Abstracts
RNA splicing is an essential, precisely regulated process that occurs after gene transcription and before mRNA translation, in which introns may be removed and exons, retained. Variability in splicing patterns is a major source of protein diversity from the genome and function to generate a tremendously diverse proteome from a relatively small number of genes. Changes in splice site choice can determine different effects on the encoded protein. Small changes in peptide sequence can alter ligand binding, enzymatic activity, allosteric regulation, or protein localization. Errors in splicing regulation have been implicated in a number of different disease states. This study reviewed the mechanisms of splicing and their repercussion in endocrinology, emphasizing its importance in some thyroid physiological and pathological conditions.
Splicing; thyroid gland; TSH; TPO; thyroid hormone receptor; TGFbeta
Após a transcrição genética e antes da tradução do mRNA, ocorre o splicing do RNA, que consiste em um processo essencial, precisamente regulado, por meio do qual podem ocorrer excisões de íntrons e retenções de éxons. A variabilidade dos padrões de splicing é a principal fonte de diversidade de proteínas geradas por um pequeno número de genes. Alterações na escolha do sítio de splicing podem determinar efeitos diferentes nas proteínas codificadas. Pequenas alterações na sequência peptídica podem alterar o seu sítio de ligação de substratos, sua atividade enzimática, a regulação alostérica ou a localização proteica. Erros na regulação do splicing têm sido implicados em grande número de doenças. Nessa revisão, foram descritos os mecanismos de splicing enfatizando sua importância em algumas condições fisiológicas e patológicas envolvendo a tireoide.
Splicing; glândula tireoide; TSH; TPO; receptor do hormônio tiroidiano; TGF-beta
REVIEW
Splicing variants impact in thyroid normal physiology and pathological conditions
Variantes de splicing e seu impacto em condições fisiológicas e patológicas da tireoide
Elizabete Rosária de MirandaI; Luiz De MarcoII; Maria Marta Sarquis SoaresIII
ICurso de Pós-Graduação em Ciências Aplicadas à Saúde do Adulto, Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, MG, Brasil; Hospital Luxemburgo, Belo Horizonte, MG, Brasil
IIDepartamento de Cirurgia, UFMG, Belo Horizonte, MG, Brasil
IIIDepartamento de Clínica Médica, UFMG, Belo Horizonte, MG, Brasil; pesquisadora, Instituto Felício Rocho de Pesquisa e Educação Continuada (IFERPEC), Belo Horizonte, MG, Brasil
Correspondence to Correspondence to: Maria Marta Sarquis Soares Rua Uberaba, 370/705 - Barro Preto 30180-080 - Belo Horizonte, MG, Brasil martasarquis@hotmail.com
ABSTRACT
RNA splicing is an essential, precisely regulated process that occurs after gene transcription and before mRNA translation, in which introns may be removed and exons, retained. Variability in splicing patterns is a major source of protein diversity from the genome and function to generate a tremendously diverse proteome from a relatively small number of genes. Changes in splice site choice can determine different effects on the encoded protein. Small changes in peptide sequence can alter ligand binding, enzymatic activity, allosteric regulation, or protein localization. Errors in splicing regulation have been implicated in a number of different disease states. This study reviewed the mechanisms of splicing and their repercussion in endocrinology, emphasizing its importance in some thyroid physiological and pathological conditions.
Keywords: Splicing; thyroid gland; TSH; TPO; thyroid hormone receptor; TGFbeta
RESUMO
Após a transcrição genética e antes da tradução do mRNA, ocorre o splicing do RNA, que consiste em um processo essencial, precisamente regulado, por meio do qual podem ocorrer excisões de íntrons e retenções de éxons. A variabilidade dos padrões de splicing é a principal fonte de diversidade de proteínas geradas por um pequeno número de genes. Alterações na escolha do sítio de splicing podem determinar efeitos diferentes nas proteínas codificadas. Pequenas alterações na sequência peptídica podem alterar o seu sítio de ligação de substratos, sua atividade enzimática, a regulação alostérica ou a localização proteica. Erros na regulação do splicing têm sido implicados em grande número de doenças. Nessa revisão, foram descritos os mecanismos de splicing enfatizando sua importância em algumas condições fisiológicas e patológicas envolvendo a tireoide.
