Abstracts
Tumor-induced osteomalacia (TIO) is a rare paraneoplasic syndrome with overproduction of fibroblast growth factor 23 as a phosphaturic agent, leading to chronic hyperphosphaturia and hypophosphatemia, associated with inappropriately normal or low levels of 1,25-dihydroxyvitamin D. Diagnosis of this disease is often challenging. The following case report described a middle-aged man with symptoms of bone pain and severe muscle weakness, who was found to have TIO. The tumor responsible for the symptoms was localized on his thigh and its resection resulted in normalization of blood chemistry and complaints. Subsequent microscopic examination revealed a phosphaturic mesenchymal tumor, mixed connective tissue type. The authors reinforce the importance of recognition of this disease, as severe disability and even death can be avoided with the surgical removal of the causative tumor.
Osteomalacia; growth factor fibroblasts; phosphorus metabolism disorder; hypophosphatemia; hypophosphatemic rickets
Osteomalácia induzida por tumor (OIT) é uma síndrome paraneoplásica rara, causada por hiperprodução do agente fosfatúrico, levando a hipofosfatemia e hiperfosfatúria crônicas, associadas a níveis reduzidos ou inapropriadamente normais de 1,25-dihidroxivitamina D. O diagnóstico dessa doença é, geralmente, desafiador. O relato de caso aqui apresentado descreveu um homem de meia-idade, com quadro inicial de dor óssea, fraqueza muscular extrema e hipofosfatemia, com diagnóstico tardio de OIT. O tumor responsável pelos sintomas foi localizado em membro inferior, e sua exérese resultou em normalização das alterações bioquímicas e dos sintomas. O exame microscópico da lesão revelou tumor mesenquimal fosfatúrico, tecido conectivo misto. Os autores reforçam a importância do reconhecimento dessa entidade, uma vez que a remoção do tumor responsável pelos sintomas pode evitar sérias complicações ou mesmo a morte.
Osteomalácia; fatores de crescimento de fibroblastos; distúrbios do metabolismo do fósforo; hipofosfatemia; raquitismo hipofosfatêmico
CLINICAL CASE REPORT
Tumor-induced osteomalacia: a case report
Osteomalácia induzida por tumor: relato de caso
Daniel Dutra Romualdo-SilvaI; Bárbara Campolina Carvalho SilvaI; Cristiane Vasconcelos CaetanoI; Angélica Maria França Paiva TibúrcioI; Maurício Buzelin NunesII; Sérgio Almeida Pinheiro ChagasII; Elizandra Tomazela Laurenti PolitoIII; Antônio Rodrigues FerreiraI; Saulo PurischI
IDepartamento de Doenças Osteometabólicas, Serviço de Endocrinologia, Hospital Santa Casa, Belo Horizonte, MG, Brazil
IIDepartamento de Anatomia Patológica, Hospital Santa Casa, Belo Horizonte, MG, Brazil
IIIServiço de Reumatologia, Hospital Santa Casa, Belo Horizonte, MG, Brazil
Correspondence to Correspondence to: Bárbara Campolina Carvalho Silva Centro de Estudos e Pesquisa Clínica de Endocrinologia e Metabologia (CEPCEM) Av. Francisco Sales, 1.111, 5º andar, Ala D Santa Efigênia 30150-221 Belo Horizonte, MG, Brasil barbaracampolina@mac.com
ABSTRACT
Tumor-induced osteomalacia (TIO) is a rare paraneoplasic syndrome with overproduction of fibroblast growth factor 23 as a phosphaturic agent, leading to chronic hyperphosphaturia and hypophosphatemia, associated with inappropriately normal or low levels of 1,25-dihydroxyvitamin D. Diagnosis of this disease is often challenging. The following case report described a middle-aged man with symptoms of bone pain and severe muscle weakness, who was found to have TIO. The tumor responsible for the symptoms was localized on his thigh and its resection resulted in normalization of blood chemistry and complaints. Subsequent microscopic examination revealed a phosphaturic mesenchymal tumor, mixed connective tissue type. The authors reinforce the importance of recognition of this disease, as severe disability and even death can be avoided with the surgical removal of the causative tumor.
