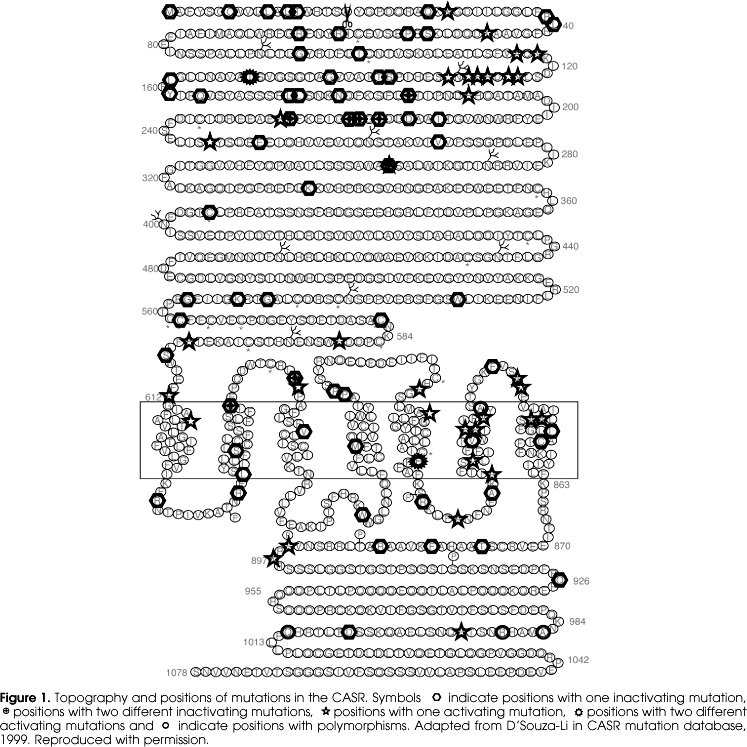
One hundred and twelve mutations (98 missense, 6 nonsense, 8 insertion and or deletion, and 1 splice mutation) have been described in the CASR mutation database (http://www.casrdb.mcgill.ca) related to FHH, NSHPT, ADH families or as de novo disease (figure 1) (96). In addition, 6 polymorphisms were found in samples from a normal population or in families with FHH and ADH in which this base pair change was present in affected and unaffected members and did not segregate with the disease. Fifteen mutations were found more than once in the CASR gene in apparently unrelated families. In several positions two different mutations were described in the same codon with the same receptor phenotype, with one exception. At position 297 an activation mutation (E297D) and an inactivating mutation (E297K) were described (26,70) confirming the crucial role of this position on receptor activation. Most of the mutations found in the ECD are located in the first third of the N-terminus suggesting the importance of this region in ligand binding. Activating mutations in the proximal 1/3 of the ECD may facilitate the ligand-binding interaction in the different binding sites, increasing the receptor affinity to the ligand, whereas inactivating mutations may have the opposite effect, disrupting the ligand binding pockets. This is supported by in vitro functional analyses of mutations in this location that show ligand-dependent changes in the affinity of the receptor to extracellular calcium (67).
Mutation in the TM domain may abrogate constraints, tilting the TM and locking the receptor in either an inactivating or activating conformation, as residues in the TM7 are critical for maintaining the receptor in an inactive conformation (97). From functional analyses of receptors with gain of function, only A843E showed the ability to activate the receptor in the absence of the ligand (92). The other activating mutations all showed a ligand-dependent shift of the dose-response curves to the left. This suggests that the mechanism of activation of the receptor in most of the TM domain mutations is to facilitate the TMD activation, with the exception of A843E that most likely locks the TMD in an active conformation. Inactivating mutations resulting in total loss of function of the receptor may also be associated to total loss of ability of the ligand bind and activate the receptor, even though the receptor is well expressed in the plasma membrane or due to misfolding and retention within the cytoplasm resulting in lack of receptor at the membrane (98).
The ICD seems to be important for receptor trafficking to the cell membrane and for the interaction with intracellular proteins. Large deletion of the c-terminal tail was associated to gain of function in an ADH family (99). In vitro functional studies showed gain of function and increase mutant receptor cell surface expression level. Mutagenesis in the ICD confirms its involvement in degradation and processing of the receptor (99). Residues 962-981 in the c-terminal tail are critical for its interaction with filamin A and this interaction prevents the receptor degradation and facilitates MAPK signaling (100). In contrast, interaction of the ICD with dorfin targets the receptor for degradation (101).