Descritores: Splicing; glândula tireoide; TSH; TPO; receptor do hormônio tiroidiano; TGF-beta
THE SPLICE AND SPLICING ALTERNATIVE PROCESSES
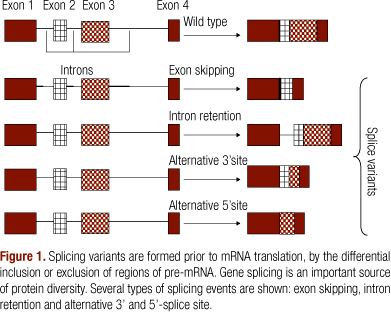
A gene is first transcribed into a pre-mRNA, which is a copy of the genomic DNA containing intronic and exonic regions. RNA splicing is an essential, precisely regulated process, that occurs after gene transcription and before mRNA translation. It is essential for the generation of protein diversity and can also have regulatory functions (1). During splicing, alternative usage of splice donor or acceptor sites can lead to various exons and introns being skipped or retained and creating alternative usage of splice donor or acceptor sites, in order to generate a diverse array of mRNAs from a single pre-mRNA, a process referred to as alternative RNA splicing (2). Alternative splicing is, therefore, the joining of different 5' and 3' splice sites, allowing individual genes to express multiple mRNAs that encode proteins with diverse and even antagonistic functions (3).
It is estimated that at least 55% of all human genes encodes alternatively spliced transcripts and some of these are alternatively spliced to generate an amazingly large number of distinct mRNA isoforms (1,4,5). Many gene transcripts have multiple splicing patterns and some have thousands. Hence, alternative splicing generates a tremendously diverse proteome from a relatively small number of genes.
In a typical multi-exon mRNA, the splicing pattern can be altered in many ways (Figure 1). Most exons are constitutive; they are always spliced or included in the final mRNA. A regulated exon that is sometimes included and sometimes excluded from the mRNA is called a cassette exon (6). Exons can also be lengthened or shortened by altering the position of one of their splice sites. The 5'-terminal exons of an mRNA can be switched through the use of alternative promoters and alternative splicing (1). Similarly, the 3'-terminal exons can be switched by combining alternative splicing with alternative polyadenylation sites. Alternative promoters are primarily an issue of transcriptional control. Finally, some important regulatory events are controlled by the failure to remove an intron, a splicing pattern called intron retention (7).
The excision of the introns from a pre-mRNA and joining of the exons are directed by special sequences at the intron/exon junctions called splice sites (8). The 5' splice site marks the exon/intron junction at the 5' end of the intron. This includes a GU (guanine-uracil) dinucleotide at the intron end encompassed within a larger, less conserved consensus sequence (8). At the other end of the intron, the 3' splice site region has three conserved sequence elements: the branch point, followed by a polypyrimidine tract, followed by a terminal AG at the extreme 3' end of the intron (7).
The precise excision of introns from pre-mRNA is performed by the spliceosome, a macromolecular machine containing five small mRNAs and numerous proteins (Figure 2). The splice site consensus sequences generally lack sufficient information to determine whether a site will assemble a spliceosome and play a role in splicing. Other information and interactions are necessary to activate their use (9). Usually, spliceosomal components binding on opposite sides of an exon can interact to stimulate excision of the flanking introns (10). This process is called exon definition and, apparently, occurs in most internal exons. On top of this process, there are many non-splicing site regulatory sequences that strongly affect spliceosome assembly (10). RNA elements that act positively to stimulate spliceosome assembly are called splicing enhancers (11). Exonic splicing enhancers are commonly found even in constitutive exons. Intronic enhancers also occur and appear to differ from exonic enhancers (11). Conversely, other RNA sequences act as splicing silencers or repressors to block spliceosome assembly and certain splicing choices. Again, these silencers have both exonic and intronic varieties. Some regulatory sequences create an RNA secondary structure that affects splice site recognition, but most seem to be protein binding sites (11).
