Abstracts
Mutations in the vitamin D receptor (VDR) are associated to the hereditary 1,25-dihydroxivitamin D-resistant rickets. The objectives of this work are: search for mutations in the VDR and analyze their functional consequences in four Brazilian children presented with rickets and alopecia. The coding region of the VDR was amplified by PCR e direct sequenced. We identified three mutations: two patients had the same mutation in exon 7 at aminoacid position 259 (p.Q259E); one patient had a mutation in exon 8 at codon 319 (p.G319V) and another one had a mutation in exon 3 leading to a truncated protein at position 73 (p.R73X). Functional studies of the mutant receptors of fibroblast primary culture, from patients' skin biopsy treated with increasing doses of 1,25(OH)2 vitamin D showed that VDR mutants were unable to be properly activated and presented a reduction in 24-hydroxylase expression level.
Rickets; Bone deformities; Hypocalcemia; Vitamin D
Mutações no receptor de vitamina D (VDR) são associadas a raquitismo hereditário resistente a 1,25-dihidroxivitamina D. Os objetivos deste trabalho foram procurar mutações no VDR e analisar suas conseqüências funcionais em quatro pacientes com raquitismo e alopécia. A região codificadora do VDR foi amplificada por PCR e seqüenciada diretamente. Identificamos três mutações: dois pacientes apresentavam a mesma mutação no éxon 7 na posição protéica 259 (p.Q259E); um paciente apresentava uma mutação no éxon 8 no códon 319 (p.G319V) e o outro apresentava uma mutação no exon 3 resultando em uma proteína truncada na posição 73 (p.R73X). O estudo funcional dos receptores mutados nos extratos de culturas de fibroblasto primárias obtidas de biópsia de pele dos pacientes, tratados com doses crescentes de 1,25(OH)2 vitamina D demonstraram que os receptores mutantes não apresentam ativação adequada apresentando expressão reduzida de 24-hidroxilase.
Raquitismo; Deformidades ósseas; Hipocalcemia; Vitamina D
CLINICAL CASE REPORT
Mutations in the vitamin D receptor gene in four patients with hereditary 1,25-dihydroxyvitamin D-resistant rickets
Mutações no gene do receptor de vitamin D em quatro pacientes com raquitismo hereditário resistente a 1,25-dihydroxyvitamin D
Luciana Cosentino de MacedoI; Fernanda Caroline Soardi; Nayla AnaniasI; Vera Maria Santoro BelangeroII; Sumara Zuazani Pinto RigattoII; Maricilda Palandi De-MelloIII; Lília D'Souza-LiI, II
IPediatric Endocrinology Laboratory, Center for Investigation in Pediatrics. Faculty of Medical Science, Campinas, SP, Brazil
IIDepartment of Pediatrics. Faculty of Medical Science, Campinas, SP, Brazil
IIICenter of Molecular Biology and Genetic Engineering (CBMEG) State University of Campinas (Unicamp), Campinas, SP, Brazil
Correspondence to Correspondence to: Lília D'Souza-Li Center for Investigation in Pediatrics Rua Tessália Vieira de Camargo, 126 Cidade Universitária "Zeferino Vaz" Caixa postal 6111 13083-887 Campinas, SP, Brazil. E-mail: Idesouza@globo.com
ABSTRACT
Mutations in the vitamin D receptor (VDR) are associated to the hereditary 1,25-dihydroxivitamin D-resistant rickets. The objectives of this work are: search for mutations in the VDR and analyze their functional consequences in four Brazilian children presented with rickets and alopecia. The coding region of the VDR was amplified by PCR e direct sequenced. We identified three mutations: two patients had the same mutation in exon 7 at aminoacid position 259 (p.Q259E); one patient had a mutation in exon 8 at codon 319 (p.G319V) and another one had a mutation in exon 3 leading to a truncated protein at position 73 (p.R73X). Functional studies of the mutant receptors of fibroblast primary culture, from patients' skin biopsy treated with increasing doses of 1,25(OH)2 vitamin D showed that VDR mutants were unable to be properly activated and presented a reduction in 24-hydroxylase expression level.
