Abstracts
OBJECTIVE: Congenital hypothyroidism (CH) may be caused by defects in the thyroid or in one of the stages in the synthesis of thyroid hormones. Thyroid dysgenesis may be associated with mutation in the paired box transcription factor 8 (PAX8) gene. We attempted to screen PAX8 gene mutation in 50 CH patients with thyroid dysgenesis. SUBJECTS AND METHODS: The patients were classified in two groups as agenesis and ectopic based on biochemical and para clinical tests. By employing PCR, Single Strand Conformation Polymorphism (SSCP) and sequencing, exons 3 to 12 of PAX8 gene with their exon-intron boundaries were studied. RESULTS: No mutation was found in these patients in any of the exons. CONCLUSION: Our results, once again, indicate that the PAX8 mutation rate is very low and can only explain a minority of the cases. Therefore, it is highly needed to further investigate the genes controlling development and function of thyroid.
Congenital hypothyroidism; thyroid dysgenesis; PAX8; gene mutation
OBJETIVO: O hipotireoidismo congênito (HC) pode ser causado por defeitos na formação da tireoide ou em uma das etapas da síntese dos hormônios tireoidianos. A disgenesia da tireoide pode ser associada a mutações no fator de transcrição PAX8. Neste estudo, foram rastreadas mutações no gene PAX8 em 50 pacientes com CH com disgenesia da tireoide. SUJEITOS E MÉTODOS: Os pacientes foram classificados em dois grupos, com agenesia ou com ectopia, segundo os testes bioquímicos e paraclínicos. Foram empregadas as técnicas de SSCP (Single Strand Conformation Polymorphism) e sequenciamento para analisar os éxons 3 a 12 do gene PAX8 e suas bordas éxon-intron. RESULTADOS: Nenhuma mutação foi encontrada nesses pacientes, em qualquer um dos éxons. CONCLUSÃO: Nossos resultados, mais uma vez, indicam que a taxa de mutação PAX8 é muito baixa e só pode explicar a minoria dos casos. Portanto, é altamente necessário investigar outros genes que controlam o desenvolvimento e as funções tireoideanas.
Hipotireoidismo congênito; disgenesia da tireoide; PAX8; mutação genética
ORIGINAL ARTICLE
Mutations in the gene encoding paired box domain (PAX8) are not a frequent cause of congenital hypothyroidism (CH) in Iranian patients with thyroid dysgenesis
As mutações no gene PAX8 não constituem uma causa frequente de hipotireoidismo congênito em pacientes iranianos com disgenesia tiroidiana
Frouzandeh MahjoubiI; Mona Malek MohammadiI; Maryam MontazeriI; Masoud AminiiII; Mahin HashemipourII
IClinical Genetic Department, National Institute of Genetic Engineering and Biotechnology (NIGEB), Tehran, Iran
IIIsfahan Endocrine & Metabolism Research Center, Isfahan University of Medical Sciences, Isfahan, Iran
Correspondence to Correspondence to: Frouzandeh Mahjoubi, Clinical Genetics Department, National Institute of Genetic Engineering and Biotechnology (NIGEB) Pajoohesh Blvd. Karaj High Way P.O. Box 14155-6343, Tehran, Iran frouz@nigeb.ac.ir
ABSTRACT
OBJECTIVE: Congenital hypothyroidism (CH) may be caused by defects in the thyroid or in one of the stages in the synthesis of thyroid hormones. Thyroid dysgenesis may be associated with mutation in the paired box transcription factor 8 (PAX8) gene. We attempted to screen PAX8 gene mutation in 50 CH patients with thyroid dysgenesis.
SUBJECTS AND METHODS: The patients were classified in two groups as agenesis and ectopic based on biochemical and para clinical tests. By employing PCR, Single Strand Conformation Polymorphism (SSCP) and sequencing, exons 3 to 12 of PAX8 gene with their exon-intron boundaries were studied.
RESULTS: No mutation was found in these patients in any of the exons.
CONCLUSION: Our results, once again, indicate that the PAX8 mutation rate is very low and can only explain a minority of the cases. Therefore, it is highly needed to further investigate the genes controlling development and function of thyroid.
Keywords: Congenital hypothyroidism; thyroid dysgenesis; PAX8; gene mutation
RESUMO
OBJETIVO: O hipotireoidismo congênito (HC) pode ser causado por defeitos na formação da tireoide ou em uma das etapas da síntese dos hormônios tireoidianos. A disgenesia da tireoide pode ser associada a mutações no fator de transcrição PAX8. Neste estudo, foram rastreadas mutações no gene PAX8 em 50 pacientes com CH com disgenesia da tireoide.