The spliceosome performs the two primary functions of splicing: it recognizes splicing signals in the pre-mRNA and catalyses the removal of the intron and the joining of the exons (12) (Figure 2). The spliceosome catalyzes the two transesterification steps of the splicing reaction. In the first step, the 2'-hydroxyl group of a special "A" (adenine) residue at the branch-point attacks the phosphate at the 5' splice site. This leads to cleavage of the 5'-exon from the intron and the concerted ligation of the intron 5'-end to the branch-point 2'-hydroxyl. This step produces two reaction intermediates, a detached 5'-exon and an intron/3'-exon fragment in a lariat configuration containing a branched "A" nucleotide at the branch-point (12). The second transesterification step is the attack on the phosphate at the 3'-end of the intron by the 3'-hydroxyl of the detached exon. This ligates the two exons and releases the intron, still in the form of a lariat (7,12). The mechanism of splice site selection in constitutive and alternative splicing are closely connected, since components of the splicing machinery, essential for constitutive splicing, also have a role in the regulation of alternative splicing (13).
The physiological activity of splicing variant products, compared to the wild-type protein, may be completely different (14). Changes in splice site choice can determine different effects on the encoded protein. Whole functional domains may be added or deleted from the protein coding sequence. Small changes in peptide sequence can alter ligand binding, enzymatic activity, allosteric regulation, or protein localization (13). Genetic switches based on alternative splicing are important in many cellular and developmental processes, including sex determination, apoptosis, axon guidance, cell excitation and contraction, and many others (15). Errors in splicing regulation have been implicated in a number of different disease states (13,14).
SPLICING VARIANTS AND THYROID PHYSIOLOGICAL AND PATHOLOGICAL CONDITIONS
Thyroid hormone action regulation
The primary mechanism of thyroid hormone (TH) action is to modulate target gene transcription via nuclear thyroid hormone receptors (TRs). Nuclear receptors, including TRs, are hormone-regulated transcription factors (16). TRs control growth, development, and homeostasis by binding to TH response elements (TREs) in target promoters and modulating transcription. Multiple TR isoforms exist in human, encoded by c-erbAα and c-erbAβ genes. Alternative splicing of the c-erbAα gene produces mRNAs encoding two proteins, TRα1 and c-erbAα2. A further splicing variant, c-erbA-α3, has been identified in rats. Of these products, only TRα1 binds TH (17) and this isoform has an important role in mediating TH action during first trimester of human fetal brain development (17). The c-erbAβ gene gives rise to TRβ1 and TRβ2, both of which bind to TH.
TRs have bidirectional regulatory properties and can either repress or activate target gene expression by alternatively recruiting either corepressors (CoRs) or coactivators (CoAs) (18). It is these auxiliary coregulator proteins that mediate, in turn, the actual molecular events necessary for transcriptional regulation. CoRs typically place repressive marks in chromatin and mediate inhibitory interactions with the general transcriptional machinery, whereas CoAs do the opposite (18).
The most extensively characterized CoRs for the nuclear receptors are silencing mediator of retinoic acid and TRs (SMRT) and nuclear receptor corepressor (N-CoR) (18). SMRT is regulated by MAPK kinase kinase (MAPKKK) cascades, that induce its release from its receptor partners, its export from nucleus to cytoplasm, and derepression of target gene expression (18). Intriguingly, the otherwise closely related N-CoR is refractory to MAPKKK signaling under the same conditions.
Interestingly unliganded TRs repress TH activated genes by recruiting CoRs, such as N-CoR and SMRT/TRs-associated corepressor 2 (TRAC2) (16). For example, unliganded TRs activate the thyroid-stimulating hormone (TSH) and pro-TSH releasing hormone promoters, and T3 represses transcription of both genes to mediate feedback inhibition of TH synthesis.
It has recently become appreciated that both SMRT and N-CoR are subject to extensive alternative mRNA splicing events that generate a diverse series of distinct CoR variants from each locus (18). These CoR splicing variants have not the same molecular architecture, are expressed at different abundances in distinct tissues, and display distinct affinities for different nuclear receptor partners (18). Alternative mRNA splicing can generate forms of SMRT that are either sensitive or resistant to inhibition by MAPK cascades. On the other hand, N-CoR variants are generically resistant to this regulatory network, and the interaction properties versus subcellular distribution properties of these CoRs are independently regulated by distinct elements in the MAPKKK hierarchy (18).