Keywords: Osteomalacia; growth factor fibroblasts; phosphorus metabolism disorder; hypophosphatemia; hypophosphatemic rickets
RESUMO
Osteomalácia induzida por tumor (OIT) é uma síndrome paraneoplásica rara, causada por hiperprodução do agente fosfatúrico, levando a hipofosfatemia e hiperfosfatúria crônicas, associadas a níveis reduzidos ou inapropriadamente normais de 1,25-dihidroxivitamina D. O diagnóstico dessa doença é, geralmente, desafiador. O relato de caso aqui apresentado descreveu um homem de meia-idade, com quadro inicial de dor óssea, fraqueza muscular extrema e hipofosfatemia, com diagnóstico tardio de OIT. O tumor responsável pelos sintomas foi localizado em membro inferior, e sua exérese resultou em normalização das alterações bioquímicas e dos sintomas. O exame microscópico da lesão revelou tumor mesenquimal fosfatúrico, tecido conectivo misto. Os autores reforçam a importância do reconhecimento dessa entidade, uma vez que a remoção do tumor responsável pelos sintomas pode evitar sérias complicações ou mesmo a morte.
Descritores: Osteomalácia; fatores de crescimento de fibroblastos; distúrbios do metabolismo do fósforo; hipofosfatemia; raquitismo hipofosfatêmico
INTRODUCTION
Hypophosphatemic osteomalacia comprises hereditary and acquired disturbances leading to impairment of matrix mineralization in remodeling sites of mature bones. Many syndromes have been described as causing this disease and the main are X-linked hypophosphatemic rickets (XHR), autosomal-dominant hypophosphatemic rickets (ADHR) and tumor-induced osteomalacia (TIO). Although these conditions exhibit different etiopathogenic mechanisms, they share the same pathophysiologic pathway: reduction in the phosphate reabsorption by the renal tubules, leading to chronic hyperphosphaturia and hypophosphatemia, associated with inappropriately normal or low levels of 1,25-dihydroxyvitamin D or calcitriol (1).
Literature suggests that one of the determinants for this pathophysiologic change is the high serum levels and increased activity of fibroblast growth factor 23 (FGF23) by overproduction or impairment to its degradation. FGF23 is produced by osteogenic cells, osteoblasts and osteocytes and exerts inhibitory activity on type IIa and IIc sodium-phosphate (NaPiT-IIa and IIc) co-transport system in proximal tubules, promoting hyperphosphaturia, beyond inhibit renal 1α-hydroxylation of 25-hydroxyvitamin D (1-7). Circulating FGF23 detectable in healthy adults and children increases the likelihood that this growth factor may be involved in the physiological regulation of phosphate homeostasis (3).
TIO is a rare paraneoplasic syndrome that leads to acquired hypophosphatemic osteomalacia and is related to mesenchymal or mixed connective tissues tumors that markedly overexpress the FGF23 messenger RNA and protein, which cannot be adequately degraded by specific endopeptidases (1,3,8,9). Tumors less frequently involved are fibromas, condrosarcomas, hystiocytomas, neuroblastomas, osteosarcomas and soft tissue tumors (angiosarcomas, prostate carcinoma, schwannoma, osteoblastoma and mixed neuroendocrine tumors). Other clinical syndromes have been also associated with FGF23 overproduction, such as neurofibromatosis type 1 (NF-1), epidermic nevus syndrome, McCune-Albright syndrome (MAS) and fibrous dysplasia (1,10). Two other proteins seem to be overexpressed by these tumors, contributing to the mineralization disturbance: matrix extracellular phosphoglycoprotein (MEPE) e secreted frizzled-related protein-4 (sFRP-4), but their mechanisms of action have not yet been elucidated (1,11).
The diagnostic management and clinical-laboratorial definition of TIO are delayed in most of cases, due to the small size and slow growth of these tumors, which frequently are not detected in clinical examinations. As clinical and biochemical changes of TIO are potentially reversible by tumor removal, its localization is essential (1,8,12).