Key words: Rickets; Bone deformities; Hypocalcemia; Vitamin D.
RESUMO
Mutações no receptor de vitamina D (VDR) são associadas a raquitismo hereditário resistente a 1,25-dihidroxivitamina D. Os objetivos deste trabalho foram procurar mutações no VDR e analisar suas conseqüências funcionais em quatro pacientes com raquitismo e alopécia. A região codificadora do VDR foi amplificada por PCR e seqüenciada diretamente. Identificamos três mutações: dois pacientes apresentavam a mesma mutação no éxon 7 na posição protéica 259 (p.Q259E); um paciente apresentava uma mutação no éxon 8 no códon 319 (p.G319V) e o outro apresentava uma mutação no exon 3 resultando em uma proteína truncada na posição 73 (p.R73X). O estudo funcional dos receptores mutados nos extratos de culturas de fibroblasto primárias obtidas de biópsia de pele dos pacientes, tratados com doses crescentes de 1,25(OH)2 vitamina D demonstraram que os receptores mutantes não apresentam ativação adequada apresentando expressão reduzida de 24-hidroxilase.
Descritores: Raquitismo; Deformidades ósseas; Hipocalcemia; Vitamina D.
INTRODUCTION
Hereditary 1,25-dihydroxivitamin D-resistant rickets (HVDRR) is a rare autosomal recessive disorder associated with mutation in the vitamin D (vit D) receptor (VDR), leading to impairment of its function (1). The human VDR is a member of the nuclear receptor superfamily and consists of a 427 amino acid protein. At the amino terminal portion of the molecule there is a Cysteine rich region that interacts with the VDRE in the DNA, through two zinc fingers motifs (2). The ligand binding domain has high affinity to the 1,25 dihydroxyvitamin D (1,25(OH)2vit D) and is located at the carboxi-terminal portion of the VDR. The 1,25(OH)2vit D activates the VDR, which by heterodimerazing with the Retinoic acid receptor X (RXR) interacts with specific sequences in the DNA of target genes denominated VDR response elements (VDRE) (2). The VDRE is composed of two sequences of direct repeats (RGKTCA) separated by three base pairs. More than 40 mutations in the VDR have been described in approximately seventy families presenting HVDRR (3). Clinically, patients presented bone deformities in the lower limbs, due to rickets during the first years of life. In addition, the majority of affected individuals have alopecia that appears soon after birth denoting the importance of the VDR in the natural development of hair follicle. Mutations that affects the DNA ligand domain usually is associated with a more severe phenotype. The objective of this study is to perform a molecular analysis of the VDR in four patients with rickets and alopecia.
SUBJECTS AND METHODS
Patients
We studied four patients with clinical diagnosis of HVDRR presenting alopecia and rickets, followed at the Pediatrics Nephrology Outpatient Clinics from the Department of Pediatrics, at the Faculty of Medical Science of the State University of Campinas. Patient 1 is a 9.5 years old boy followed since he is two years-old. He presented alopecia since 2 months old and develop deformities of the lower limbs at the age of 1 year. He responded remarkably well to the treatment with Calcitriol (0.25µg/day), improving bone deformities. Patient 2 is a 19.2 years old man that at age of 2 years presented severe malnourishment, rickets, alopecia and several upper and lower respiratory infections. He responded well to the treatment with vitamin D3 (vit D) (3500 IU/day), latter changed to Calcitriol (0.25 µg/day), associated with calcium and phosphate solution, normalizing his weight and growth. Patient 3 is a 20.6 years old woman that presented alopecia since birth and coarse bone deformities of lower limbs. She started the treatment at the age of seven with vitamin D3 (7000 IU/day), latter changed to Calcitriol (0.25µg/day) and phosphate solution. Patient 4 is a 6.7 years old boy, who presented alopecia since 2 months of age and by the age of 2 he had severe rickets with coarse bone deformities, of upper and lower limbs not deambulating. His parents are first degree cousins. He underwent treatment with increasing doses of Calcitriol up to 1.5 µg/day and phosphate solution. After 6 months of treatment,his condition improved, become able to walk but persisted hypocalcemic and with high serum alkaline phosphatase levels. Patients 1 and 3 were apparently not related, although both are from the same region (Bahia State). Both patients refer cousins presenting similar features, with alopecia and rickets. This study was approved by the local Ethical Committee of the Faculty of Health Science (CEP nº 330/02) and to the National Ethics in Research Council (CONEP nº 4623).