Activating mutations
Forty activating mutations in the CASR gene have been described. The majority is missense mutation, with 2 deletions described. Most ADH affected individuals are heterozygous for the activating mutation. In one family, homozygous mutation is described but it is not associated to a more severe phenotype (99). Clinical data from affected individuals with activating mutations are abundant and, despite the spectrum of severity of the phenotype for the same genotype, similar symptoms are found in different families.
Inactivating mutations
Of the 72 inactivating mutations in the CASR gene, 59 are missense, 6 are nonsense, 6 are insertions and/or deletion including an Alu element insertion (102) and one splice mutation (103). The gene dosage effect is clear in most FHH cases, with one mutated gene copy resulting in FHH with mild hypercalcemia and two mutated copies resulting in NSHPT, a more severe phenotype that manifests very early in life with severe hypercalcemia, bone demineralization and failure to thrive (68). However, the three cases of de novo NSHTP reported in the literature were heterozygous for missense mutations located in the extracellular domain, with only one mutated allele and no mutation found in the parents (79,104). One individual with de novo NSHPT was heterozygous for a previously described mutation in a FHH family (79).
Polymorphisms
Six polymorphisms were found in the CASR gene: one in intron 5 just before exon 6 (IVS 5 -88 t/c) and the remaining five in exon 7 in the coding region (one in the 6th TM [A/T826], one in the 7th TM [C/S851], and three in the ICD [A/S986, R/G990 and Q/E1011]). The polymorphism in intron 5, IVS 5 -88 t/c, is very common (105) and, when analyzed in a large group of normal and affected individuals, no correlation was found between this mutation and the incidence of parathyroid adenoma or diabetes (106). The A/T 826 mutation was initially found in 4 parathyroid adenomas (107). Further analysis showed the same change in 16% of 50 normal subjects' samples (108). The C/S 851 was found in an ADH family in both affected and unaffected members (109). They also found another mutation in this family (A116T), which segregates with the disease, and concluded that C/S 851 was a rare polymorphism. The frequency of the 3 common polymorphisms in the cytoplasmic tail varies in different populations. In a large series in a Caucasian population the incidence for A/S986 was 24%, for R/G990 was 4% and for Q/E1011 was 3% within 377 unrelated DNA samples (110). In addition, a study analyzing serum calcium levels in samples from a normal population found that the homozygous polymorphism 986S was associated to higher serum calcium levels when compared to the heterozygous form, while the homozygous 986A had the lowest calcium levels (110,111).
INTERACTION OF THE CASR WITH PHARMACOLOGICAL COMPOUNDS
Calcimimetics
The CASR is also a target for pharmacological compounds that act synergistically with calcium as positive allosteric modulators of the receptor, such as NPS R-568 and cinalcalcet (112,113). However, these compounds require the presence of calcium and act by increasing the sensitivity of the receptor to [Ca2+o] (114). Their interaction sites are in the TMD of the receptor with Glu 837 critical for their action (115). Clinical studies using a calcimimetic in secondary hyperparathyroidism showed significant reduction on PTH, and in the calcium x phosphate product levels in patients treated for 26 weeks (116). Cinacalcet was approved by the US FDA for the treatment of secondary hyperparathyroidism (113). Other potential uses for calcimimetics are co-adjuvant in the treatment of hyperparathyroidism and parathyroid carcinoma.
Calcilytic
Compounds that interact with the CASR as negative allosteric modulators have also been developed, such as NPS-2143 (117) and compound 1 (118). These compounds are potent CASR antagonists resulting in transient increase PTH secretion and bone formation (117). Calcilytic compounds are potential therapeutic agents for the treatment of osteoporosis, since they can reproduce the anabolic effect in bone of transitory increase in PTH.
CONCLUSION
The CASR plays an important role in regulating [Ca+2o]. The receptor is more versatile than ever expected, being involved in a variety of cellular function. It is also target to new pharmacological compounds that modifies its function with potential therapeutic applications.
ACKNOWLEDGEMENTS
Work supported by FAPESP (grant research #00/ 08587-0, fellowship 00/14775-4).
Received in 03/08/06
Accepted in 03/20/03
- 1. Brown EM. Extracellular Ca2+ sensing, regulation of parathyroid cell function, and role of Ca2+ and other ions as extracellular (first) messengers. Physiol Rev 1991;71(2):371-411.
- 2. Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 1993;366(6455):575-80.
- 3. Hauache OM. Extracellular calcium-sensing receptor: Structural and functional features and association with diseases. Braz J Med Biol Res 2001;34(5):577-84.