TSHβ splicing variant and immunology
TSH, as a member of glycoprotein hormones, consists of a common α-subunit and unique β-subunits, the latter being responsible for hormone specificity. TSH is synthesized by the anterior pituitary and regulates TH output, which, in turn, controls metabolic activity. Currently, the pituitary is believed to be the only source of TSH used by the thyroid. Recent studies have identified a TSHβ isoform that is expressed in the pituitary, in peripheral blood leukocytes (PBL), bone marrow (BM) and in the thyroid (19). This splicing variant consists of a portion of intron 4 and it includes the entire coding region of exon 5, but none of exon 4, thereby coding for a polypeptide that corresponds to 71.2% of the mature native TSH molecule. Interestingly, exon 5 of TSH, the coding portion retained in the novel TSH slice variant, is important for the biological function of TSH as it includes an 18-amino-acid 'seatbelt' region (CNTDNSDCIHEAVRTNYC), used for attachment to the α-subunit. This suggests that the splicing variant may retain the ability to function as a heterodimeric complex (19). In fact, it has been demonstrated that the polypeptide derived from the TSHβ splicing variant is biologically active with regard to its ability to induce a cAMP signal (20).
The fact that there are extra-pituitary sources of TSH, including TSH produced by cells of the immune system, has been known for over 20 years (19). Although TSH may have some activity as an immunoregulatory/cytokine like molecule within the immune system, several observations now point to a more direct link between the immune system, TSH and the hypothalamus-pituitary-thyroid axis. These findings may have implications for immune-endocrine interactions in the thyroid, if intrathyroidal production of the TSHβ splicing variant protein leads to enhanced immunogenicity due to unique physicochemical properties, resulting from misfolding, anomalous dimerization, or high output of the splicing variant in the thyroid (20). A system such as this could have negative consequences for the host, particularly if the TSHβ splicing variant displays an enhanced potential for immunogenicity relative to native TSHβ. It has been shown that TSHβ splicing variant expression increases in the thyroid following systemic virus infection (19). Intrathyroidal host response is linked to the TSHβ splicing variant, but not the native form of TSHβ (19). Local intrathyroidal production of the TSHβ splicing variant might also inadvertently lead to the generation of anti-TSH autoantibodies and autoimmune thyroiditis, as it can occur following systemic virus infection (20).
As the BM cells appear to be a primary source of TSHβ in the thyroid, it is possible that the higher level of TSHβ gene expression is either because of an increase in TSH synthesis by resident BM cells in the thyroid, or because of the increased trafficking of BM cells to the thyroid during infection. On the one hand, it could function agonistically to elicit a TH response leading to the increases in T3 and T4 synthesis and an upregulation in cellular and physiologic metabolic activity. The fact that the TSHβ splicing variant was capable of inducing a cAMP response provides some support for this possibility. Conversely, it is possible that the TSHβ splicing variant has an antagonistic activity, which serves to restrict TH synthesis by binding to and competing for TSH receptor signaling. Clearly, considerably more work is needed to discriminate between these two possibilities in the context of both physiological and pathological states.
The expression of RET and its multiple splice forms
The c-RET proto-oncogene (RET, Rearranged during Transfection) encodes a member of the receptor tyrosine kinase (RTK) family (21) with roles in migration, development and survival of neural crest cells and their derivatives (22,23). Multiple RET transcripts, generated by alternative splicing of RET exons encoding the extracellular ligand-binding and C-terminal domains, are expressed in cells and tissues derived from the neural crest, branchial arches and ureteric bud (22,24).
RET mutations are responsible for the development of several human diseases, including papillary thyroid carcinoma (PTC), multiple endocrine neoplasia types 2A and 2B and familial medullary thyroid carcinoma (25,26). On the other hand, loss of function RET mutations are associated with Hirschsprung's disease (congenital megacolon), which is the most common congenital defect affecting the development of the enteric nervous system (25).
RET is alternatively spliced to produce three isoforms (designated RET9, RET43 and RET51) that differ in the carboxy-terminal sequence (24). RET isoforms have distinct signaling properties and suggest that RET51 is dispensable during embryogenesis, whereas RET9 is necessary and sufficient for normal development of the enteric nervous system and the excretory system (27).