Sequence analysis of the VDR gene
Leukocyte DNA was isolated from blood from the four patients after written informed consent, and used as a template for the PCR reactions. All coding and flanking regions from exons 2 to 9 from the VDR gene were PCR amplified using 7 pairs of primers. Due to its proximity and small sizes, a single pair of primers were used to amplify exon 7 and 8, with the forward primer located upstream exon 7 and the reverse primer downstream exon 8. The PCR products were gel purified and directly sequenced using a semi-automated laser detection system (ABI Prism 377sequencer, Applied Biosystems). The same primers from the PCR amplification were used in the Big Dye reaction (Applied Biosystem, version 2). All sequences were compared to the human VDR sequence (GenBank gi|3617739|gb|AC004466.1). The mutations were confirmed by digesting the PCR fragments with specific restriction enzymes.
Fibroblast primary culture
Using tissues from patients'skin biopsy, fibroblast primary cultures were performed and used for functional analyses. The skin sample was scraped to remove fat tissue and blood, cut into small fragments and treated with trypsin for 3 hours at 37 ºC to separate the epidermis and to dissociate the cells individually. The cells were grown in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen) and penicillin/streptomycin (complete medium).
1,25(OH)2 vit D induction of 24-hydroxylase mRNA
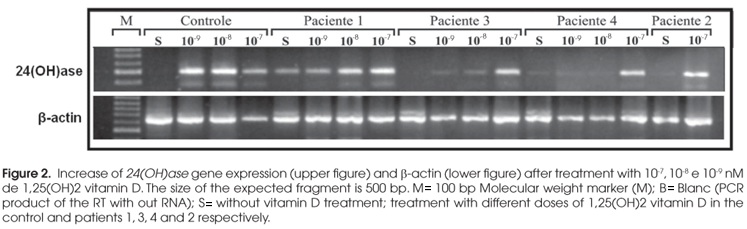
For VDR functional studies, we analyzed the 24-hydroxylase [24(OH)ase] expression from the patient's fibroblast primary culture. When the primary cultures were confluent they were treated with different doses of 1,25(OH)2vit D (10-7, 10-8 and 10-9 M) in a serum free media supplemented with 1% BSA for 6 hours. Controls, without treatment were also included. At the end of 6 hours, cells were harvested for total RNA and nuclear extraction.
RT-PCR
Total RNA was extracted from the fibroblast primary cultures by TRIzol (Invitrogen) as manufacturer's protocol. Five-microgram RNA samples were reverse-transcribed with recombinant SuperScript IIITM , reverse transcriptase (Invitrogen) using random primers in a total volume of 20 µl. Five microliters of the RT mixture were used in the PCR with primers specific to amplify the 24(OH)ase gene. A water control from the RT and RNA from fibroblast primary culture from normal individual were used as negative and positive controls, respectively. The PCR products were analyzed by agarose gel electrophoresis.
Real time PCR
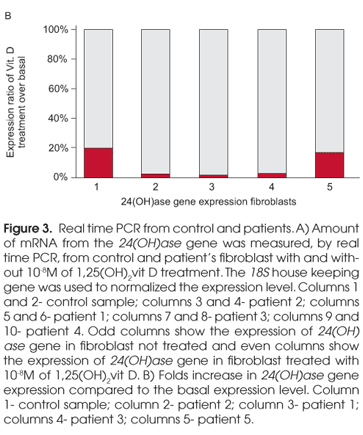
For a quantitative measurement of mutant receptor response, 100 ng of cDNA from control and patient's fibroblast with and without 10-8M of 1,25(OH)2vit D treatment, were used to perform a real time PCR experiment. The amount of mRNA from the 24(OH)ase gene were measured using specific primers labeled with FAM, and controlling its expression with the 18S house keeping gene. The degree of expression was quantified by real time PCR using the detection system ABI Prism sequencer 5700 (PE).