SUJEITOS E MÉTODOS: Os pacientes foram classificados em dois grupos, com agenesia ou com ectopia, segundo os testes bioquímicos e paraclínicos. Foram empregadas as técnicas de SSCP (Single Strand Conformation Polymorphism) e sequenciamento para analisar os éxons 3 a 12 do gene PAX8 e suas bordas éxon-intron.
RESULTADOS: Nenhuma mutação foi encontrada nesses pacientes, em qualquer um dos éxons.
CONCLUSÃO: Nossos resultados, mais uma vez, indicam que a taxa de mutação PAX8 é muito baixa e só pode explicar a minoria dos casos. Portanto, é altamente necessário investigar outros genes que controlam o desenvolvimento e as funções tireoideanas.
Descritores: Hipotireoidismo congênito; disgenesia da tireoide; PAX8; mutação genética
INTRODUCTION
Permanent congenital hypothyroidism is a common disease with the prevalence of 1 of 3,000-4,000 newborns worldwide (1). This prevalence has been represented to be much higher in Iran compared to the world wide report (1 in 914 and 1 in 748 newborns in Tehran and Isfahan, respectively) (1,2).
Congenital hypothyroidism can be caused by either a defect in thyroid development (thyroid dysgenesis, in 85% of cases) or inborn errors of thyroid hormone biosynthesis (dyshormonogenesis in 15% of CH patients). The main group of CH patients with thyroid dysgenesis is also divided in 3 groups with different rates of occurrence. Thyroid dysgenesis can be due to agenesis (in 40% of cases), ectopic (in 40% of cases) or hypoplasia (in 5% of cases) (3).
In a few patients with thyroid dysgenesis, some mutations in transcription factors, such as transcription factor-1 (TTF1) (4-9); transcription factor-2 (TTF2) (10-12); paired box transcription factor-8 (PAX8) (1,13-21); or TSH receptor genes have been reported (22-23).
PAX8 is a member of a family of transcription factors characterized with recognizing specific DNA sequence through highly conserved 128- amino acids, called paired box domain (7,24). It has a key role in mammalian embryogenesis and it is expressed in adult thyroid as a transcription factor for TPO, TG and NIS (2-27). The PAX8 gene is located on chromosome 2q12-q14 and contains a 4-kb transcript sequence divided into 12 exons (28-30).
To date, 15 mutations have been identified in 5 exons of PAX8 (1,13-21). The exon number 3 and 4 which make the paired box domain are the hotspot with 9 mutations recognized. Exon 7 with 3 and exon 9 and 12, each with 1 mutation recognized. In Italy 8 mutations out of 14 have been found (1,13,17,18,20-21), 3 in French (13,15-16,18-19) and 1 in each Japanese (15), German (19) and American patients with thyroid dysgenesis (14).
In the current study, we aimed to screen the presence of mutations in the PAX8 gene in patients with thyroid dysgenesis employing PCR-SSCP and sequencing.
SUBJECTS AND METHODS
Patients
One hundred fifty samples, including 50 CH patients caused by thyroid dysgenesis (13 ectopic, 47 agenesis) and their parents were collected from Isfahan Endocrine and Metabolism Research
Center of Isfahan University. On 7th-28th days of birth, neonates were considered as hypothyroid if T4 was < 6.5 ug/dL and TSH was > 10 mIU/L.
The study was approved by the Research and Ethics Committee of National Institute of Genetic Engineering and Biotechnology, Iran (NIGEB).
Written informed consents were obtained from the parents of the patients.
PCR SSCP analysis
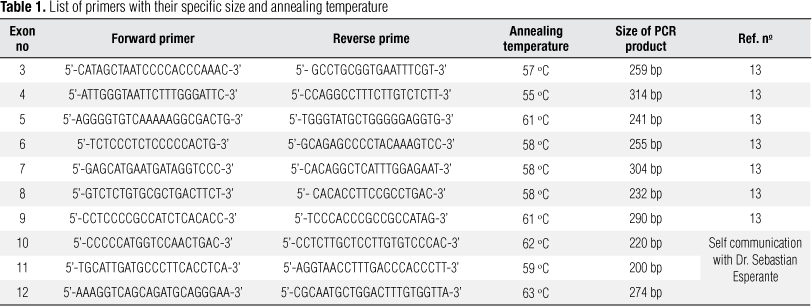
SSCP was used to screen the presence of mutations in each of the exons of PAX8 gene (exons 3-12). DIAtom DNA Prep 100 kit (Isogen Laboratory, Russia) was used to extract Genomic DNA from peripheral blood. Exons 3 to 12 with their flanking intronic regions were amplified in a 25 μL reaction using some of the primers shown in table 1. PCR was performed in 25 μL, using 200 ng of genomic DNA, 0.2 μM of each deoxy (d)-NTP (dATP, dCTP, dTTP, dGTP), 1.5 mM MgCl2, 1X buffer, 1.5 U Taq polymerase (Cynagen, Tehran, Iran) and 5 pmol of each forward and reverse primer. Samples were denatured at 94 °C for 3 minutes followed by 35 cycles of amplification. Each cycle consisted of denaturation at 95 °C for 40 seconds, primer specific annealing temperature for 40 seconds, and primer extension at 72 °C for 50 seconds. After the last cycle, the samples were incubated for an additional 6 minutes at 72 °C to ensure that the final extension step was complete. The amplified products were analyzed in 2% agarose gel.