There are 16 tyrosines in the intracellular domain of RET9 and RET43 and 18 tyrosines in that of RET51. Amongst these, tyrosines 1015 and 1062 (Y1015 and Y1062) are important for cell signaling mediated by both ligand-dependent and independent activation of RET (28). Y1015 represents a binding site for PLCc, and Y1062 represents a binding site for several adaptor proteins, such as SHC, SNT/FRS2, IRS1, DOK1, 4 and 5, and Enigma (28). As a result, various signaling pathways including the RAS/ERK, PI-3K/AKT, p38MAPK, JNK and ERK5 pathways are activated, mainly via phosphorylated Y1062 in RET (29). Consistent with these findings, mutation of Y1062 markedly impaired the transforming activity of all types of MEN2 mutant proteins (30).
The splicing site of the three RET isoforms is present just downstream of Y1062, and their different roles were recently demonstrated by NIH 3T3 transfection assay (31). RET51-MEN2A and RET51-MEN2B mutant proteins have stronger transforming activity than RET9-MEN2A and RET9-MEN2B mutant proteins, respectively (32). In addition, the activity of RET43 was very low (33). These findings suggested that the sequences downstream of Y1062 are important for binding of adaptor proteins and that RET51 may contribute more significantly to tumor development associated with MEN2 than RET9 and RET43. On the contrary, RET9 was shown to be critical for the development of the kidney and the enteric nervous system using targeted mutagenesis mice expressing RET9 or RET51 only (34), suggesting the possibility that RET9 and RET51 are involved in the activation of different signaling pathways in vivo (35). It appears that the transcripts coding for RET9 and RET51 are more abundant in MEN 2-associated tumors than mRNAs coding for the other RET isoform (36). Furthermore, RET51 can induce more neuritis outgrowth in pheochromocytoma PC12 cells than the corresponding active mutants of RET9 (37,38). These findings provide evidence that the biological properties of RET-MEN2 mutants depend on the interplay between the RET isoforms and the nature of the activating MEN2 mutation.
ALTERNATIVE SPLICING AND THYROID TUMORIGENESIS: CANDIDATES FOR TUMOR-SPECIFIC ALTERNATIVE SPLICING IN THE THYROID
Thyroid cancer is the most common neoplasia from the endocrine system with an estimated annual incidence of 122,800 worldwide. Most of the incidence variations are probably due to ethnic or environmental factors, such as radiation, dietary habits, or other external factors (39).
Thyroid cancers derived from follicular cells are divided into three subtypes: PTC, follicular thyroid carcinoma (FTC), and undifferentiated thyroid carcinoma (UTC). PTC and its variants are by far the most common thyroid malignancies, accounting for about 80% of all thyroid tumors, followed by FTC, which accounts for 10% to 20% of cases (40). Although key molecular events of thyroid tumorigenesis, such as RET/PTC and PAX8-PPARG rearrangements and BRAF (the gene for the B-type Raf kinase) point mutations, have been identified (41), other molecular mechanisms, such as alternative splicing, might be a natural source of disease-related alterations in gene expression (42).
Attempting to validate cancer-specific alternative splicing in the thyroid, using the thyroid libraries generated through the Human Cancer Genome Project (HCGP), it has been demonstrated that five genes (PTPN18, ABI3BP, PFDN5, SULF2, and ST5) presented isoforms differentially expressed between malignant and benign or normal thyroid (43). It is not clear, however, what kind of role these isoforms play in thyroid tumors. Many of the tumor-specific splice forms may not be associated with thyroid tumor progression but, instead, may represent events that occur early in the pathogenesis of thyroid tumors (43).
Thyroperoxidase (TPO) is the key enzyme in TH synthesis. The human TPO (hTPO) gene consists of 17 exons and 16 introns and spans about 150kb. Several single spliced variants have been described: TPO2, with exon 10 deleted (44); TPO3, with exon 16 deleted (45); TPO4, with exon 14 deleted; and TPO5, with exon 8 deleted (44-46). In a study analyzing the rates of hTPO mRNA with alternatively spliced exons 10, 14, and 16 in normal, benign, and malignant thyroid tissues, it has been found that spliced forms of exons 10 and 16 were more expressed by at least 50% in thyroid carcinomas. This event might partly explain the decrease in both the quantity and the level of activity of hTPO observed in thyroid cancer due to the loss of stability of the spliced isoforms (47).
