Resumos
O diabetes melito e suas complicações apresentam origem multifatorial. Mecanismos bioquímicos e patológicos estão associados com hiperglicemia crônica no diabetes e o aumento do estresse oxidativo tem sido postulado com papel central nestas desordens. Evidências sugerem que a lesão celular oxidativa causada pelos radicais livres contribuem para o desenvolvimento das complicações no diabetes tipo 1 (DM1) e a diminuição das defesas antioxidantes (enzimáticas e não-enzimáticas) parecem correlacionar-se com a gravidade das alterações patológicas no DM1. Nesta revisão, relata-se como o estresse oxidativo pode exercer efeitos deletérios no diabetes e são apresentadas as opções terapêuticas em estudo para modulação da injúria vascular.
Diabetes tipo 1; Estresse oxidativo
Diabetic complications appear to be multifactorial process. The biochemical and pathological mechanisms are associated with chronic hyperglycemia of diabetes and the increased oxidative stress which has been postulated to play a central role in these disorders. Accumulating evidence suggests that oxidative cell injury caused by free radicals contributes to the development of type 1 diabetes (DM1) complications and decreased efficiency of antioxidant defenses (both enzymatic and nonenzymatic) seems to correlate with the severity of pathological tissue changes in DM1. In this review, we report as oxidative stress may exert deleterious effects in diabetes, as well as address current strategies in study to down-regulating vascular injury.
Type 1 diabetes; Oxidative stress
REVISÃO
Estresse oxidativo: revisão da sinalização metabólica no diabetes tipo 1
Oxidative stress: a review on metabolic signaling in type 1 diabetes
Janice Sepúlveda ReisI, II; Clara Araújo VelosoI; Rafael Teixeira MattosI; Saulo PurishII; José Augusto Nogueira-MachadoI
INúcleo de Pesquisa e Pós-graduação (NPPG) da Santa Casa de Belo Horizonte, Belo Horizonte, MG
IICentro de Pesquisa em Endocrinologia da Santa Casa de Belo Horizonte, Belo Horizonte, MG
Endereço para correspondência Endereço para correspondência: Janice Sepúlveda Reis Centro de Estudos e Pesquisa da Clínica de Endocrinologia e Metabologia (CEPCEM) Av. Francisco Sales, 1111, 5º andar, ala D - Santa Efigênia 30150-221, Belo Horizonte MG E-mail: janicesepulveda@terra.com.br
RESUMO
O diabetes melito e suas complicações apresentam origem multifatorial. Mecanismos bioquímicos e patológicos estão associados com hiperglicemia crônica no diabetes e o aumento do estresse oxidativo tem sido postulado com papel central nestas desordens. Evidências sugerem que a lesão celular oxidativa causada pelos radicais livres contribuem para o desenvolvimento das complicações no diabetes tipo 1 (DM1) e a diminuição das defesas antioxidantes (enzimáticas e não-enzimáticas) parecem correlacionar-se com a gravidade das alterações patológicas no DM1. Nesta revisão, relata-se como o estresse oxidativo pode exercer efeitos deletérios no diabetes e são apresentadas as opções terapêuticas em estudo para modulação da injúria vascular.
Descritores: Diabetes tipo 1; Estresse oxidativo
ABSTRACT
Diabetic complications appear to be multifactorial process. The biochemical and pathological mechanisms are associated with chronic hyperglycemia of diabetes and the increased oxidative stress which has been postulated to play a central role in these disorders. Accumulating evidence suggests that oxidative cell injury caused by free radicals contributes to the development of type 1 diabetes (DM1) complications and decreased efficiency of antioxidant defenses (both enzymatic and nonenzymatic) seems to correlate with the severity of pathological tissue changes in DM1. In this review, we report as oxidative stress may exert deleterious effects in diabetes, as well as address current strategies in study to down-regulating vascular injury.