The authors reported a case of chronic evolution hypophosphatemic osteomalacia, characterized by severe muscle weakness, pain and difficult diagnosis. The development in adult age and negative family history led us to suspect the existence of TIO, which was confirmed by pathological testing. Beyond registering this uncommon case, this report aimed to discuss the diagnostic and therapeutic management of TIO.
CASE REPORT
A 45-year-old male sought medical attention complaining of pain in his knees, ankles and feet, extended to thighs and lumbosacral region, which became progressive and debilitating. Four years later, he was referred to medical assistance at our institution to a different medical staff, while experiencing severe muscle weakness. At that time, he was unable to walk without support. He had a medical history of hypertension and suffered a traumatic fracture of his right femur several years before. He denied any family history of metabolic bone disease.
Upon physical examination, an accentuation of thoracic kyphosis was found, diminished muscle strength of upper and lower limbs and normal sensorial neurological examination. His gait was significantly limited, subordinated to pain and muscle weakness.
Initial laboratory testing revealed a normal calcium level of 8.8 mg/dL (normal range: 8,4-10,2 mg/dL), a low phosphorus level of 1,5 mg/dL (normal range: 2,54,5 mg/dL), an elevated alkaline phosphatase level of 893 U/L (normal range: 50-250 U/L), a normal parathyroid hormone (PTH) of 15 pg/mL (normal range: 7-53 pg/mL), and a insufficient 25-hydroxyvitamin D level of 19,9 ng/mL (normal range: 30-80 ng/mL). His 1,25-dihydroxyvitamin D levels were not assessed. Complete blood count, liver and kidney function tests, thyroid-stimulating hormone, serum protein electrophoresis, myelogram, creatine phosphokinase, antinuclear antibody, aldolase and rheumatoid factor were all normal. A 24-hour urine sample revealed a normal phosphorus level of 470 mg/24 hours (normal range: 300-1.000mg/24 hours). Electromyography and nerve conduction studies were normal.
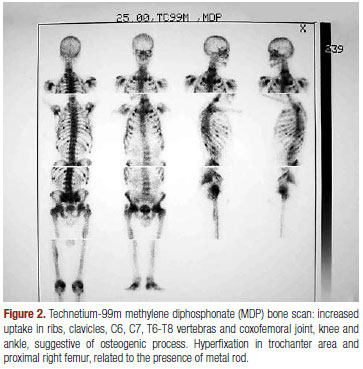
Radiological images showed decreased bone mineralization in ribs, vertebral bodies and hips, consolidated fractures in costal arches and left femur, multiple fractures of thoracic and lumbar vertebras, enlargement of the hip joint and Looser's zones in iliac bones and hips (Figure 1). A technetium-99m methylene diphosphonate (MDP) bone scan showed increased uptake in vertebras, ribs, clavicles and bones around almost all of the joints (Figure 2). Bone biopsy showed bone neoformation, without signs of fibrosis or neoplasm. However, a bone biopsy doubly labeled with tetracycline was not obtained as part of investigational exams.
The differential diagnosis at the time, included osteomalacia and the initial hypothesis, were malabsorption syndromes, like celiac disease and inflammatory bowel disease, since he had referred an increased defecation frequency. However, confirmatory tests were negative for these diseases. The normal urinary phosphorus in presence of low serum phosphorus, indicating a renal phosphate wasting, was overlooked, and phosphate tubular reabsorption rate was not assessed at this time. Even without a correct diagnostic, medical therapy was initiated with oral phosphorus (1 g daily), calcium (1,2 g of elemental calcium daily), and calcitriol (0,5 µg daily), and significant improvement in his symptoms followed. Two years later, he continued medical treatment, asymptomatic, able to walk without help and his levels of serum calcium and phosphorus were within the normal range, but his intact PTH was elevated at 189 pg/mL, and the curative treatment have not been attempted.
About six years after the onset of the original symptoms, he was referred to our service. Patient's records were all checked and hypophosphatemic osteomalacia was included as a differential diagnosis. This diagnosis was considered based on low phosphorus level before the treatment associated with inappropriately normal urinary phosphorus, indicating the presence of renal phosphate wasting and carrying on the suspect of hypophosphatemic rickets or osteomalacia. As symptoms developed in adulthood and there was no family history of metabolic bone disease, the main suspect was TIO.