- 4. Janicic N, Soliman E, Pausova Z, Seldin MF, Riviere M, Szpirer J, et al. Mapping of the calcium-sensing receptor gene (CASR) to human chromosome 3q13.3-21 by fluorescence in situ hybridization, and localization to rat chromosome 11 and mouse chromosome 16. Mamm Genome 1995;6(11):798-801.
- 5. Aida K, Koishi S, Tawata M, Onaya T. Molecular cloning of a putative Ca(2+)-sensing receptor cDNA from human kidney. Biochem Biophys Res Commun 1995;214(2):524-9.
- 6. Garrett JE, Capuano IV, Hammerland LG, Hung BC, Brown EM, Hebert SC, et al. Molecular cloning and functional expression of human parathyroid calcium receptor cDNAs. J Biol Chem 1995;270(21):12919-25.
- 7. Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science 1992;258 (5082):597-603.
- 8. Kaupmann K, Huggel K, Heid J, Flor PJ, Bischoff S, Mickel SJ, et al. Expression cloning of GABA(B) receptors uncovers similarity to metabotropic glutamate receptors. Nature 1997;386(6622):239-46.
- 9. Herrada G, Dulac C. A novel family of putative pheromone receptors in mammals with a topographically organized and sexually dimorphic distribution. Cell 1997;90(4):763-73.
- 10. Hoon MA, Adler E, Lindemeier J, Battey JF, Ryba NJ, Zuker CS. Putative mammalian taste receptors: A class of taste-specific GPCRs with distinct topographic selectivity. Cell 1999;96(4):541-51.
- 11. Wellendorph P, Hansen KB, Balsgaard A, Greenwood JR, Egebjerg J, Brauner-Osborne H. Deorphanization of GPRC6A: A promiscuous L-alpha-amino acid receptor with preference for basic amino acids. Mol Pharmacol 2005;67(3):589-97.
- 12. Brauner-Osborne H, Krogsgaard-Larsen P. Sequence and expression pattern of a novel human orphan G-protein-coupled receptor, GPRC5B, a family C receptor with a short amino-terminal domain. Genomics 2000;65(2):121-8.
- 13. Brauner-Osborne H, Jensen AA, Sheppard PO, Brodin B, Krogsgaard-Larsen P, O'Hara P. Cloning and characterization of a human orphan family C G-protein coupled receptor GPRC5D. Biochim Biophys Acta 2001;1518(3): 237-48.
- 14. Bai M, Trivedi S, Brown EM. Dimerization of the extracellular calcium-sensing receptor (CaR) on the cell surface of CaR-transfected HEK293 cells. J Biol Chem 1998;273(36):23605-10.
- 15. Ward DT, Brown EM, Harris HW. Disulfide bonds in the extracellular calcium-polyvalent cation-sensing receptor correlate with dimer formation and its response to divalent cations in vitro. J Biol Chem 1998;273(23):14476-83.
- 16. Bai M, Trivedi S, Kifor O, Quinn SJ, Brown EM. Intermolecular interactions between dimeric calcium-sensing receptor monomers are important for its normal function. Proc Natl Acad Sci USA 1999;96(6):2834-9.
- 17. Hauache OM, Hu J, Ray K, Spiegel AM. Functional interactions between the extracellular domain and the seven-transmembrane domain in Ca2+ receptor activation. Endocrine 2000;13(1):63-70.
- 18. Bai M, Quinn S, Trivedi S, Kifor O, Pearce SHS, Pollak MR, et al. Expression and characterization of inactivating and activating mutations in the human Ca2+o-sensing receptor. J Biol Chem 1996;271(32):19537-45.
- 19. Oda Y, Tu CL, Pillai S, Bikle DD. The calcium sensing receptor and its alternatively spliced form in keratinocyte differentiation. J Biol Chem 1998;273 (36):2 3344-52.
- 20. Fan G, Goldsmith PK, Collins R, Dunn CK, Krapcho KJ, Rogers KV, et al. N-linked glycosylation of the human Ca2+ receptor is essential for its expression at the cell surface. Endocrinology 1997;138(5):1916-22.
- 21. Kubo Y, Miyashita T, Murata Y. Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science 1998;279(5357):1722-5.
- 22. Hebert SC, Brown EM, Harris HW. Role of the Ca(2+)-sensing receptor in divalent mineral ion homeostasis. J Exp Biol 1997;200(Pt 2):295-302.
- 23. Conklin BR, Bourne HR. Homeostatic signals. Marriage of the flytrap and the serpent [news]. Nature 1994;367 (6458):22.
- 24. O'Hara PJ, Sheppard PO, Thogersen H, Venezia D, Haldeman BA, McGrane V, et al. The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron 1993;11(1):41-52.
