Resumos
Neste artigo, descrevemos o caso de um paciente com diagnóstico de macroprolactinoma, que apresentava valores discordantes entre IGF-1 (elevado) e GH (hormônio de crescimento) pós-teste de tolerância oral à glicose (normal) realizados para pesquisa de co-secreção de GH pelo tumor. Houve normalização dos níveis de prolactina após uso de bromocriptina, porém, durante o acompanhamento, o paciente persistiu com níveis elevados de IGF-1 sem clínica aparente, sugerindo tratar-se de possível forma subclínica de acromegalia. Após o desenvolvimento de novos métodos laboratoriais, mais sensíveis, para a dosagem de GH, casos de discordância entre esses testes vêm sendo observados, levando alguns autores a sugerir que o nadir de GH pós-teste de tolerância à glicose oral (TTGO) para diagnóstico e tratamento da acromegalia pode ser bem menor do que o limite considerado atualmente (1,2). Assim, se isto for confirmado, casos subclínicos ou oligossintomáticos de acromegalia serão diagnosticados mais precocemente.
Acromegalia; Prolactinoma; Hormônio de crescimento; IGF-1
We describe a patient with macroprolactinoma and discrepant insulin-like growth factor (IGF-1) concentration (elevated) and growth hormone (GH) values during a 75 g oral glucose tolerance test (normal), that were measured to evaluate the co-secretion of GH by tumor. With the bromocriptin use, the patient achieved normalization of prolactin, but persisted with high levels of IGF1, suggesting to be subclinical acromegaly. After the development of new more sensitive GH assays, cases of discrepant GH and IGF-1 results have been observed and taken to some authors to suggest that GH nadir concentration during 75 g OGTT used to acromegaly diagnosis and treatment could be lower than values considered currently normal. Thus, if this is confirmed, subclinical and oligosymptomatic acromegaly cases could have earlier diagnoses.
Acromegaly; Prolactinoma; Growth hormone; IGF-1
APRESENTAÇÃO DE CASO
Discordância entre IGF-1 e GH pós-sobrecarga de glicose no rastreamento de acromegalia em paciente com macroprolactinoma relato de caso e revisão sobre o tema
Discrepancy between IGF-1 and GH during 75 g oral glucose tolerance test measures to acromegaly screening in a patient with macroprolactinoma case report and literature review
Fabrícia Torres Gonçalves; Taciana C. Maia Feibelmann; Maria Luiza M. P. Fernandes; Alessandra R.C.Fonseca; Henrique Pierotti Arantes; Paulo Tannús Jorge
Serviço de Endocrinologia do Hospital de Clínicas da Universidade Federal de Uberlândia, MG
Endereço para correspondência Endereço para correspondência: Paulo Tannús Jorge Rua Geraldo Morais 1115, ap. 403 38400-020 Uberlândia, MG E-mail: pauloj@triang.com.br
RESUMO
Neste artigo, descrevemos o caso de um paciente com diagnóstico de macroprolactinoma, que apresentava valores discordantes entre IGF-1 (elevado) e GH (hormônio de crescimento) pós-teste de tolerância oral à glicose (normal) realizados para pesquisa de co-secreção de GH pelo tumor. Houve normalização dos níveis de prolactina após uso de bromocriptina, porém, durante o acompanhamento, o paciente persistiu com níveis elevados de IGF-1 sem clínica aparente, sugerindo tratar-se de possível forma subclínica de acromegalia. Após o desenvolvimento de novos métodos laboratoriais, mais sensíveis, para a dosagem de GH, casos de discordância entre esses testes vêm sendo observados, levando alguns autores a sugerir que o nadir de GH pós-teste de tolerância à glicose oral (TTGO) para diagnóstico e tratamento da acromegalia pode ser bem menor do que o limite considerado atualmente (1,2). Assim, se isto for confirmado, casos subclínicos ou oligossintomáticos de acromegalia serão diagnosticados mais precocemente.