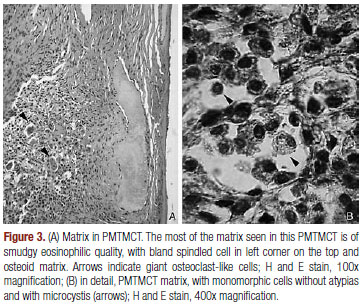
Based on this hypothesis, palpable masses were sought on physical examination, with special attention to the extremity inspection. A 3-cm, mobile, non-tender and firm mass was noted on his right thigh. Computed tomography scan of thorax, abdomen and paranasal sinuses were normal. Given the possibility that the thigh tumor was associated with the osteomalacia, it was removed and following microscopic examination showed a neoplasia with small cells - some in a bland spindled shape with microcystis and amorphous calcified deposits, beyond vessels with hyalinized walls, giant cells osteoclast-like and osteoid matrix, compatible with phosphaturic mesenchymal tumor, mixed connective tissue type (PMTMCT) (Figure 3). One week later, levels of serum calcium and phosphorus were within the normal range without medical treatment, confirming the diagnosis. At 12-month follow-up, he continued well, and his serum biochemistry remained normal.
DISCUSSION
This study presented a patient with phosphaturic mesenchymal TIO, confirmed by pathologic exam and with metabolic disturbance resolution after tumor removal.
TIO is a rare paraneoplasic syndrome with overproduction of FGF23 as a phosphaturic agent, frequently offering difficulty in diagnostic confirmation and clinical management (1,2,8,9,11,13). Until 2005, 120 cases have been reported in literature (9). Its radiological and clinical manifestations are equal to that found in other kinds of osteomalacia. However, bone pain and fractures are more severe; muscle weakness and fatigue are more frequent and it is generally found in persons with advanced age in comparison with hereditary types. Bone and muscle complications are the main features of these tumors - mostly small and benign and of difficult diagnosis by physical exam, requiring extensive radiological investigation to locate them (1,2,14-16).
Hypophosphatemic osteomalacia diagnosis requires the detection of hyperphosphaturia, hypophosphatemia, increased levels of alkaline phosphatase, normocalcemia and normal or reduced calciuria. A phosphorus tubular reabsorption rate less than 85% is indicative of hyperphosphaturia. Calcitriol levels are inappropriately normal or low, justifying the slight increase in PTH levels in these patients. It must be remembered that PTH also exerts phosphaturic effect due to promotion of internalization of sodium-phosphate cotransporters and its lisossomal degradation (1,2,9).
Laboratory exams, in this case presented, were compatible with the usual findings described above. Initial evaluation revealed a normal calcium and PTH levels, a hyperphosphatasia, and a normal urinary phosphorus concentration in spite of low serum phosphorus, indicating a renal phosphorus wasting. Unfortunately, 1,25-dihydroxyvitamin D levels and tubular reabsorption of phosphate were not assessed, and renal phosphorus wasting was overlooked, which could have delayed the syndrome diagnosis.
The occult nature of TIO delays its recognition, and the average time from onset of symptoms to a correct diagnosis often exceeds 2.5 years. Once the syndrome is recognized, inability to locate the underlying tumor further delays definitive treatment by an average of five years (9).