The various isoforms of transforming growth factor-beta (TGF-β) are growth-inhibiting cytokines for cells of epithelial origin. In thyroid carcinomas and benign tumors, TGF-β1, TGF-β2, and TGF-β3 isoforms are expressed (48). In contrast to the abundant and homogeneous expression in differentiated thyroid carcinomas, TGF-β expression displays a highly variable interfollicular and intrafollicular pattern in multinodular goiters (48). TGF-β1 and activin A (ActA) induce similar intracellular signaling mediated by the SMAD (mothers against decapentaplegic homolog) proteins and have an antiproliferative effect on thyroid papillary carcinoma cell, exerting an important role in the control of thyroid tumorigenesis (49). Both, TGF-β1 and ActA, generate SMADs signaling with each regulating the expression of their target genes, SMAD7 and c-MYC. Since TGF-β and activin generate their intracellular signaling through the same components of the SMAD pathway, the unbalance of this pathway impairs both of anti-mitogenic signals in the cell (50).
Destruction of the TGF-β signaling network at the level of Smad genes is common in human carcinomas (51). Several sporadic epithelial tumors, in fact, show missense, nonsense and frameshift Smad4 point mutations. In a recent study of 56 different benign and malignant thyroid tumors Smad4 mutations were found in 27% of the lesions (51). The authors demonstrated the presence of a number of alternatively spliced Smad4 forms that are unable to resume the transcriptional activity of TGF-β. Smad4 is both frequently mutated and deregulated by aberrant splicing in thyroid tumors and these alterations may contribute as an early event to thyroid tumorigenesis. These data confer further relevance to the statement that escaping from TGF-β control is almost an obligate requirement in thyreocyte multistep transformation process (51).
The connection between cancer and novel splice variations has been well established but still needs more clarification. Identification of these specific splice variations will provide important insight into the molecular mechanism of normal cellular physiology as well as the disease processes in the thyroid.
CONCLUSIONS
The complete comprehension of the mechanism involved in the alternative splicing, its correlation with normal or mutant genes, and the differential downstream signaling may not only add to the fundamental understanding of complex pathways in health and disease, but also may help us to understand the resultant phenotype of the different syndromes associated with a particular gene. For the majority of alternative splicing events, however, we still need further investigations to clarify their functional significance. We could potentially use these splicing variant expressions as markers differentiating malignant from benign tumors, as markers of poor or good prognosis or targeting more precisely the diseases mechanisms as a therapeutic option.
Disclosure: no potential conflict of interest relevant to this article was reported.
Received in June/28/2009
Accepted in July/20/2009
- 1. Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103(3):367-70.
- 2. Bracco L, Kearsey J. The relevance of alternative RNA splicing to pharmacogenomics. Trends Biotechnol. 2003;21(8):346-53.
- 3. Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17(4):419-37.
- 4. Kan Z, Rouchka EC, Gish WR, States DJ. Gene structure prediction and alternative splicing analysis using genomically aligned ESTs. Genome Res. 2001;11(5):889-900.
- 5. Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17(2):100-7.
- 6. Smith CW, Nadal-Ginard B. Mutually exclusive splicing of alpha-tropomyosin exons enforced by an unusual lariat branch point location: implications for constitutive splicing. Cell. 1989;56(5):749-58.
- 7. Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291-336.
- 8. Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14(5):802-13.
- 9. Black DL. Finding splice sites within a wilderness of RNA. RNA. 1995;1(8):763-71.
- 10. Hoffman BE, Grabowski PJ. U1 snRNP targets an essential splicing factor, U2AF65, to the 3' splice site by a network of interactions spanning the exon. Genes Dev. 1992;6(12B):2554-68.
- 11. Libri D, Stutz F, McCarthy T, Rosbash M. RNA structural patterns and splicing: molecular basis for an RNA-based enhancer. RNA. 1995;1(4):425-36.
- 12. Dauksaite V, Akusjärvi G. The second RNA-binding domain of the human splicing factor ASF/SF2 is the critical domain controlling adenovirus E1A alternative 5'-splice site selection. Biochem J. 2004;381(Pt2):343-50.
- 13. Cáceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18(4):186-93.