Nuclear protein extraction
For the nuclear protein extraction, cells were harvested in solution A (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.5 mM PMSF, 2.5 µg/ml of aprotinin), centrifuged for 5 minutes at 1,000 rpm, at 4ºC and the pellet was re-suspended in 200µl of solution A plus 0.1% Nonidet P-40, passed through and syringe gauge with fine needle for 15 times and centrifuged 5 min, at 12,000 x g at 4 ºC. The nuclear membrane in the pellet was lysed with osmotic shock with solution B (20 mM HEPES, pH 7.9, 25% glycerol, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF, 2.5 µg/ml of aprotinin and leupeptin, and 1 µg/ml of pepstatin). After 30 min incubation at 4 ºC under agitation, cells were centrifuged for 10 min, 4ºC, 12,000 x g. The supernatants, containing the nuclear fraction, were collected and stored at -70 ºC. The protein concentration were determined by Bradford assay.
Western Blotting
The expression of the mutant receptor compared to wild type was analyzed by Western Blot. Nuclear protein extract was subjected to 12% SDS-PAGE and the separated proteins were electrotransferred and immobilized on PVDF membranes. After blocking unspecific binding with PBS plus Tween 20 (PBS-T) and 5% skim milk, the membranes were incubated with primary antibody VDR specific diluted 1:100 (Santa Cruz) for twelve hours followed by a secondary antibody (anti-mouse) conjugated to horseradish peroxidase (Biorad; diluted 1:5,000) for two hours. For detection of the proteins, ECL chemiluminescence system (GE Healthcare) was used.
Electrophoretic Mobility Shift Assay (EMSA)
Double-strand oligonucleotides containing the VDRE binding site was used as a probe in gel shift mobility assays. The double-strand oligonucleotides were obtained from annealing synthesized oligonucleotides (Integrated DNA Technologies - IDT®, USA): VDR-F: 5'- GAG CTT CAG TCA AGG AGG TCA GAG AGC -3' and VDR-R: 5'- GCT CTC TGA CCT CCT TGA CTG AAG CT -3'. The double-stranded probe was labeled with the [a32P]-dCTP (PerkinElmer, USA) using the Klenow DNA polymerase fill-in reaction (Invitrogen, USA) (4). The probe was purified by Qiaquick nucleotide remove kit (Qiagen, USA). For EMSA, 10 mg of nuclear protein extract were pre-incubated with DNA binding buffer 2X (DNA binding buffer 10X: 50 mM MgCl2, 2.5 mM EDTA, 2.5 mM DTT, 250 mM NaCl, 50 mM Tris-HCl pH7.5, 20% glycerol) followed by 30 minutes incubation with 25 fmoles of VDRE probe (~ 1x105 cpm) at 20ºC. Reaction mixtures were separated on 6% non-denaturing polyacrylamide gels using 0.5x Tris borate/EDTA buffer and the complexes were visualized in Hyperfilm MP autoradiographies (Amersham Biosciences/GE, USA).
RESULTS
Patients
The biochemical levels of all patients at the beginning of the treatment and the most recent evaluations are in table 1. Except for patient 4, all patients responded well to the treatment with supra physiological doses of vitamin D and or Calcitriol, calcium and phosphate. In patient 1 and 2 there was a better control of the disease between the age of six to eight years of age, requiring discontinuation of the vitamin D due to hypercalciuria. Even in patient 4 that had a more severe form of rickets compared to the other patients, became at the age of six clinically and biochemically stable. All patients have normal renal functions and after correction of the phosphate depletion, presented a normal phosphate excretion rate.