Descritores: Acromegalia; Prolactinoma; Hormônio de crescimento; IGF-1
ABSTRACT
We describe a patient with macroprolactinoma and discrepant insulin-like growth factor (IGF-1) concentration (elevated) and growth hormone (GH) values during a 75 g oral glucose tolerance test (normal), that were measured to evaluate the co-secretion of GH by tumor. With the bromocriptin use, the patient achieved normalization of prolactin, but persisted with high levels of IGF1, suggesting to be subclinical acromegaly. After the development of new more sensitive GH assays, cases of discrepant GH and IGF-1 results have been observed and taken to some authors to suggest that GH nadir concentration during 75 g OGTT used to acromegaly diagnosis and treatment could be lower than values considered currently normal. Thus, if this is confirmed, subclinical and oligosymptomatic acromegaly cases could have earlier diagnoses.
Keywords: Acromegaly; Prolactinoma; Growth hormone; IGF-1
A CO-SECREÇÃO DE HORMÔNIO de crescimento deve ser pesquisada em todos os pacientes com prolactinoma, e o GH após teste de tolerância oral à glicose (TTGO) com 75 g é considerado o padrão-ouro para o diagnóstico e seguimento da acromegalia (3). No entanto, casos de acromegalia com GH pós-teste de supressão normal e IGF-1 elevado têm sido descritos (1,2). Neste artigo, descrevemos um caso de um paciente com diagnóstico de macroprolactinoma que apresentava valores discordantes entre IGF-1 e GH pós-TTGO realizados para pesquisa de co-secreção de hormônio de crescimento.
CASO CLÍNICO
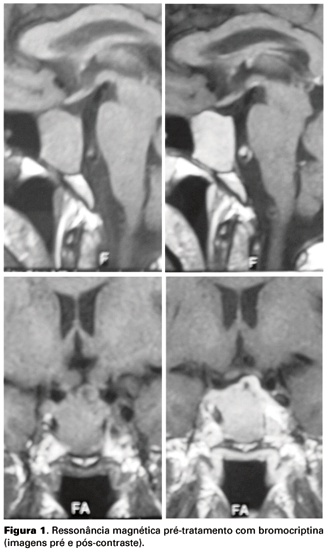
Paciente do sexo masculino, 54 anos, foi admitido com clínica de cefaléia crônica e diminuição de libido há um ano de evolução. Sem queixas no campo visual. Antecedentes pessoais: hipertensão arterial sistêmica (há 12 anos) e litíase renal de repetição. Antecedentes familiares: filho e irmã com litíase renal; pai diabético e hipertenso falecido por AVC e mãe falecida por infarto do miocárdio aos 55 anos. Ao exame: fácies com traços grosseiros, mas não característicos de acromegalia. Comparação com fotos antigas não mostrava mudanças significativas. Tireóide normal à palpação. Trazia exames: prolactina 2.969 ng/ml, campimetria sem alterações e imagem em ressonância nuclear magnética (RNM) mostrando volumoso adenoma hipofisário com dimensões de 3,0 cm x 2,9 cm x 1,8 cm, invadindo seio esfenoidal e cavernoso à direita e comprimindo quiasma óptico (figura 1). O paciente foi internado para investigação diagnóstica e, no quarto dia de internação, em uso de bromocriptina 1,25 mg/d, apresentou quadro de cefaléia intensa holocraniana, porém sem sinais neurológicos. Tomografia computadorizada (TC) de sela sem contraste mostrava apenas um adenoma gigante da hipófise. No entanto, em TC realizada 1 ano depois, havia realce heterogêneo intra-selar compatível com áreas de necrose. Diante de boa evolução clínica nos dias subseqüentes, teve alta hospitalar com bromocriptina, usando doses progressivas até 10 mg ao dia. A evolução dos exames laboratoriais e de doses da medicação estão dispostas nas tabelas 1 e 2. A RNM pós-tratamento com bromocriptina mostrou redução importante do tamanho do tumor (figura 2). O paciente encontrava-se em acompanhamento ambulatorial com normalização dos níveis de prolactina, quando, 2 anos após admissão inicial no Serviço, teve morte súbita, em domicílio.
DISCUSSÃO
A co-secreção de hormônio de crescimento deve ser pesquisada em todos os pacientes com macroprolactinomas, devido à possibilidade, não rara, de tumor misto.