- 25. Brauner-Osborne H, Jensen AA, Sheppard PO, O'Hara P, Krogsgaard-Larsen P. The agonist-binding domain of the calcium-sensing receptor is located at the amino-terminal domain. J Biol Chem 1999;274(26):18382-6.
- 26. Silve C, Petrel C, Leroy C, Bruel H, Mallet E, Rognan D, et al. Delineating a Ca2+ binding pocket within the Venus flytrap module of the human calcium-sensing receptor. J Biol Chem 2005;280(45):37917-23.
- 27. Quinn SJ, Kifor O, Trivedi S, Diaz R, Vassilev P, Brown E. Sodium and ionic strength sensing by the calcium receptor. J Biol Chem 1998;273(31):19579-86.
- 28. Doroszewicz J, Waldegger P, Jeck N, Seyberth H, Waldegger S. pH dependence of extracellular calcium sensing receptor activity determined by a novel technique. Kidney Int 2005;67(1):187-92.
- 29. Hawkins D, Enyedi P, Brown E. The effects of high extracellular Ca2+ and Mg2+ concentrations on the levels of inositol 1,3,4,5-tetrakisphosphate in bovine parathyroid cells. Endocrinology 1989;124(2):838-44.
- 30. Dare E, Kifor O, Brown EM, Weber G. Characterization of the phosphatidylinositol-specific phospholipase C isozymes present in the bovine parathyroid and in human kidney HEK293 cells stably transfected with the human parathyroid Ca2+-sensing receptor. J Mol Endocrinol 1998;21(1):7-17.
- 31. Brown EM, Hebert SC. Calcium-receptor-regulated parathyroid and renal function. Bone 1997;20(4):303-9.
- 32. Ruat M, Snowman AM, Hester LD, Snyder SH. Cloned and expressed rat Ca2+-sensing receptor. J Biol Chem 1996;271(11):5972-5.
- 33. Emanuel RL, Adler GK, Kifor O, Quinn SJ, Fuller F, Krapcho K, et al. Calcium-sensing receptor expression and regulation by extracellular calcium in the AtT-20 pituitary cell line. Mol Endocrinol 1996;10(5):555-65.
- 34. Arthur JM, Collinsworth GP, Gettys TW, Quarles LD, Raymond JR. Specific coupling of a cation-sensing receptor to G protein alpha-subunits in MDCK cells. Am J Physiol 1997;273(1 Pt 2):129-35.
- 35. Chattopadhyay N, Ye CP, Yamaguchi T, Vassilev PM, Brown EM. Evidence for extracellular calcium-sensing receptor mediated opening of an outward K+ channel in a human astrocytoma cell line (U87). Glia 1999;26 (1):64-72.
- 36. Chattopadhyay N, Ye CP, Yamaguchi T, Kifor O, Vassilev PM, Nishimura R, et al. Extracellular calcium-sensing receptor in rat oligodendrocytes: Expression and potential role in regulation of cellular proliferation and an outward K+ channel. Glia 1998;24(4):449-58.
- 37. Chattopadhyay N, Ye C, Yamaguchi T, Nakai M, Kifor O, Vassilev PM, et al. The extracellular calcium-sensing receptor is expressed in rat microglia and modulates an outward K+ channel. J Neurochem 1999;72(5):1915-22.
- 38. Chattopadhyay N, Ye C, Singh DP, Kifor O, Vassilev PM, Shinohara T, et al. Expression of extracellular calcium-sensing receptor by human lens epithelial cells. Biochem Biophys Res Commun 1997;233(3):801-5.
- 39. McNeil SE, Hobson SA, Nipper V, Rodland KD. Functional calcium-sensing receptors in rat fibroblasts are required for activation of SRC kinase and mitogen-activated protein kinase in response to extracellular calcium. J Biol Chem 1998;273(2):1114-20.
- 40. Kallay E, Kifor O, Chattopadhyay N, Brown EM, Bischof MG, Peterlik M, et al. Calcium-dependent c-myc proto-oncogene expression and proliferation of Caco-2 cells: A role for a luminal extracellular calcium-sensing receptor. Biochem Biophys Res Commun 1997;232(1):80-3.
- 41. Mithal A, Kifor O, Kifor I, Vassilev P, Butters R, Krapcho K, et al. The reduced responsiveness of cultured bovine parathyroid cells to extracellular Ca2+ is associated with marked reduction in the expression of extracellular Ca(2+)-sensing receptor messenger ribonucleic acid and protein. Endocrinology 1995;136(7):3087-92.
- 42. Hebert SC. Extracellular calcium-sensing receptor: Implications for calcium and magnesium handling in the kidney. Kidney Int 1996;50(6):2129-39.