Keywords: Type 1 diabetes; Oxidative stress
INTRODUÇÃO
O diabetes melito (DM) é uma doença multifatorial associada ao aumento no risco cardiovascular quando comparado a não-diabéticos (1). Os resultados do The Diabetes Control and Complications Trial Research Group (DCCT) (2) e do UK Prospective Diabetes Study (UKPDS) (3) demonstraram relação direta entre hiperglicemia cronicamente mantida e as complicações micro e macrovasculares. Entretanto, hiperglicemia crônica ou intermitente tem sido identificada na patogênese da lesão endotelial no diabetes (4) e, em particular, no diabetes tipo 1 (DM1), disfunção endotelial tem sido demonstrada mesmo quando a normoglicemia é alcançada (5,6) sendo sugerido que o estresse oxidativo tenha papel central na patogênese das complicações do diabetes (7).
O estresse oxidativo é um estado de desequilíbrio entre a produção de espécies reativas de oxigênio (ROS) e a capacidade antioxidante endógena (8) e o seu papel como determinante principal do início e da progressão das complicações cardiovasculares associadas ao DM tem sido alvo de grande interesse.
Mecanismos bioquímicos têm sido propostos para explicar as anormalidades estruturais e funcionais associadas com a exposição prolongada dos tecidos vasculares à hiperglicemia com indícios de que a capacidade antioxidante endógena esteja prejudicada nos indivíduos diabéticos,dificultando a remoção dos radicais livres (9).
Neste artigo foram revisados os mecanismos patogênicos relacionados ao estresse oxidativo no diabetes, com ênfase nos conhecimentos recentes em relação ao DM1 e aos avanços terapêuticos direcionados ao mecanismo celular de injúria vascular.
ALTERAÇÕES BIOQUÍMICAS
Espécies reativas de oxigênio
As espécies reativas de oxigênio são átomos, íons ou moléculas que contêm oxigênio com um elétron não-pareado em sua órbita externa. São caracterizadas por grande instabilidade e elevada reatividade e tendem a ligar o elétron não-pareado com outros presentes em estruturas próximas de sua formação, comportando-se como receptores (oxidantes) ou doadores (redutores) de elétrons. Os radicais livres podem reagir com as principais classes de biomoléculas, sendo os lipídeos os mais suscetíveis (10) (Tabela 1).
Aumento da atividade da aldose redutase - via dos polióis
A hiperglicemia, com conseqüente aumento de ROS, reduz os níveis de óxido nítrico (NO) ativando a aldose redutase. O aumento do fluxo pela via dos polióis, induzido pelo aumento de ROS, determina maior conversão de glicose a sorbitol, reduzindo NADPH e glutationa (antioxidante intracelular) e aumentando o estresse oxidativo induzido pela hiperglicemia. O sorbitol é convertido à frutose, resultando aumento da relação NADH: NAD+, o que aumentaria a síntese "de novo" de diacilglicerol (DAG), principal ativador fisiológico da proteína kinase C (PKC) (Figura 1) (11).
Formação de produtos avançados da glicosilação não-enzimática (AGEs)
Os AGEs são proteínas ou lipídios que se tornam glicados após a exposição a açúcares oxidados e contribuem para o desenvolvimento de arteriosclerose. Acredita-se que atuem modificando (12,13): proteínas intracelulares envolvidas na regulação gênica; moléculas da matriz extracelular vizinha, interferindo na sinalização entre a matriz e a célula, causando disfunção celular; proteínas, como a albumina, que então ativam os receptores de AGEs (RAGEs), estimulando a produção de citocinas inflamatórias como a interleucina 1 e 6, fator de crescimento I, fator de necrose tumoral alfa, prostaglandinas e fator estimulador de colônias de granulócitos (Figura 2).
Os AGEs se acumulam na maioria dos órgãos-alvo que podem ser acometidos no diabetes, como rim e retina e, ainda, nas placas ateroscleróticas (14,15). O rim é o principal alvo dos danos mediados por AGEs, tendo em vista que representa o maior sítio de clearence destes produtos (16).
O N (carboximetil) lisina (CML) e outros AGEs têm sido encontrados em vasos retinianos de portadores de diabetes e se correlacionam com o grau de retinopatia (17). Elevados níveis de AGEs têm também sido documentados em nervos periféricos de portadores de diabetes e em modelos animais, demonstrou-se que AGEs pioram a neuropatia diabética por reduzirem a velocidade de condução nervosa e o fluxo sangüíneo para os nervos periféricos (18).