A dosagem do GH basal < 0,4 ng/ml (métodos imunométricos) e IGF-1 sérico normal para idade e sexo (método imunorradiométrico) exclui o diagnóstico de acromegalia, estando indicada dosagem de GH no TTGO caso um desses valores esteja alterado (3,4). Segundo o Guideline para Diagnóstico e Tratamento da Acromegalia, 2004, da AACE, nos últimos 40 anos, a medida do GH pós-TTGO com 75 g tem sido considerada o exame padrão-ouro para o diagnóstico e seguimento dos pacientes com acromegalia, considerando um teste alterado a não supressão do GH para níveis abaixo de 1 ng/dl; apesar disso, os autores deixam evidente que o diagnóstico deve ser feito em conjunto com a clínica e os valores séricos de IGF-1 (3). O IGF-1 tem sido considerado um importante parâmetro de atividade de acromegalia por ter boa correlação com os níveis médios de GH de 24 h e com as manifestações clínicas da doença, além disso, a sua normalização está associada a melhora nas taxas de sobrevida dos pacientes tratados (5).
No entanto, os casos de acromegalia com teste de supressão normal e IGF-1 elevado descritos na literatura têm levantado algumas questões sobre o tema. No trabalho de Dimaraki e cols., 16 pacientes foram avaliados devido a níveis plasmáticos médios de GH em 24 h normais, IGF-1 elevado e alterações clínicas leves. Os pacientes apresentavam um nadir de 24 h mais elevado e com um padrão mais tônico do que pulsátil em relação a um grupo controle, e 8 suprimiram o GH para níveis < 1 ng/ml pós-TTGO. A maioria apresentava microadenoma (10 de 16 pacientes), havendo melhora clínica após tratamento cirúrgico. Os autores acreditam que a contínua exposição a níveis tônicos de GH minimamente elevados ou uma maior sensibilidade tecidual ao GH seriam suficientes para levar a uma produção supra-normal de IGF-1, e concluem que, apesar de ser um bom teste, os valores do GH durante TTGO devem ser revisados (2). Freda e cols., em estudo de pacientes pós-cirúgicos, mostraram que a supressão do GH pós-TTGO para níveis menores do que 1 ng/ml não indica necessariamente uma dinâmica de secreção normal de GH, e também sugerem que o ponto de corte atual do GH durante o TTGO pode ser alto para o diagnóstico e seguimento dos pacientes com acromegalia (1).
Minuto e col. compararam dois valores de corte, 1 µg/l e 0,3 µg/l, de GH durante TTGO e IGF-1 e concluem que se o GH for menor que 1 µg/l no tempo 120 minutos pós-TTGO e os níveis de IGF-1 estão normais, o paciente pode ser considerado "controlado" ou "não-ativo"; enquanto que níveis de IGF-1 elevados é um sinal de doença ativa independente dos valores de GH (6).
Outros autores levantam a possibilidade de problemas metodológicos serem os responsáveis pela discrepância entre GH e IGF-1 (7,8). Os novos ensaios utilizados para dosagem do GH (por métodos imunométricos) são mais sensíveis em relação aos antigos radioimunoensaios (RAI), permitindo maior acurácia na detecção do limite inferior desse hormônio. Devido a essa mudança metodológica, os valores de normalidade de GH vêm historicamente reduzindo e pode ser que os níveis atualmente aceitos ainda não sejam os mais apropriados (8). Por outro lado, a maioria dos ensaios imunométricos é direcionada para a isoforma de GH 22 kDa, deixando de detectar outras isoformas que também podem ser bioativas. Na acromegalia, pode ocorrer uma proporção aumentada das isoformas não-22 kDa do GH e presença de moléculas anormais ou "GH-like". Boguszewski e col. observaram uma alta proporção de moléculas não-22 kDa em pacientes pós-cirúrgicos e sugerem que as diferenças biológicas e de imunorreatividade destas isoformas podem ser responsáveis pela ausência de correlação GH/IGF1 nesses pacientes. Também houve, neste estudo, uma correlação direta das formas não-22 kDa com os níveis de prolactina em pacientes acromegálicos não-tratados, e os autores afirmam que pacientes que secretam mais GH e prolactina parecem secretar mais isoformas não-22 kDa (7).
O último Statement from a Joint Consensus Conference of the Growth Hormone Research Society and The Pituitary Society (2004), considerando os aspectos acima discutidos, não estabelece uma definição bioquímica de cura em pacientes acromegálicos tratados. De acordo com os autores, uma secreção normal de GH pode corresponder a valores de GH pós-TTGO tão baixos quanto 0,3 µg/l medido por ensaios altamente sensíveis (atualmente utilizados), em adição à dosagem de IGF-1 dentro da variação normal ajustada para sexo e idade (8).