PCR products were diluted 1 time in a buffer containing 95% formamide, 100 mL NaOH, 0.25% xylene cyanol, and 0.25% bromophenol blue and denatured at 95 °C for 10 minutes and cooled down on ice and loaded on non-denaturing gel containing 8% acrylamide/bis-acrylamide (39:1), 5% glycerol and 0.5X TBE. The gel was electrophoresed at 4 °C with constant power of 200W for 20 h. DNA was visualized by silver staining.
Direct DNA sequencing
Samples showing aberrant pattern and few samples of normal pattern in SSCP analysis were directly sequenced. The results were analyzed using the Chromas and Sequencher and Nucleotide BLAST software programs.
RESULTS
SSCP analysis and direct sequencing of PAX8
Exons 3 to 12 of PAX8 from all 50 unrelated CH patients were successfully amplified with PCR and screened by SSCP followed by direct sequencing of suspicious ones and a few samples with normal patterns. Our study group included 13 CH patients with ectopic and 37 patients with agenesis. Fourteen samples for exon 3, fifteen samples for exon 4, two for exon 5, seven for exon 6, eleven for exon 7, five for exon 8, twelve for exon 9, six for exon 10, six for exon 11 and five for exon 12 were selected to be sent for sequencing. SSCP analysis of some PCR products showed different banding patterns (an example is illustrated in Figure 1).
The resulting sequences were compared with the sequences of the most complete and the longest PAX8 mRNA (GenBank accretion nº NM_003466). No mutation or polymorphism was found in any of the samples.
DISCUSSION
In this study, the entire coding regions of PAX8 of 50 unrelated CH patients were analyzed by SSCP followed by direct sequencing. The patients were composed by two groups (ectopic and agenesis). CH patients with ectopic and agenesis were 13 (26%) and 32 (74%), respectively. No mutation in PAX8 gene was found among any of these patients.
In only 5% of CH patients a mutation in one of the transcription factors (TTF1, TTF2, PAX8) or the TSH receptor was found to be the cause of thyroid dysgenesis. This means that there is almost 1% of chance for each gene. This indicates that the defect in the TTF1/TTF2/TSH receptor or even other unknown gene(s) can be responsible for thyroid dysgenesis in this group of patients.
Reviewing literature revealed that 60% of the PAX8 mutation in this gene were found in Italian patients (1,17,20-21), 20% in French (13,16-18), almost 7% in US (14), 7% In Germany (19) and 7% in Japan (15).
Only 9 out of 302 (3%) CH patients studied in Italy had a mutation in the PAX8 gene. In France 5% (3/57), in US 25% (1/4) and in Germany 0.6% (1/170) had mutation in PAX8.
Sixty four percent (9/14) of the reported PAX8 mutations were found in the paired box domain causing reduction of the DNA binding activity of this gene (1,13-21). About 21.4% (3/14) of the PAX8 mutations were in exon 7 that codes a residual paired type homeodomain in C-terminal region, which is necessary for transcriptional activity of PAX8 (17,21). One mutation is found in exon 9 known to have an antagonistic role for the activating domain which can influence the normal thyroid development or transcriptional control of several thyroid specific genes not tested yet (28). A mutation found in exon 12 seems to be synonymous with no change of any amino acids.
Interestingly, although, only 5% of the CH patients with thyroid dysgenesis have thyroid hyperplasia (1) but, most of the mutation found in PAX8 (64%) are from this group of patients.
Among 14 mutations recognized in PAX8, 10 mutations are inactivating (71%) and all of them are located in the paired box domain except one which is in exon 7 (30,31). Two mutations (14%) are synonymous which are showing the wild type or comparable protein as the wild type. These two mutations are located in C-terminal region and it is possible to influence the normal thyroid development or the transcriptional control of several thyroid specific genes. Two mutations (14%) are nonsynonymous substitutions that might have inhibitory role on an unknown particular function.
This result, once again, indicates the very low rate mutation in PAX8 gene in CH patients
The result presented in this report shows that no mutation in PAX8 gene is responsible for the thyroid dysgenesis in this cohort of Iranian CH patients. All these data, once again, highlights the need for further study in higher level to find the cause of CH in other genes.