Ativação da PKC por DAG induzido pela hiperglicemia
A NAD(P) H-oxidase tipo fagocítica é a maior fonte de produção de ROS em muitas células não-fagocíticas, incluindo fibroblastos, células musculares lisas, endoteliais, mesangiais renais e tubulares renais (19). A produção de ROS pela oxidase, em pequenas quantidades, pode funcionar na sinalização metabólica e, em grandes quantidades, pode originar dano oxidativo. Os tipos celulares encontrados nas paredes dos vasos possuem NAD(P) H-oxidase semelhantes às fagocíticas, as quais são ativadas em condições fisiológicas. Essas oxidases parecem ter múltiplas utilidades, controlando funções vasculares, respostas à expressão gênica, sinalização em processos celulares, como crescimento, apoptose, migração e remodelação da matriz extracelular. No entanto, em determinadas patologias, como DM e hipertensão arterial, ocorre aumento na produção de ROS que ultrapassa o limiar fisiológico, causando danos celulares, principalmente às células endoteliais (20).
A NAD(P) H-oxidase é formada por subunidades de membrana, como gp91phox, p22phox e subunidades citosólicas, como p47phox e p67phox. Quando ativadas, algumas subunidades são fosforiladas por várias quinases, incluindo a PKC, sendo translocadas para a membrana, formando a oxidase ativa cataliticamente (Figura 3) (21,22).
A PKC é uma das três principais quinases serina-treonina (fosforilam proteínas em resíduos de serina e treonina), estando envolvida em eventos de transdução de sinais, respondendo a estímulos específicos hormonais, neuronais e de fatores de crescimento. Atua catalisando a transferência de um grupo fosfato do ATP (adenosina tri-fosfato) a várias proteínas. Da mesma forma, a PKC também sofre fosforilações antes de ser ativada, o que ocorre durante sua translocação do citosol para a membrana da célula. Sua ativação e translocação do citosol à membrana plasmática ocorrem em resposta a aumento transitório de DAG ou exposição a agentes exógenos, como os ésteres de forbol (mimetiza a hiperglicemia in vitro) (23).
O DAG é o principal ativador fisiológico da PKC; deriva de múltiplas fontes, incluindo a hidrólise de fosfatidilinositídeos, metabolismo da fosfatidilcolina por ação de fosfolipases ou pela síntese "de novo". Também é possível que a ativação da via DAG-PKC induzida pela hiperglicemia seja resultado de glico-oxidação, já que existem evidências de que alguns oxidantes, como H2O2, podem ativar a PKC (Figura 4) (7).
A via DAG-PKC é importante na regulação da permeabilidade vascular, contratilidade, proliferação celular, angiogênese, ação de citocinas, adesão leucocitária, sendo todas as alterações descritas no diabetes (Figura 5) (24).
Aumento da atividade da via das hexosaminas
Por este mecanismo, o aumento da glicose intracelular tem como conseqüência a metabolização final de frutose-6-fosfato a uridina difosfato-N-acetil glucosamina (UDPGIcNAc), resultando alterações patológicas na expressão gênica, aumentando a produção de citocinas inflamatórias e fatores de transcrição (Figura 6) (25).
MECANISMO UNIFICADO DE LESÃO CELULAR
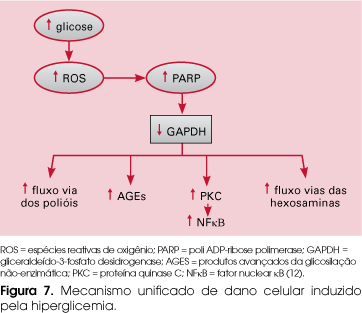
O mecanismo que parece ser comum a todas as células lesadas, como conseqüência da hiperglicemia, é a produção aumentada de ROS, sendo esta hipótese capaz de unificar todas as vias. A hiperglicemia leva a aumento da PARP (poli ADP-ribose polimerase), enzima envolvida no reparo de danos ao DNA, e conseqüentemente, à diminuição da GAPDH (gliceraldeído-3-fosfato desidrogenase), responsável pela metabolização final da glicose, ativando todas as vias (12) (Figura 7).