No entanto, os dados sobre níveis de GH pós-TTGO e IGF-1 especificamente em pacientes com macroprolactinomas, ou seja, possíveis tumores co-secretores, são mais escassos. Kageyiama e cols. descreveram um caso de prolactinoma com produção de hormônio de crescimento subclínica, que apresentava níveis de prolactina e IGF-1 aumentados, mas GH basal normal, com elevação após teste do TRH. No entanto, tratava-se de paciente com níveis glicêmicos alterados, diferentemente do caso que estamos aqui relatando, o que poderia ter afetado a dosagem do GH. A imuno-histoquímica e hibridização in situ revelaram um tumor com células que secretavam ao mesmo tempo prolactina e GH (adenoma de células acidofílicas) (9). Os adenomas que co-secretam prolactina e GH podem ser de três tipos histológicos: os mamossomatotróficos, que têm crescimento lento e apresentam imuno-reatividade intensa para GH e variável para prolactina e cujo quadro clínico é tipicamente de acromegalia com hiperprolactinemia leve a moderada; os adenomas que possuem lactotrofos e somatotrofos, que também possuem quadro clínico evidente de acromegalia e graus variados de hiperprolactinemia, porém são muito invasivos em 30% dos casos; e os adenomas de células acidofílicas, que são tumores de células imaturas que secretam GH e prolactina, habitualmente são macroprolactinomas invasivos com grande grau de proliferação, onde predominam os sintomas decorrentes da hiperprolactinemia, com poucos efeitos decorrentes do GH e até mesmo ausência de acromegalia clássica (4,10).
No trabalho de Andersen e cols., entre 78 pacientes com prolactinoma acompanhados por um período de 5 anos, 3 desenvolveram sinais clínicos e laboratoriais de acromegalia, 2 tiveram apenas diagnóstico laboratorial e 3 apresentaram elevação isolada do IGF-1 (11). Estes resultados demonstram que, durante o seguimento de pacientes com prolactinoma, acromegalia clínica ou subclínica pode se desenvolver, sendo importante realizar o rastreamento de hipersecreção de GH periodicamente, mesmo diante de uma investigação inicial negativa.
No caso relatado, algumas questões precisam ser discutidas. Primeiro, poderia tratar-se de um tumor de células acidofílicas co-secretor, no entanto apenas hipóteses serão feitas sobre o tipo histológico tumoral, visto que o paciente não teve indicação cirúrgica. Segundo, acreditamos que tenha existido apoplexia tumoral no quarto dia após iniciada a dose da bromocriptina, que, por ter se tratado de uma dose inicial pequena (meio comprimido de 2,5 mg por dia), parece mais uma coincidência temporal com início do tratamento. Os dados que nos fazem acreditar em apoplexia são a clínica sugestiva, as imagens analisadas e os baixíssimos níveis atuais de prolactina (2,0 ng/ml) em uso de dose pequena de bromocriptina (2,5 mg/d). Por fim, apesar de suprimir o GH durante o teste do TTGO em duas ocasiões e de uma clínica não muito compatível com acromegalia, o paciente tinha dados que corroboravam com o diagnóstico de hipersecreção de GH visto que apresentava níveis persistentemente muito altos de IGF-1.
A última questão a ser avaliada seria qual a melhor opção de tratamento diante do caso apresentado: o impacto clínico do excesso de GH não foi evidente, portanto não justificaria uma indicação cirúrgica, além disso tratava-se de um tumor co-secretor de prolactina, cujas opções de terapia farmacológica apresentam bons resultados. A bromocriptina foi o primeiro agonista dopaminérgico utilizado no tratamento da acromegalia (10), no entanto as doses necessárias para o controle do excesso de GH são muito altas e trata-se de medicamento com muitos efeitos colaterais quando utilizado em doses elevadas. A cabergolina é uma opção terapêutica nos pacientes com acromegalia, cujos melhores resultados foram obtidos com pacientes co-secretores de GH e prolactina, com níveis de IGF-1 < 750 ng/ml (12) e poderia ter sido a escolha mais adequada para o paciente. Em caso de resistência uso deste agonista dopaminérgico, bioquímica ou pela não redução do tamanho tumoral, a associação com análogos da somatostatina poderia ser uma alternativa (13).