Most of TIO cases are due to currently recognized phosphaturic mesenchymal tumor (mixed connective tissue variant), an extremely rare tumor that is found mainly in soft tissues and bone in lower limbs as small and clinically unapparent lesions. As proposed by Weidner and Santa Cruz (17), in 1987, and recently reaffirmed in a review (8), these tumors have been considered a distinct histopathological entity. According to Jan de Beur (9), 70 to 80% of tumors in TIO correspond to PMTMCT, including hemagiopericytomas. Many of these tumors appear to have been largely misunderstood, ignored, or unknown by most pathologists and clinicians. It is likely that the rarity of PMTMCTs and their polymorphous histological appearance account, in large part, for their frequent misdiagnoses (8). The defined histological features of PMTMCT includes low cellularity, myxoid changes, bland spindled cells, distinctive "grungy" calcified matrix, fat, hemangiopericytoma-like vessels, microcystis, hemorrhage, osteoclasts and an incomplete rim of membranous ossification. They are generally benign, but malignant variants have already been described (8,9). Expression of FGF23 by Real Time Polymerase Chain Reaction (RT-PCR) and by immunohistochemistry indicates the involvement of these tumors on TIO pathogenesis (8,11). It is not clear, however, if FGF23 is the only (or even main) mediator of phosphaturia (11).
Treatment of TIO involves localization and surgical removal of tumor, promoting reversal of its clinical and biochemical changes. Thus, tumor localization is essential and, once suspected, requires detailed clinical examination with special attention to lower limbs and craniofacial mass and careful imaging study with computer tomography (CT), magnetic resonance image (MRI) or even PET-CT (1,9,14). In vitro studies showed that some of these tumors express somatostatin receptors (SSTRs) and successful localization of causative tumors with the use of 111Indio pentetreotide or octreotide scintigraphy has been demonstrated (9,14). Recent studies have described that reliable FGF23 assays have the potential to be clinically useful in the diagnosis and management of TIO and may help to establish whether tumors have been completely removed by surgery (3,11,13).
The serum levels of phosphate and calcitriol as well as the tubular phosphate reabsorption become normal in hours or days after surgical removal of tumor. Serum biochemical markers of bone turnover, such as the osteocalcin level and alkaline phosphatase activity tend to take longer to normalize (1,3,14). Serum FGF23 levels fall is also detected. According to a recent study, the plasma half-life of serum FGF23 is in the range of 46-58 minutes in patients with PMTMCT surgically removed (2).
Clinical treatment of hypophosphatemic osteomalacia aims at minimizing the metabolic disturbance, reducing bone deformities and improving symptoms, although the hyperphosphaturia persists, once this treatment does not alter the impaired tubular phosphate reabsorption. It includes phosphate administration (elementary phosphorus at dose of 30-60 mg/kg/day, divided in 4-6 fractions) and calcitriol (dose of 3070 ng/kg/day). With adequate treatment, beyond the biochemical normalization, the serum levels of alkaline phosphatase decrease, reaching normal or slightly elevated levels. Doses of phosphate and calcitriol need to be followed as they increase the risk of secondary and tertiary hyperparathyroidism and nephrolitiasis, respectively (1,9,14,15).
In the case presented, there was an increase of PTH levels after starting medical treatment with phosphate and calcitriol. The augmenting of PTH could be explained by phosphorus administration itself, since phosphate can induce PTH stimulation and secondary hyperparathyroidism. When it happens, calcitriol dose should be increased and phosphorus dose should be decreased (1). However, secondary hyperparathyroidism can persist for long periods of time, leading to autonomous function of parathyroid, characterizing the tertiary hyperparathyroidism, which can be severe and hard to treat (15).
Differential diagnosis of this patient was ADHR that can exhibit a variable and delayed age at the onset. Patients with adult onset ADHR may present severe bone pain, pseudofractures and weakness (1,9). The absence of family history, as well the severity and rapid progression of symptoms, made this hypothesis improbable. Serum levels of FGF23 would help in preoperative diagnostic definition as suggested by Takeuchi and cols. (18). However, this exam was not available in our institution. The identification of a thigh mass, common location of PMTMCT, strengthened our suspicion of TIO. The following tumor histopathology exam and the resolution of hypophosphatemia a week after surgery have confirmed the initial suspicion of TIO.
The case presented here, in opposition to the most cases related in literature, revealed an easily located tumor, showing us that if the diagnosis of TIO had been made before, the treatment and cure of this patient would have been anticipated. Although TIO represents a rare disease, we reinforce the importance of its recognition, as severe disability and even death can be avoided with the surgical removal of the causative tumor.