DEFESAS ANTIOXIDANTES
O organismo possui sistemas de defesas antioxidantes que incluem as moléculas degradadoras de ROS (ROS scavengers), ácido úrico, ácido ascórbico, α-tocoferol, moléculas que contêm sulfidril e enzimas antioxidantes, como catalase, glutationa peroxidase (GPx) e superóxido dismutases (26,27). Em condições patológicas, em que a produção excessiva de ROS ultrapassa a defesa antioxidante, o estresse oxidativo pode modificar irreversivelmente macromoléculas biológicas, como DNA, proteínas, carboidratos e lipídeos, contribuindo para a aterogênese (25). Acredita-se que a capacidade antioxidante do plasma deva-se principalmente à presença de albumina, por meio de seus grupos tióis, sendo esta considerada a principal molécula antioxidante extracelular (28,29).
ESTRESSE OXIDATIVO E DIABETES TIPO 1
Poucos estudos avaliaram o estresse oxidativo em DM1. Diferentes metodologias para quantificação do status oxidante e antioxidante vêm sendo utilizadas e aperfeiçoadas (Tabela 2).
Dominguez e cols. (30) avaliando 24 pacientes pré-púberes dentro de sete a dez dias após início clínico do diabetes, quando o controle metabólico já estava restaurado, demonstraram concentrações elevadas de malonaldeído (MDA) plasmático, produto final da oxidação de ácidos graxos poliinsaturados, em relação ao grupo-controle (p < 0,0001), sugerindo que radicais livres do oxigênio possam exercer seus efeitos tóxicos em estágios precoces da doença, mantendo-se elevados no curso dela, ao serem comparados com um grupo de 30 DM1 com dois a 22 anos de doença e sem complicações. Demonstraram ainda baixos níveis de glutationa peroxidase no grupo recém-diagnosticado em relação aos controles (p < 0,0001), com progressivo declínio no curso da doença. Estes mesmos autores não encontraram correlação entre estes achados e os parâmetros de controle glicêmico e lipídico.
Vessby e cols. (31) avaliando 38 pacientes DM1, com 16,1 ± 10,3 anos desde o diagnóstico, não demonstraram diferença significativa nas variáveis de peroxidação lipídica (8-iso-prostaglandina F2 e MDA) em relação ao grupo-controle. Neste grupo, a capacidade antioxidante total do plasma (quantificada por quimioluminescência) (36) foi 16% menor no grupo de diabéticos (p < 0,0005), a despeito do simultâneo aumento nos níveis de tocoferol (p < 0,05), sem correlação com o controle glicêmico.
Em estudo mais recente, Hata e cols. (32) demonstraram, em DM1 com 5,5 ± 4,4 anos de doença, correlação direta entre aumento de 8-OhdG (8-hidroxi-2'-deoxiguanosina), outro marcador de estresse oxidativo, com o controle glicêmico e a presença de microalbuminúria, achados confirmados em outros estudos (5,30).
Gleisner e cols. (33), avaliando um grupo de DM1 pré-púberes, com menos de cinco anos de diagnóstico, não observou diferença estatisticamente significante nos marcadores do estresse oxidativo em relação aos controles, com parâmetros bioquímicos similares entre os dois grupos.
Na avaliação do estresse oxidativo em um grupo de DM1 brasileiros (34), com 2,62 ± 2,24 anos de doença, sem comorbidades associadas e em tratamento intensivo com insulina, observou-se diferença estatisticamente significante na produção de ROS por granulócitos de DM1 em relação aos não-diabéticos (p < 0,05), quantificados por quimioluminescência dependente de luminol (o método se fundamenta na amplificação pelo luminol da luminescência natural emitida pelas espécies reativas de oxigênio). O status antioxidante do plasma foi avaliado pela redução direta do MTT (sal de tetrazólio: [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]) pelo plasma (37), ensaio utilizado na medida de viabilidade e proliferação celular, que se fundamenta na redução do MTT por meio de redutases, que agem como doadora de elétrons na redução do MTT. Esta redução, descrita inicialmente como fenômeno intracelular, envolve NADH2 e FADH2 e é primariamente uma medida da taxa de produção de NADHna hiperglicemia. Não foi observada diferença significativa na capacidade de redução direta do MTT pelo plasma entre DM1 e não-diabéticos (p > 0,05), caracterizando manutenção do poder antioxidante do plasma neste grupo (Figura 8). Nenhuma correlação entre controle glicêmico e lipídico e os parâmetros avaliados foi encontrada.