CONCLUSÕES
A presença de doença ativa deve ser considerada nos pacientes com níveis altos de IGF-1, mesmo se suprimem o GH pós-sobrecarga de glicose. Os casos de discrepância entre estes dois exames podem alertar para o fato de que o ponto de corte do nadir de GH pós-TTGO deva ser reavaliado. No entanto, estudos são necessários para avaliar o real impacto clínico da doença nos pacientes com aumento isolado do IGF-1, assim como qual a melhor conduta terapêutica nesses casos.
Recebido em 18/01/06
Revisado em 12/07/06
Aceito em 22/09/06
- 1. Freda PU, Post KD, Powell JS, Wardlaw SL. Evaluation of disease status with sensitive measures of growth hormone secretion in 60 postoperative patients with acromegaly. J Clin Endocrinol Metab 1998;83(11):3808-16.
- 2. Dimaraki EV, Jaffe CA, DeMott-Friberg R, Chandler WF, Barkan AL. Acromegaly with apparently normal GH secretion: implications for diagnosis and folow-up. J Clin Endocrinol Metab 2002;87(8):3537-42.
- 3. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the Diagnosis and Treatment of Acromegaly. Endocr Pract 2004;10(2):214-25.
- 4. Donangelo I, Une K, Gadelha M. Diagnóstico e tratamento da acromegalia no Brasil. Arq Bras Endocrinol Metab 2003;47(4):331-46.
- 5. Swearingen B, Barker FG 2nd, Katznelson L, Biller BM, Grinspoon S, Klibanski A, et al. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab 1998;83(10):3419-26.
- 6. Minuto F, Resmini E, Boschetti M, Arvigo M, Sormani MP, Giusti M, et al. Assessment of disease activity in acromegaly by means of a single blood sample: comparison of the 120th minute post glucose value with spontaneous GH secretion and with the IGF system. Clin Endocrinol (Oxf) 2004;61(1):138-44.
- 7. Boguszewski CL, Johannsson G, Bengtsson B, Johansson A, Carlsson B, Carlsson LMS. Circulating non-22-kilodalton growth hormone isoforms in acromegaly men before and after transsphenoidal surgery. J Clin Endocrinol Metab 1997;82(5):1516-21.
-
8Biochemical assessment and long-term monitoring in patients with acromegaly: statement from a Joint Consensus Conference of a Growth Hormone Research Society and the Pituitary Society. J Clin Endocrinol Metab 2004;89:3099-102.
- 9. Kageyama K, Nigawara T, Kamata Y, Takahashi T, Anzai J, Suzuki S, et al. A case of macroprolactinoma with subclinical growth hormone production. Endocr J 2002;49(1):41-7.
- 10. Czepielewski MA, Rollin GAFS. Acromegalia e gigantismo. In: Liberman B, Cukiert A (eds). Fisiologia e Fisiopatologia do Hormônio de Crescimento São Paulo: Lemos editorial, 2004 pp. 337-60.
- 11. Andersen M, Hagen C, Frystyk J, Schroeder HD, Hagen C. Development of acromegaly in patients with prolactinomas. Eur J Endocrinol 2003;149:17-22.
- 12. Abs R, Verhelst J, Maiter D, Acker KV, Nobels F, Coolens J, et al. Cabergoline in the treatment of acromegaly: a study of 64 patients. J Clin Endocrinol Metab 1998;83(2):374-8.
- 13. Ferone D, Pivonello R, Lastoria S, Faggiano A, Del Basso de Caro ML, Cappabianca P. In vivo and in vitro effects of octreotide, quinagolide and cabergoline in four hyperprolactinaemic acromegalics: correlation with somatostatin and dopamine D2 receptor scintigraphy. Clin Endocrinol (Oxf) 2001;54(4):469-77.
Datas de Publicação
-
Publicação nesta coleção
31 Maio 2007 -
Data do Fascículo
Abr 2007
Histórico
-
Aceito
22 Set 2006 -
Revisado
12 Jul 2006 -
Recebido
18 Jan 2006