- 14. Sarquis MS, Agrawal S, Shen L, Pilarski R, Zhou XP, Eng C. Distinct expression profiles for PTEN transcript and its splice variants in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome. Am J Hum Genet. 2006;79(1):23-30.
- 15. Baker BS. Sex in flies: the splice of life. Nature. 1989;340(6234): 521-4.
- 16. Meng X, Webb P, Yang YF, Shuen M, Yousef AF, Baxter JD, et al. E1A and a nuclear receptor corepressor splice variant (N-CoRI) are thyroid hormone receptor coactivators that bind in the corepressor mode. Proc Natl Acad Sci U S A. 2005;102(18):6267-72.
- 17. Iskaros J, Pickard M, Evans I, Sinha A, Hardiman P, Ekins R. Thyroid hormone receptor gene expression in first trimester human fetal brain. J Clin Endocrinol Metab. 2000;85(7):2620-3.
- 18. Jonas BA, Varlakhanova N, Hayakawa F, Goodson M, Privalsky ML. Response of SMRT (silencing mediator of retinoic acid and thyroid hormone receptor) and N-CoR (nuclear receptor corepressor) corepressors to mitogen-activated protein kinase kinase kinase cascades is determined by alternative mRNA splicing. Mol Endocrinol. 2007;21(8):1924-39.
- 19. Vincent BH, Montufar-Solis D, Teng BB, Amendt BA, Schaefer J, Klein JR. Bone marrow cells produce a novel TSHbeta splice variant that is upregulated in the thyroid following systemic virus infection. Genes Immun. 2009;10(1):18-26.
- 20. Schaefer JS, Klein JR. A novel thyroid stimulating hormone beta-subunit isoform in human pituitary, peripheral blood leukocytes, and thyroid. Gen Comp Endocrinol. 2009;162(3):241-4.
- 21. Takahashi M, Cooper GM. Ret transforming gene encodes a fusion protein homologous to tyrosine kinases. Mol Cell Biol. 1987;7(4):1378-85.
- 22. Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119(4):1005-17.
- 23. Avantaggiato V, Dathan N, Grieco M, Fabien N, Lazzaro D, Fusco A, et al. Developmental expression of the RET protooncogene Cell Growth Differ. 1994;5(3):305-11.
- 24. Tahira T, Ishizaka Y, Itoh F, Sugimura T, Nagao M. Characterization of ret proto-oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma cell line. Oncogene. 1990;5(1):97-102.
- 25. Pasini B, Ceccherini I, Romeo G. RET mutations in human disease. Trends Genet. 1996;12(4):138-44.
- 26. Edery P, Eng C, Munnich A, Lyonnet S. RET in human development and oncogenesis. Bioessays.1997;19(5):389-95.
- 27. Srinivas S, Wu Z, Chen CM, D'Agati V, Costantini F. Dominant effects of RET receptor misexpression and ligand-independent RET signaling on ureteric bud development. Development. 1999;126(7):1375-86.
- 28. Asai N, Murakami H, Iwashita T, Takahashi M. A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs their transforming activity and association with Shc adaptor proteins. J Biol Chem. 1996;271(30):17644-9.
- 29. Hayashi H, Ichihara M, Iwashita T, Murakami H, Shimono Y, Kawai K, et al. Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene. 2000;19(39):4469-75.
- 30. Hayashi Y, Iwashita T, Murakamai H, Kato Y, Kawai K, Kurowa K, et al. Activation of BMK1 via tyrosine 1062 in RET by GDNF and MEN2A mutation. Biochem Biophys Res Commun. 2001;281(3):682-9.
- 31. Kurokawa K, Kawai K, Hashimoto M, Ito Y, Takahashi M. Cell signalling and gene expression mediated by RET tyrosine kinase. J Int Med. 2003;253(6):627-33.
- 32. Borrello MG, Alberti L, Arighi E, Bongarzone I, Battistini C, Bardelli A, et al. The full oncogenic activity of Ret/ptc2 depends on tyrosine 539, a docking site for phospholipase Cgamma. Mol Cell Biol. 1996;16(5):2151-63.
- 33. Ishiguro Y, Iwashita T, Murakami H, Asai N, Iida K, Goto H, et al. The role of amino acids surrounding tyrosine 1062 in Ret in specific binding of the Shc phosphotyrosine-binding domain. Endocrinol. 1999;140(9):3992-8.