Intervenções terapêuticas
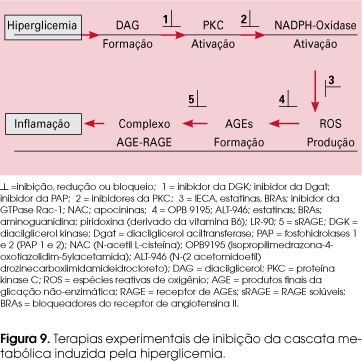
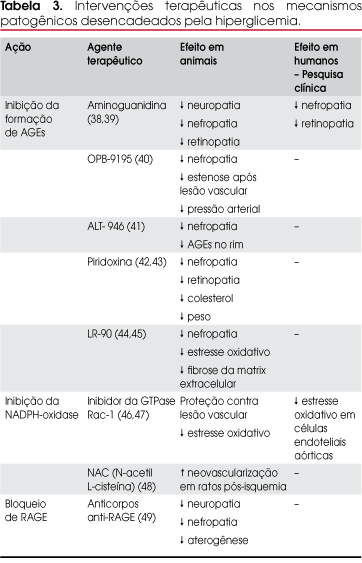
Os tratamentos atuais para o diabetes incluem controle glicêmico e de pressão arterial, agentes hipolipemiantes e orientações nutricionais, e, a despeito das múltiplas opções, deparou-se com a maioria dos pacientes não atingindo controle metabólico satisfatório, com evolução para lesões endoteliais. Estudos em andamento focam nos mecanismos patogênicos desencadeados pela hiperglicemia, a fim de reduzir ou bloquear a injúria vascular, sendo os principais objetivos reduzir a formação de DAG, AGEs e ROS, inibir o sistema NADPH-oxidase e PKC e prevenir a interação entre AGEs e seu receptor (RAGE). A seguir, os efeitos de algumas drogas em estudo que interferem na cascata metabólica induzida pela hiperglicemia (Figura 9 e Tabela 3).
CONCLUSÃO
O papel do estresse oxidativo nas complicações diabéticas tem sido amplamente estudado, principalmente no diabetes tipo 2, com estudos envolvendo bloqueio da via oxidante e terapias antioxidantes aumentado nos últimos anos. No DM1, cujo diagnóstico é precoce, a importância do estresse oxidativo desde estágios precoces da doença e o efeito protetor do tratamento intensivo com insulina sobre este mecanismo bioquímico são inconclusivos, com mais estudos sendo necessários para o entendimento das bases moleculares do seu efeito, a fim de se implementar novas terapêuticas adjuvantes à insulina nesta patologia.
Os autores declaram não haver conflitos de interesse científico neste artigo.
Recebido em 16/3/2008
Aceito em 24/6/2008
- 1. Deckert T, Pousen JE. Prognosis of diabetes with onset before age of thirty-one. Diabetologia. 1978;14:363-77.
- 2. The Diabetes Control and Complications Trial Research Group (DCCT): The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977-86.
- 3. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352:837-53.
- 4. Thomas E, Lin YS, Dagher Z et al. Hyperglycemia and insulin resistance: possible mechanism. NY Acad Sci. 2002;967:43-8.
- 5. Huvers FC, De Leeuw PW, Houben AJ, De Haan CH, Hamulyak K, Schouten H, et al. Endothelium-dependent vasodilatation, plasma markers of endothelial function, and adrenergic vasoconstrictor responses in type 1 diabetes under near-normoglycemic conditions. Diabetes. 1999;48(6):1300-7.
- 6. Dogra G, Rich L, Stanton K, Watts GF. Endothelium-dependent and independent vasodilation studies at normoglycaemia in type I diabetes mellitus with and without microalbuminuria. Diabetologia. 2001;44(5):593-601.
- 7. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813-20.
- 8. Yoshihiro Taniyama, Kathy K. Griendling. Reactive Oxygen Species in the Vasculature: Molecular and Cellular Mechanisms. Hypertension. 2003;42:1075-108.
- 9. Santini SA, Marra G, Giardina B, Cotroneo P, Mordente A, Martorana GE, et al. Defective plasma antioxidant defenses and enhanced susceptibility to lipid peroxidation in uncomplicated IDDM. Diabetes.1997;46(11):1853-8.