- 43. Chattopadhyay N, Baum M, Bai M, Riccardi D, Hebert SC, Harris HW, et al. Ontogeny of the extracellular calcium-sensing receptor in rat kidney. Am J Physiol 1996;271(3 Pt 2):736-43.
- 44. Quamme GA. Control of magnesium transport in the thick ascending limb. Am J Physiol 1989;256(2 Pt 2):197-210.
- 45. Chabardes D, Firsov D, Aarab L, Clabecq A, Bellanger AC, Siaume-Perez S, et al. Localization of mRNAs encoding Ca2+-inhibitable adenylyl cyclases along the renal tubule. Functional consequences for regulation of the cAMP content. J Biol Chem 1996;271(32):19264-71.
- 46. Sands JM, Naruse M, Baum M, Jo I, Hebert SC, Brown EM, et al. Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Invest 1997;99(6):1399-405.
- 47. de Jesus Ferreira MC, Helies-Toussaint C, Imbert-Teboul M, Bailly C, Verbavatz JM, Bellanger AC, et al. Co-expression of a Ca2+-inhibitable adenylyl cyclase and of a Ca2+- sensing receptor in the cortical thick ascending limb cell of the rat kidney. Inhibition of hormone-dependent cAMP accumulation by extracellular Ca2+ J Biol Chem 1998;273(24):15192-202.
- 48. Attie MF, Gill JR Jr., Stock JL, Spiegel AM, Downs RW Jr., Levine MA, et al. Urinary calcium excretion in familial hypocalciuric hypercalcemia. Persistence of relative hypocalciuria after induction of hypoparathyroidism. J Clin Invest 1983;72(2):667-76.
- 49. Bikle DD, Ratnam A, Mauro T, Harris J, Pillai S. Changes in calcium responsiveness and handling during keratinocyte differentiation. Potential role of the calcium receptor. J Clin Invest 1996;97(4):1085-93.
- 50. Chakrabarty S, Wang H, Canaff L, Hendy GN, Appelman H, Varani J. Calcium sensing receptor in human colon carcinoma: interaction with Ca(2+) and 1,25-dihydroxyvitamin D(3). Cancer Res 2005;65(2):493-8.
- 51. Lin KI, Chattopadhyay N, Bai M, Alvarez R, Dang CV, Baraban JM, et al. Elevated extracellular calcium can prevent apoptosis via the calcium-sensing receptor. Biochem Biophys Res Commun 1998;249(2):325-31.
- 52. Yamaguchi T, Chattopadhyay N, Kifor O, Brown EM. Extracellular calcium (Ca2+(o))-sensing receptor in a murine bone marrow-derived stromal cell line (ST2): Potential mediator of the actions of Ca2+(o) on the function of ST2 cells. Endocrinology 1998;139(8):3561-8.
- 53. Yamaguchi T, Kifor O, Chattopadhyay N, Brown EM. Expression of extracellular calcium (Ca2+o)-sensing receptor in the clonal osteoblast-like cell lines, UMR-106 and SAOS-2. Biochem Biophys Res Commun 1998;243(3):753-7.
- 54. Kameda T, Mano H, Yamada Y, Takai H, Amizuka N, Kobori M, et al. Calcium-sensing receptor in mature osteoclasts, which are bone-resorbing cells. Biochem Biophys Res Commun 1998;245(2):419-22.
- 55. Zaidi M, Shankar VS, Tunwell R, Adebanjo OA, Mackrill J, Pazianas M, et al. A ryanodine receptor-like molecule expressed in the osteoclast plasma membrane functions in extracellular Ca2+ sensing. J Clin Invest 1995;96(3):1582-90.
- 56. Quarles LD, Hartle JE2, Siddhanti SR, Guo R, Hinson TK. A distinct cation-sensing mechanism in MC3T3-E1 osteoblasts functionally related to the calcium receptor. J Bone Miner Res 1997;12(3):393-402.
- 57. Ferry S, Chatel B, Dodd RH, Lair C, Gully D, Maffrand JP, et al. Effects of divalent cations and of a calcimimetic on adrenocorticotropic hormone release in pituitary tumor cells. Biochem Biophys Res Commun 1997;238(3):866-73.
- 58. Van Den Hurk MJ, Jenks BG, Roubos EW, Scheenen WJ. The extracellular calcium-sensing receptor increases the number of calcium steps and action currents in pituitary melanotrope cells. Neurosci Lett 2005;377(2):125-9.