- 34. de Graaff E, Srinivas S, Kilkenny C, D'Agati V, Mankoo BS, Constantini F, et al. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev. 2001;15(18):2433-44.
- 35. Tsui-Pierchala BA, Ahrens RC, Crowder RJ, Milbrandt J, Johnson EM. The long and short isoforms of Ret function as independent signalling complexes. J Biol Chem. 2002;277(37):34618-25.
- 36. Lorenzo MJ, Eng C, Mulligan LM, Stonehouse TJ, Healey CS, Ponder BA, et al. Multiple mRNA isoforms of the human RET proto-oncogene generated by alternate splicing. Oncogene.1995;10(7):1377-83.
- 37. Rossel M, Pasini A, Chappuis S, Geneste O, Fournier L, Schuffenecker I, et al. Distinct biological properties of two RET isoforms activated by MEN 2A and MEN 2B mutations. Oncogene. 1997;14(3):265-75.
- 38. Iwashita T, Kato M, Murakami H, Asai N, Ishiguro Y, Ito S, et al. Biological and biochemical properties of Ret with kinase domain mutations identified in multiple endocrine neoplasia type 2B and familial medullary thyroid carcinoma Oncogene. 1999;18(26):3919-22.
- 39. Reis EM, Ojopi EP, Alberto FL, Rahal P, Tsukumo F, Mancini UM, et al. Large-scale transcriptome analyses reveal new genetic marker candidates of head, neck, and thyroid cancer. Cancer Res. 2005;65(5):1693-9.
- 40. Gimm O. Thyroid cancer. Cancer Lett. 2001;163(2):143-56.
- 41. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer:genetic evidence for constitutive activation of the RET/PTCRAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63(7):1454-7.
- 42. Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004;64(21):7647-54.
- 43. Guimarães GS, Latini FRM, Camacho CP, Maciel RMB, Dias-Neto E, Cerutti JM. Identification of candidates for tumor-specific alternative splicing in the thyroid. Genes Chromosomes Cancer. 2006;45(6):540-53.
- 44. Elisei R, Vassart G, Ludgate M. Demonstration of the existence of the alternatively spliced form of thyroid peroxidase in normal thyroid. J Clin Endocrinol Metab. 1991;72(3):700-2.
- 45. Zanelli E, Henry M, Charvet B, Malthièry Y. Evidence for an alternate splicing in the thyroperoxidase messenger from patients with Graves' disease. Biochem Biophys Res Commun. 1990;170(2):735-41.
- 46. Ferrand M, Le Fourn V, Franc JL. Increasing diversity of human thyroperoxidase generated by alternative splicing characterized by molecular cloning of new transcripts with single- and multispliced mRNAs. J Biol Chem. 2003;278(6):3793-800.
- 47. Le Fourn V, Ferrand M, Franc JL. Differential expression of thyroperoxidase mRNA splice variants in human thyroid tumors. Biochim Biophys Acta. 2004;1689(2):134-41.
- 48. Kimura ET, Kopp P, Zbaeren J, Asmis LM, Ruchti C, Maciel RM, et al. Expression of transforming growth factor beta1, beta2, and beta3 in multinodular goiters and differentiated thyroid carcinomas: a comparative study. Thyroid. 1999;9(2):119-25.
- 49. Matsuo SE, Leoni SG, Colquhoun A, Kimura ET. Transforming growth factor-beta1 and activin A generate antiproliferative signaling in thyroid cancer cells. J Endocrinol. 2006;190(1):141-50.
- 50. Kimura ET, Matsuo SE, Ricarte-Filho JC. TGFbeta, activin and SMAD signalling in thyroid cancer. Arq Bras Endocrinol Metabol. 2007;51(5):683-9.
- 51. Lazzereschi D, Nardi F, Turco A, Ottini L, D'Amico C, Mariani-Costantini R, et al. A complex pattern of mutations and abnormal splicing of Smad4 is present in thyroid tumours. Oncogene. 2005;24(34):5344-54.
Publication Dates
-
Publication in this collection
28 Oct 2009 -
Date of issue
Aug 2009
History
-
Accepted
20 July 2009 -
Received
28 June 2009