- 10. Halliwell B, Gutteridge JMC. Free radical in biology and medicine. 2. ed. New York: Oxford University Press; 1989.
- 11. Darley-Usmar V, Wiseman H, Halliwell B. Nitric oxide and oxygen radicals: a question of balance. FEBS Lett. 1995;369(2-3):131-5.
- 12. Brownle M. The pathobiology of diabetic complications - a unifying mechanism. diabetes. 2005;56:1615-25.
- 13. Goldin BAA, Beckman JA, Schimidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597-605.
- 14. Barbosa JHP, Oliveira SL, Seara LT. O papel dos produtos finais da glicação avançada (AGEs) no desencadeamento das complicações vasculares do diabetes. Arq Bras Endocrinol Metab. 2008;52(6);940-6.
- 15. Hammes HP, Alt A, Niwa T, Clausen JT, Bretzel RG, Brownlee M, et al. Differential accumulation of advanced glycation end products in the course of diabetic retinopathy. Diabetologia. 1999;42(6):728-36.
- 16. Miyata T, Ueda Y, Horie K, Nangaku M, Tanaka S, van Ypersele de Strihou C, Kurokawa K. Renal catabolism of advanced glycation end products: the fate of pentosidine. Kidney Int. 1998;53(2):416-22.
- 17. Murata T, Nagai R, Ishibashi T, Inomuta H, Ikeda K, Horiuchi S. The relationship between accumulation of advanced glycation end products and expression of vascular endothelial growth factor in human diabetic retinas. Diabetologia. 1997;40(7):764-9.
- 18. Chen AS, Taguchi T, Sugiura M, Wakasugi Y, Kamei A, Wang MW, et al. Pyridoxal- aminoguanidine adduct is more effective than aminoguanidine in preventing neuropathy and cataract in diabetic rats. Horm Metab Res. 2004;36(3):183-7.
- 19. Li JM, Shah AM. ROS generation by nonphagocytic NADPH oxidase: potential relevance in diabetic nephropathy. J Am Soc Nephrol. 2003;14(Suppl 3):S221-6.
- 20. Zalba G, San Jose G, Moreno MU, Fortuno MA, Fortuno A, Beaumont FJ, et al. Oxidative stress in arterial hypertension: role of NAD(P)H oxidase. Hypertension. 2001;38(6):1395-9.
- 21. Kitada M, Koya D, Sugimoto T, Isono M, Araki S, Kashiwagi A, et al. Translocation of glomerular p47phox and p67phox by protein kinase c-[beta] activation is required for oxidative stress in diabetic nephropathy. Diabetes. 2003;52(10):2603-14.
- 22. El-Benna J, Dang PM, Gougerot-Pocidalo MA, Elbim C. Phagocyte NADPH oxidase: a multicomponent enzyme essential for host defenses. Arch Immunol Ther Exp (Warsz). 2005;53(3):199-206.
- 23. Idris I, Gray S, Donnelly. Protein kinase C activation: isozyme-specific effects on metabolism and cardiovascular complications in diabetes. Diabetologia. 2001;44:659-73.
- 24. Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859-66.
- 25. Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97(22):12222-6.
- 26. Stinefelt B, Leonard SS, Blemings KP, Shi X, Klandorf H. Free radical scavenging, DNA protection, and inhibition of lipid peroxidation mediated by uric acid. Ann Clin Lab Sci. 2005;35(1):37-45.
- 27. Frei B, Stocker R, Ames BN. Antioxidant defenses and lipid peroxidation in human blood plasma. Proc Natl Acad. Sci. 1988;85:9748-52.
- 28. Faure P, Troncy L, Lecomte M, Wiernsperger N, Lagarde M, Ruggiero D, et al. Albumin antioxidant capacity is modified by methylglyoxal. Diabetes Metab. 2005;31(2):169-77.
- 29. Mera K, Anraku M, Kitamura K, Nakajou K, Maruyama T, Tomita K, et al. Oxidation and carboxy methyl lysine-modification of albumin: possible involvement in the progression of oxidative stress in hemodialysis patients. Hypertens Res. 2005;28(12):973-80.