- 59. Kato M, Doi R, Imamura M, Furutani M, Hosotani R, Shimada Y. Calcium-evoked insulin release from insulinoma cells is mediated via calcium-sensing receptor. Surgery 1997;122(6):1203-11.
- 60. Canaff L, Petit JL, Kisiel M, Watson PH, Gascon-Barre M, Hendy GN. Extracellular calcium-sensing receptor is expressed in rat hepatocytes coupling to intracellular calcium mobilization and stimulation of bile flow. J Biol Chem 2001;276(6):4070-9.
- 61. Ray JM, Squires PE, Curtis SB, Meloche MR, Buchan AM. Expression of the calcium-sensing receptor on human antral gastrin cells in culture. J Clin Invest 1997;99(10):2328-33.
- 62. Foley TP Jr., Harrison HC, Arnaud CD, Harrison HE. Familial benign hypercalcemia. J Pediatr 1972;81(6):1060-7.
- 63. Marx SJ, Attie MF, Levine MA, Spiegel AM, Downs RW Jr., Lasker RD. The hypocalciuric or benign variant of familial hypercalcemia: Clinical and biochemical features in fifteen kindreds. Medicine (Baltimore) 1981;60(6):397-412.
- 64. Law WM Jr., Heath HD. Familial benign hypercalcemia (hypocalciuric hypercalcemia). Clinical and pathogenetic studies in 21 families. Ann Intern Med 1985;102(4):511-9.
- 65. Pidasheva S, Canaff L, Simonds WF, Marx SJ, Hendy GN. Impaired co-translational processing of the calcium-sensing receptor due to signal peptide missense mutations in familial hypocalciuric hypercalcemia. Hum Mol Genet 2005;14(12):1679-90.
- 66. Warner J, Epstein M, Sweet A, Singh D, Burgess J, Stranks S, et al. Genetic testing in familial isolated hyperparathyroidism: Unexpected results and their implications. J Med Genet 2004;41(3):155-60.
- 67. Hendy GN, D'Souza-Li L, Yang B, Canaff L, Cole DE. Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat 2000;16(4):281-96.
- 68. Pollak MR, Chou YH, Marx SJ, Steinmann B, Cole DE, Brandi ML, et al. Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Effects of mutant gene dosage on phenotype. J Clin Invest 1994;93(3):1108-12.
- 69. Chou YH, Brown EM, Levi T, Crowe G, Atkinson AB, Arnqvist HJ, et al. The gene responsible for familial hypocalciuric hypercalcemia maps to chromosome 3q in four unrelated families. Nat Genet 1992;1(4):295-300.
- 70. Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, et al. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 1993;75(7):1297-303.
- 71. Chou YH, Pollak MR, Brandi ML, Toss G, Arnqvist H, Atkinson AB, et al. Mutations in the human Ca(2+)-sensing-receptor gene that cause familial hypocalciuric hypercalcemia. Am J Hum Genet 1995;56(5):1075-9.
- 72. Heath HD, Jackson CE, Otterud B, Leppert MF. Genetic linkage analysis in familial benign (hypocalciuric) hypercalcemia: Evidence for locus heterogeneity. Am J Hum Genet 1993;53(1):193-200.
- 73. Lloyd SE, Pannett AA, Dixon PH, Whyte MP, Thakker RV. Localization of familial benign hypercalcemia, Oklahoma variant (FBHOk), to chromosome 19q13. Am J Hum Genet 1999;64(1):189-95.
- 74. Brown EM. Editorial: Mutant extracellular calcium-sensing receptors and severity of disease. J Clin Endocrinol Metab 2005;90(2):1246-8.
- 75. Spiegel AM. Mutations in G proteins and G protein-coupled receptors in endocrine disease. J Clin Endocrinol Metab 1996;81(7):2434-42.
- 76. Pratt EL, Geren BB, Neuhauser EBD. Hypercalcemia and idiopathic hyperplasia of parathyroid glands in an infant. J Pediat 1947;30:388-99.
- 77. Philips R. Primary diffuse parathyroid hyperplasia in an infant of four months. Pediatrics 1948;2:428-34.
- 78. Pearce SH, Bai M, Quinn SJ, Kifor O, Brown EM, Thakker RV. Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest 1996;98(8):1860-6.
- 79. Bai M, Pearce SH, Kifor O, Trivedi S, Stauffer UG, Thakker RV, et al. In vivo and in vitro characterization of neonatal hyperparathyroidism resulting from a de novo, heterozygous mutation in the Ca2+-sensing receptor gene: Normal maternal calcium homeostasis as a cause of secondary hyperparathyroidism in familial benign hypocalciuric hypercalcemia. J Clin Invest 1997;99 (1):88-96.