VDR Mutations
We identified VDR-gene mutations in all the patients studied. All the mutations were in homozygous state. In patients 1 and 3 we identified a novel missense mutation in exon 7 changing a C to a G at nucleotide 775 (c.775 C>G) (Figure 1A). Resulting in a change in amino acid from the Glutamine to Glutamic acid at protein position 259 (p.Q259E), on the ligand binding domain of the receptor. This mutation was confirmed by restriction enzyme analysis as it disrupts an Ava II site (data not shown). Patient 2 presented a novel mutation in exon 8 at nucleotide 955, resulting in a G to T transversion (c.955 G>T), changing of the amino acid from Glycine (hydrophilic) to Valine (hydrophobic) at codon 319 (p.G319V) (Figure 1B). This mutation was confirmed by restriction enzyme analysis as it creates a Rsa I site (data not shown). In patient 4, a nonsense mutation in exon 3 at nucleotide 217, changing C to T(c.217C>T) was identified, resulting in change of Arginine to a termination codon at position 73 (p.R73X) (Figure 1C). This mutation was confirmed by restriction enzyme analysis using a mutant primer that creates an Pvu II site (data not shown).
VDR functional analysis
The expression of 24(OH)ase gene increases through the activation of the VDR by vit D (5). For functional analyses, fibroblast primary culture from the patients and a normal individual were treated with increasing doses of 1,25(OH)2 vit D for 6 hours. The 24(OH)ase expression was strongly induced in control cells, even with lower doses (Figure 2). Intermediate doses showed variable responses in the fibroblast cultures from the patients while all patients responded to 10-7M 1,25(OH)2 vit D treatment, despite the different types of mutation.
Real time PCR
In order to quantify the differences in response of the mutant receptors, the amount of mRNA from the 24(OH)ase gene was measured, by real time PCR, from control and patients' fibroblast with and without 10-8M of 1,25(OH)2vit D treatment. 18S house keeping gene expression was used to normalized the 24(OH) gene expression level. Unlike the patients, the control showed 24(OH)ase expression even in the basal state, and it responded well to the vit D treatment with a five fold increase in the gene expression level (Figure 3A). Only patient 2 and 1 presented a significant increase in 24(OH)ase gene expression after 1,25(OH)2 vit D treatment (Figure 3A) showing an increase of 38 and 53 fold in 24(OH)ase gene expression after 1,25(OH)2 vit D treatment respectively (Figure 3B). Patient 3 and 4 were not able to increase 24(OH)ase gene expression to a good level after 1,25(OH)2 vit D treatment (Figure 3A). However, when compared the basal level, they presented an increase of 35 and 5 fold in 24(OH)ase gene expression respectively (Figure 3B), with the truncated receptor presenting the worse response.

Western Blotting
We analyzed the receptor expression in the nuclear extract from the patients culture comparing to the control. Three bands were visualized: a 50 kDa band corresponding to the mature VDR (427 amino acid), and a 38 and 20 kDa band. In a nuclear extract from fibroblast culture of all patients, the 50 kDa band was the most reduced when compared to the wild type (Figure 4). The VDR antibody was raised against a peptide corresponding to the VDR amino acids 344-424 of the human protein. Since the truncated receptor, with only 73 amino acids, lost its epitope for the antibody, it was not detected.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extract from control fibroblast showed two shifts: a lower shift and a more abundant upper shift (Figure 5, lane1). Interesting, the lower shift disappeared with vit D treatment (Figure 5, lane 1D). Nuclear extract from patients 1, 2 and 3 showed a similar pattern, with only the upper shift similar to the control, and the shift patterns did not changed with vit D treatment (Figure 5). As expected, for patient 4 with p.R73X mutation it was not observed a shift (Figure 5, lane 4), however when treated with vitamin D a different shift slightly higher than the control sample was detected (Figura 5, lane 4D).