Acknowledgments: we would like to acknowledge Dr. Maria Marta Sarquis Soares, from Universidade Federal de Minas Gerais (UFMG), for her text revision, and Dr. Willon Garcia Carvalho, from Serviço de Radiologia of Santa Casa de Belo Horizonte, Minas Gerais, for his assistance with the radiographic images.
Disclosure: no potential conflict of interest relevant to this article was reported.
Received in Mar/3/2008
Accepted in Nov/29/2008
- 1. Filho HM, Castro LCG, Damiani D. Hypophosphatemic rickets and osteomalacia. Arq Bras Endocrinol Metabol. 2006;50(4):802-13.
- 2. Khosravi A, Cutler CM, Kelly MH, Chang R, Royal RE, Sherry RM, et al. Determination of the elimination half-life of fibroblast growth factor-23. J Clin Endocrinol Metab. 2007;92(6):2374-7.
- 3. Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, et al. Fibroblast Growth Factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Eng J Med. 2003;348(17):1656-63.
- 4. Yoshiko Y, Wang H, Minamizaki T, Ijuin C, Yamamoto R, Suemune S, et al. Mineralized tissue cells are a principal source of FGF23. Bone. 2007;40(6):1565-73.
- 5. Yamashita H, Yamazaki Y, Hasegawa H, Yamashita T, Fukumoto S, Shigematsu T, et al. Fibroblast Growth Factor 23 (FGF-23) in patients with transient hypoparathyroidism: Its important role in serum phosphate regulation. Endocr J. 2007;54(3):465-70.
- 6. Imel EA, Hui SL, Econs MJ. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J Bone Miner Res. 2007;22(4):520-6.
- 7. Murer H, Hernando N, Forster I, Biber J. Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev. 2000;80(4):1373-409.
- 8. Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, Cho JY, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity. An analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28(1):1-30.
- 9. Jan de Beur SM. Tumor-induced osteomalacia. JAMA. 2005;294(10): 1260-7.
- 10. Kobayashi K, Imanishi Y, Koshiyama H, Miyauchi A, Wakasa K, Kawata T, et al. Expression of FGF23 is correlated with serum phosphate level in isolated fibrous dysplasia. Life Sci. 2006;78(20):2295-301.
- 11. Nelson AE, Bligh RC, Mirams M, Gill A, Au A, Clarkson A, et al. Clinical case seminar: Fibroblast growth factor 23: a new clinical marker for oncogenic osteomalacia. J Clin Endocrinol Metab. 2003;88(9):4088-94.
- 12. Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol. 2007;18(6):1637-47.
- 13. Imel EA, Peacock M, Pitukcheewanont P, Heller HJ, Ward LM, Shulman D, et al. Sensitivity of fibroblast growth factor 23 measurements in tumor-induced osteomalacia. J Clin Endocrinol Metab. 2006;91(6):2055-61.
- 14. Carpenter TO. Oncogenic osteomalacia--a complex dance of factors. N Eng J Med. 2003;348(17):1705-8.
- 15. Moreira RO, Leal CT, Lacativa PG, Figueiredo JG, Lima MB, Farias LM. Hyperparathyroidism associated with hypophosphatemic osteomalacia: case report and review of the literature. Arq Bras Endocrinol Metab. 2006;50(1):150-5.
- 16. Cheung FMF, Ma L, Wu WC, Siu TH, Choi PT, Tai YP. Oncogenic osteomalacia associated with an occult phosphaturic mesenchymal tumour: clinico-radiologico-pathological correlation and ultrastructural studies. Hong Kong Med J. 2006;12(4):319-21.
- 17. Weidner N, Santa Cruz D. Phosphaturic mesenchymal tumors: a polymorphous group causing osteomalacia or rickets. Cancer. 1987;59(8):1442-54.
- 18. Takeuchi Y, Suzuki H, Ogura S, Imai R, Yamazaki Y, Yamashita T, et al . Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. J Clin Endocrinol Metab. 2004;89(8):3979-82.
Publication Dates
-
Publication in this collection
30 June 2009 -
Date of issue
Apr 2009
History
-
Received
03 Mar 2008 -
Accepted
29 Nov 2009