- 80. Marx SJ, Fraser D, Rapoport A. Familial hypocalciuric hypercalcemia. Mild expression of the gene in heterozygotes and severe expression in homozygotes. Am J Med 1985;78(1):15-22.
- 81. Harris SS, D'Ercole AJ. Neonatal hyperparathyroidism: The natural course in the absence of surgical intervention. Pediatrics 1989;83(1):53-6.
- 82. Aida K, Koishi S, Inoue M, Nakazato M, Tawata M, Onaya T. Familial hypocalciuric hypercalcemia associated with mutation in the human Ca(2+)-sensing receptor gene. J Clin Endocrinol Metab 1995;80(9):2594-8.
- 83. Winter WE, Silverstein JH, Maclaren NK, Riley WJ, Chiaro JJ. Autosomal dominant hypoparathyroidism with variable, age-dependent severity. J Pediatr 1983;103 (3):387-90.
- 84. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med 1996;335(15):1115-22.
- 85. Finegold DN, Armitage MM, Galiani M, Matise TC, Pandian MR, Perry YM, et al. Preliminary localization of a gene for autosomal dominant hypoparathyroidism to chromosome 3q13. Pediatr Res 1994;36(3):414-7.
- 86. Pollak MR, Brown EM, Estep HL, McLaine PN, Kifor O, Park J, et al. Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat Genet 1994;8(3):303-7.
- 87. Heath D. Familial hypocalcemia not hypoparathyroidism. N Engl J Med 1996;335(15):1144-5.
- 88. Sato K, Hasegawa Y, Nakae J, Nanao K, Takahashi I, Tajima T, et al. Hydrochlorothiazide effectively reduces urinary calcium excretion in two Japanese patients with gain-of-function mutations of the calcium-sensing receptor gene. J Clin Endocrinol Metab 2002;87(7):3068-73.
- 89. Mittelman SD, Hendy GN, Fefferman RA, Canaff L, Mosesova I, Cole DE, et al. A hypocalcemic child with a novel activating mutation of the calcium-sensing receptor gene: Successful treatment with recombinant human parathyroid hormone. J Clin Endocrinol Metab 2006;91(7):2474-9.
- 90. Vargas-Poussou R, Huang C, Hulin P, Houillier P, Jeunemaitre X, Paillard M, et al. Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J Am Soc Nephrol 2002;13(9):2259-66.
- 91. Watanabe S, Fukumoto S, Chang H, Takeuchi Y, Hasegawa Y, Okazaki R, et al. Association between activating mutations of calcium-sensing receptor and Bartter's syndrome. Lancet 2002;360(9334):692-4.
- 92. Zhao XM, Hauache O, Goldsmith PK, Collins R, Spiegel AM. A missense mutation in the seventh transmembrane domain constitutively activates the human Ca2+ receptor. FEBS Lett 1999;448(1):180-4.
- 93. Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med 1990;322(26):1829-36.
- 94. Li Y, Song YH, Rais N, Connor E, Schatz D, Muir A, et al. Autoantibodies to the extracellular domain of the calcium sensing receptor in patients with acquired hypoparathyroidism. J Clin Invest 1996;97(4):910-4.
- 95. Pallais JC, Kifor O, Chen YB, Slovik D, Brown EM. Acquired hypocalciuric hypercalcemia due to autoantibodies against the calcium-sensing receptor. N Engl J Med 2004;351(4):362-9.
- 96. Pidasheva S, D'Souza-Li L, Canaff L, Cole DE, Hendy GN. CASRdb: Calcium-sensing receptor locus-specific database for mutations causing familial (benign) hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat 2004;24(2):107-11.
- 97. Hu J, McLarnon SJ, Mora S, Jiang J, Thomas C, Jacobson KA, et al. A region in the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+ J Biol Chem 2005;280(6):5113-20.
- 98. D'Souza-Li L, Yang B, Canaff L, Bai M, Hanley DA, Bastepe M, et al. Identification and functional characterization of novel calcium-sensing receptor mutations in familial hypocalciuric hypercalcemia and autosomal dominant hypocalcemia. J Clin Endocrinol Metab 2002;87(3):1309-18.
- 99. Lienhardt A, Garabedian M, Bai M, Sinding C, Zhang Z, Lagarde JP, et al. A large homozygous or heterozygous in-frame deletion within the calcium-sensing receptor's carboxylterminal cytoplasmic tail that causes autosomal dominant hypocalcemia. J Clin Endocrinol Metab 2000;85(4):1695-702.