DISCUSSION
We identified mutations in all four patients studied. Patient 1 and 3 presented a novel mutation at protein position 259 (p.Q259E). Both patients presented very similar clinical symptoms. The vitamin D resistance in these patients was discrete and responded to calcitriol treatment. Despite not apparently related, both families are from the same area in the State of Bahia. In addition, both patients share the same genotyping in the different polymorphic regions (data not shown) suggesting a founder effect. Besides the possible reduction in the affinity of the receptor to the vitamin D, this mutation also reduces the receptor expression when compared to the wild type. However, it did not showed reduction in the DNA interaction. This was expected since the mutation is located in the ligand binding domain. The change in the amino acid charge from hydrophobic to acid negatively charged, probably changed the ligand binding pocket impairing agonist-receptor interaction. This mutation is located in a very conserved region in the E1 domain involved in the VDR-RXR heterodimerization (6). The presence of the alopecia also suggests that the mutation impairs the receptor signaling and this was confirmed by the poor response to vit D treatment. Despite having the same mutation, patient 1 presented a better treatment response than patient 3. One of the reasons is probably the difference in age of the treatment onset. Patient 1 started the treatment with 1 year old, while patient 3 at 7 years of age. Furthermore, on the in vitro functional analyses patient 1 responded better to vit D treatment than patient 3. This suggests that other factors are also involved in the VDR activation and function. In the same position another mutation is described (p.Q259P) in a consanguineous Indian family where the affected members presented alopecia (5). Interesting, the affected brothers of this family also responded well to iv calcium with complete healing of the rickets and correction of the biochemical abnormalities (5).
The novel mutation p.G319V found in patient 2 is also located on the ligand binding domain and in the three-dimensional model based on the structure of the RXRα apo-ligand binding domain, exon 8 represents the α-helix 7 and 8 (7). Upon ligand binding, a conformational change and a reorganization of the helices occured, allowing heterodimer formation (7). The presence of mutation in one of these helices may alter its organization and compromise the interaction of the receptor with the ligand (8). This patient presented with severe clinical manifestations, however, responded to a modest dose of vitamin D (3500 UI/day) with normalization of the biochemical levels and of the growth. This is in accordance to the functional studies, which showed a good VDR-DNA interaction as well as a good response to the vit D treatment.
These three patients with mutations located in the ligand binding domain showed less severe phenotypes, and responded to treatment with vit D, calcium and phosphate. Correction of the inorganic phosphorus with phosphate solution was crucial since its depletion may impair vit D treatment, specially under high metabolic state such as the intense growth during the first years of life and during puberty. In addition, the vit D deficiency induces an increase in PTH which in turn will worse the renal phosphate excretion. Although most of the patients with HVDRR do not respond even to supraphysiologic doses of calcitriol, there are several reports in the literature of patients responding well to treatment, with complete healing of the rickets and correction of biochemical abnormalities (5,8-13).
In patient 4, the mutation results in a truncated receptor at position 73 (p.R73X) in the DNA binding domain just after the second zinc finger. This portion of the receptor is highly conserved and mutations in this region result in reduction of the DNA affinity binding. Clinically, this patient presents a much more severe clinical manifestations than the other patients, confirming that the VDR in this patient has little if any activity. However, he improved upon supraphysiological doses of calcitriol, suggesting that some of the disease manifestations are in part VDR independent (14). Functional studies confirmed the inability of this receptor to interact with DNA under basal conditions. In fact it is debatable if this truncated protein is produced, as cells efficiently identify nonsense codons and induce mRNA decay. As expected, under basal conditions, the truncated receptor did not presented DNA interaction. Surprisingly, after vit D treatment, a shift was detected, indicating that in the presence of vit D, a interaction of a protein with the VDRE occurs. However, the complex size of the shift was slightly above the expected shift, suggesting that this truncated receptor may form a different complex and that the vit D allowed some kind of binding. Whether this binding is dependent of the mutant VDR remains unclear. This mutation was described in the literature in a Greek boy (5) with no family history of consanguinity and also in a Marocan patient (15), both with poor response to alfacalcidol and oral calcium with persistence of severe rickets. In another Brazilian family a nonsense mutation at position 30 was described, resulting in a truncated receptor in the DNA binding domain in the first zinc finger (16).