- 30. Dominguez C, Ruiz E, Gussinye M, Carrascosa A. Oxidative stress at onset and in early stages of type 1 diabetes in children and adolescents. Diabetes Care. 1998;21(10):1736-42.
- 31. Vessby J, Basu S, Mohsen R, Berne C, Vessby B. Oxidative stress and antioxidant status in type 1 diabetes mellitus. J Intern Med. 2002;251(1):69-76.
- 32. Hata I, Kaji M, Hirano S, Shigematsu Y, Tsukahara H, Mayumi M. Urinary oxidative stress markers in young patients with type 1 diabetes. Pediatr Int. 2006;48(1):58-61.
- 33. Gleisner A, Martinez L, Pino R, Rojas IG, Martinez A, Asenjo S, et al. Oxidative stress markers in plasma and urine of prepubertal patients with type 1 diabetes mellitus. J Pediatr Endocrinol Metab. 2006;19(8):995-1000.
- 34. Reis, JS. Diabetes tipo 1: Estudo da associação entre o balanço oxidante/antioxidante com parâmetros clínicos e bioquímicos [dissertação]. Belo Horizonte, Programa de Pós-graduação e Pesquisa da Santa Casa de Belo Horizonte, 2006.
- 35. Mera K, Anraku M, Kitamura K, Nakajou K, Maruyama T, Tomita K, et al. Oxidation and carboxy methyl lysine-modification of albumin: possible involvement in the progression of oxidative stress in hemodialysis patients. Hypertens Res. 2005;28(12):973-80.
- 36. Whitehead TP, Thorpe CHG, Maxwell SRJ. Enhanced chemiluminescent assay for antioxidant capacity in biological fluids. Anal Chim Acta. 1992;226:265-77.
- 37. Berridge M, Wang R. The biochemical and cellular basis of cell proliferation assays that use terazolium salts. Biochemica. 1996;4.
- 38. Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, et al. ACTION I Investigator Group. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004;24:32-40.
- 39. Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science. 1986;232:1629-32.
- 40. Nakamura S, Makita Z, Ishikawa S, Yasumura K, Fujii W, Yanagisawa K, et al. Progression of nephropathy in spontaneous diabetic rats is prevented by OPB-9195, a novel inhibitor of advanced glycation. Diabetes. 1997;46:895-9.
- 41. Edelstein D, Brownlee M. Mechanistic studies of advanced glycosylation end product inhibition by aminoguanidine. Diabetes. 1992;41:26-9.
- 42. Degenhardt TP, Alderson NL, Arrington DD, Beattie RJ, Basgen JM, Steffes MW, et al. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002;61:939-50.
- 43. Stitt A, Gardiner TA, Alderson NL, Canning P, Frizzell N, Duffy N, et al. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes. 2002;51:2826-32.
- 44. Figarola JL, Scott S, Loera S, Tessler C, Chu P, Weiss L, et al. LR-90 a new advanced glycation endproduct inhibitor prevents progression of diabetic nephropathy in streptozotocin-diabetic rats. Diabetologia. 2003;46:1140-52.
- 45. Figarola JL, Shanmugam N, Natarajan R, Rahbar S. Anti-inflammatory effects of the advanced glycation end product inhibitor LR-90 in human monocytes. Diabetes. 2007;56:647-55.
- 46. Vecchione C, Arentini A, Mauno G, et al. Selective Rac-1 inhibition protects from diabetes-induced vascular injury. Circ Res. 2006;98:218-25.
- 47. Vecchione C, Gentile MT, Aretini A, et al. A novel mechanism of action for statin against diabetes-induced oxidative stress. Diabetologia. 2007;50:874-80.
- 48. Benrahmonoune M, Therond P, Abedinzadeh Z. The activation of superoxide radical with N-acetylcysteine. Free Radic Biol Med. 2000;24:775-82.
- 49. Goova MT, Li J, Kislinger T, et al. Blockade of receptor for advanced glycation end-products restores effectives wound healing in diabetic mice. Am J Pathol. 2001;159:513-25.
Datas de Publicação
-
Publicação nesta coleção
03 Dez 2008 -
Data do Fascículo
Out 2008
Histórico
-
Aceito
24 Jun 2008 -
Recebido
16 Mar 2008