- 100.Zhang M, Breitwieser GE. High affinity interaction with filamin A protects against calcium-sensing receptor degradation. J Biol Chem 2005;280(12):11140-6.
- 101.Huang Y, Niwa JI, Sobue G, Breitwieser GE. Calcium sensing receptor ubiquitination and degradation mediated by the E3 ubiquitin ligase dorfin. J Biol Chem 2006
- 102.Janicic N, Pausova Z, Cole DE, Hendy GN. Insertion of an Alu sequence in the Ca(2+)-sensing receptor gene in familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Am J Hum Genet 1995;56(4):880-6.
- 103.D'Souza-Li L, Canaff L, Janicic N, Cole DE, Hendy GN. An acceptor splice site mutation in the calcium-sensing receptor (CASR) gene in familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Hum Mutat 2001;18(5):411-21.
- 104.Pearce SH, Trump D, Wooding C, Besser GM, Chew SL, Grant DB, et al. Calcium-sensing receptor mutations in familial benign hypercalcemia and neonatal hyperparathyroidism. J Clin Invest 1995;96(6):2683-92.
- 105.Lovlie R, Eiken HG, Sorheim JI, Boman H. The Ca(2+)-sensing receptor gene (PCAR1) mutation T151M in isolated autosomal dominant hypoparathyroidism. Hum Genet 1996;98(2):129-33.
- 106.Koishi S, Aida K, Tawata M, Onaya T. Polymorphism of the human Ca(2+)-sensing receptor gene in Japanese individuals: No relation to non-insulin dependent diabetes mellitus. Horm Metab Res 1996;28(10):541-4.
-
107Mutational analysis of the extracellular calcium-sensing receptor gene in human parathyroid tumors. 1997;97.
- 108.Cetani F, Pinchera A, Pardi E, Cianferotti L, Vignali E, Picone A, et al. No evidence for mutations in the calcium-sensing receptor gene in sporadic parathyroid adenomas [In Process Citation]. J Bone Miner Res 1999;14 (6):878-82.
- 109.Baron J, Winer KK, Yanovski JA, Cunningham AW, Laue L, Zimmerman D, et al. Mutations in the Ca(2+)-sensing receptor gene cause autosomal dominant and sporadic hypoparathyroidism. Hum Mol Genet 1996;5 (5):601-6.
- 110.Scillitani A, Guarnieri V, De Geronimo S, Muscarella LA, Battista C, D'Agruma L, et al. Blood ionized calcium is associated with clustered polymorphisms in the carboxyl-terminal tail of the calcium-sensing receptor. J Clin Endocrinol Metab 2004;89(11):5634-8.
- 111.Cole DE, Peltekova VD, Rubin LA, Hawker GA, Vieth R, Liew CC, et al. A986S polymorphism of the calcium-sensing receptor and circulating calcium concentrations [see comments]. Lancet 1999;353(9147):112-5.
- 112.Nemeth EF, Steffey ME, Hammerland LG, Hung BC, Van Wagenen BC, DelMar EG, et al. Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc Natl Acad Sci USA 1998;95(7):4040-5.
- 113.Nagano N, Nemeth EF. Functional proteins involved in regulation of intracellular Ca(2+) for drug development: The extracellular calcium receptor and an innovative medical approach to control secondary hyperparathyroidism by calcimimetics. J Pharmacol Sci 2005;97(3):355-60.
- 114.Hammerland LG, Garrett JE, Hung BCP, Levinthal C, Nemeth EF. Allosteric activation of the Ca2+ receptor expressed in Xenopus laevis oocytes by NPS 467 or NPS 568. Mol Pharmacol 1998;53(6):1083-8.
- 115.Petrel C, Kessler A, Dauban P, Dodd RH, Rognan D, Ruat M. Positive and negative allosteric modulators of the Ca2+-sensing receptor interact within overlapping but not identical binding sites in the transmembrane domain. J Biol Chem 2004;279(18):18990-7.
- 116.Block GA, Martin KJ, de Francisco AL, Turner SA, Avram MM, Suranyi MG, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med 2004;350(15):1516-25.
- 117.Nemeth EF, DelMar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL, et al. Calcilytic compounds: Potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther 2001;299(1):323-31.
- 118.Arey BJ, Seethala R, Ma Z, Fura A, Morin J, Swartz J, et al. A novel calcium-sensing receptor antagonist transiently stimulates parathyroid hormone secretion in vivo. Endocrinology 2005;146(4):2015-22.
Publication Dates
-
Publication in this collection
14 Nov 2006 -
Date of issue
Aug 2006
History
-
Received
08 Mar 2006 -
Accepted
20 Mar 2006