Functional analysis of the mutations showed that all the mutant receptors function was impaired. The receptor truncated in the DNA ligand domain was with worse response to vit D treatment. In vitro studies confirmed the loss of function of the truncated receptor. When we evaluated the receptors with mutations in the ligand binding domain their expression and their function were impaired, however, their ability to bind to DNA was preserved. In conclusion we report three mutations in the VDR. Two novel mutations in the ligand binding domain and one mutation in the DNA binding domain that resulted in a truncated receptor. All mutations result in reduction of receptor expression and function.
Acknowledgments: Thank you Jussara Rehder, for technical assistance. Special acknowledgments to the patients who had collaborated with the research and to the grant agency Fapesp Disclosure: None. No potential conflict of interest relevant to this article was reported.
Received in 25/8/2008
Accepted in 14/10/2008
- 1. Hughes MR, Malloy PJ, Kieback DG, Kesterson RA, Pike JW, Feldman D, O'Malley BW. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science. 1988;242:1702-5.
- 2. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell 1995;83:841-50.
- 3. Liberman UA. Vitamin D-resistant diseases. J Bone Miner Res. 2007;22 Suppl 2:V105-7.
- 4. Sambrook J, Fritsch EF, Maniatis TE. Molecular cloning, a laboratory manual. New York: Cold Spring Harbor 1989.
- 5. Cockerill F, Hawa NS, Yousaf N, Hewison M, O'Riordan LH, Farrow SM. Mutations in the vitamin D receptor gene in three kindreds associated with hereditary vitamin D resistant rickets. J Clin Endocrinol Metab. 1997;82:3156-60.
- 6. Jin CH, Kerner SA, Hong MH, Pike JW. Transcriptional activation and dimerization functions in the human vitamin D receptor. Mol Endocrinol. 1996;10:945-57.
- 7. Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand binding domain of the human nuclear receptor RXR-α. Nature. 1995;375:377-82.
- 8. Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr Rev. 1999;20:156-88.
- 9. Malloy PJ, Xu R, Peng L, Peleg S, Al-Ashwal A, Feldman D. Hereditary 1,25-dihydroxyvitamin D resistant rickets due to a mutation causing multiple defects in vitamin D receptor function. Endocrinology. 2004;145(11):5106-14.
- 10. Rosen JF, Fleischman AR, Finberg L, Hamstra A, DeLuca HF. Rickets with alopecia: An inborn error of vitamin D metabolism. J Pediat. 1979;94(5): 729-35.
- 11. Liberman UA, Halabe A, Samuel R, Kauli R. End-organ resistance to 1,25-Dihydroxicholecalciferol. Lancet. 1980;8:504-7.
- 12. Hochberg Z, Gilhar A, Haim S, Friedman-Birnbaum R, Levy J, Benderly A. Calcitriol-resistance rickets with alopecia. Arch Dermatol. 1985;121: 646-7.
- 13. Takeda E, Yokota I, Kawakami I, Hashimoto T, Kuroda Y, Arase S. Two siblings with vitamin-D-dependent rickets type II: no recurrence of rickets for 14 years after cessation of therapy. Eur J Pedatr. 1989;149: 54-7.
- 14. Panda DK, Miao D, Bolivar I, Li J, Huo R, Hendy GN, Goltzman D. Inactivation of the 25-hydroxyvitamin D 1α-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279:16754-66.
- 15. Wiese RJ, Goto H, Prahl JM, Marx SJ, Thomas M, al-Aqeel A, DeLuca HF. Vitamin D-dependency rickets type II: truncated vitamin D receptor in three kindreds. Mol Cell Endocrinol. 1993;90(2):197-201.
- 16. Mechica JB, Leite MO, Mendonca BB, Frazzatto ES, Borelli A, Latronico AC. A novel nonsense mutation in the first zinc finger of the vitamin D receptor causing hereditary 1,25-dihydroxyvitamin D3-resistant rickets. J Clin Endocrinol Metab. 1997;82:3892-4.
Correspondence to:
Publication Dates
-
Publication in this collection
22 Jan 2009 -
Date of issue
Nov 2008
History
-
Received
25 Aug 2008 -
Accepted
14 Oct 2008