Funding: This research was financially supported by NIGEB grant No. 354, 2008 and Endocrine and Metabolism Research Center of Isfahan University.
Acknowledgments: We would like to thank Dr. Sebastian A. Esperante from Laboratorio de Biología Molecular, Cátedra de Genética y Biología Molecular, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, Buenos Aires, Argentina for his valuable guidance.
Disclosure: no potential conflict of interest relevant to this article was reported.
Received on Apr/10/2010
Accepted on July/21/2010
- 1. Loeber JG. Neonatal screening in Europe; the situation in 2004. J Inherit Metab Dis. 2007;30:430-8.
- 2. Hashemipour M, Hovsepian S, Kelishadi R, Iranpour R, Hadian R, Haghighi S, et al. Permanent and transient congenital hypothyroidism in Isfahan-Iran. J Med Screen. 2009;16(1):11-6.
- 3. Ordookhani A, Mirmiran P, Moharamzadeh M, Hedayati M, Azizi F. A high prevalence of consanguineous and severe congenital hypothyroidism in an Iranian population. J Pediatr Endocrinol Metab. 2004;17(9):1201-9.
- 4. Park SM, Chatterjee VK. Genetics of congenital hypothyroidism. J Med Genet. 2005;42:379-89.
- 5. Devriendt K, Vanhole C, Matthijs G, de Zegher F. Deletion of thyroid transcription factor-1 gene in an infant with diseases. Clinical Endocrinology. 1998;55:143-58.
- 6. Iwatani N, Mabe H, Devriendt K, Kodama M, Miike T. Deletion of NKX2.1 gene encoding thyroid transcription factor-1 in two siblings with hypothyroidism and respiratory failure. J Pediatr. 2000;137(2):272-6.
- 7. Krude H, Schütz B, Biebermann H, von Moers A, Schnabel D, Neitzel H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Investigation. 2002;109:475-80.
- 8. Plachov D, Chowdhury K, Walther C, Simon D, Guenet JL, Gruss P. PAX8, a murine paired box gene expressed in the developing excretory system and thyroid gland. Development. 1990;110:643-51.
- 9. Doyle DA, Gonzalez I, Thomas B, Scavina M. Autosomal dominant transmission of congenital hypothyroidism, neonatal respiratory distress, and ataxia caused by a mutation of NKX2-1. J Pediatr. 2004;145(2):190-3.
- 10. Moya CM, Perez de Nanclares G, Castaño L, Potau N, Bilbao JR, Carrascosa A, et al. Functional study of a novel single deletion in the TITF1/NKX2.1 homeobox gene that produces congenital hypothyroidism and benign chorea but not pulmonary distress. J Clin Endocrinol Metab. 2006;91:1832-41.
- 11. Clifton-Bligh RJ, Wentworth JM, Heinz P, Crisp MS, John R, Lazarus JH, et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet. 1998;19:399-401.
- 12. Castanet M, Park SM, Smith A, Bost M, Leger J, Lyonnet S, et al. A novel loss-of-function mutation in TTF-2 is associated with congenital hypothyroidism, thyroid agenesis and cleft palate. Hum Mol Genet. 2002;11:2051-9.
- 13. Baris I, Arisoy AE, Smith A, Agostini M, Mitchell CS, Park SM, et al. A novel missense mutation in human TTF-2 (FKHL15) gene associated with congenital hypothyroidism but not athyreosis. J Clin Endocrinol Metab. 2006;91:4183-7.
- 14. Vilain C, Rydlewski C, Duprez L, Heinrichs C, Abramowicz M, Malvaux P, et al. Autosomal dominant transmission of congenital thyroid hypoplasia due to loss-of-function mutation of PAX8. J Clin Endocrinol Metab. 2001;86:234-8.
- 15. Congdon T, Nguyen LQ, Nogueira CR, Habiby RL, Medeiros-Neto G, Kopp P. A novel mutation (Q40P) in PAX8 associated with congenital hypothyroidism and thyroid hypoplasia: evidence for phenotypic variability in mother and child. J Clinl Endocrinol Metab. 2001;86:3962-7.
- 16. Komatsu M, Takahashi T, Takahashi I, Nakamura M, Takahashi I, Takada G. Thyroid dysgenesis caused by PAX8 mutation: the hypermutability with CpG dinucleotides at codon 31. J Pediatr. 2001;139(4):597-9.
- 17. Meeus L, Gilbert B, Rydlewski C, Parma J, Roussie AL, Abramowicz M, et al. Characterization of a novel loss of function mutation of PAX8 in a familial case of congenital hypothyroidism with in-place, normalsized thyroid. J Clin Endocrinol Metab. 2004;89:428-91.
- 18. De Sanctis L, Corrias A, Romagnolo D, Di Palma T, Biava A, Borgarello G, et al. Familial PAX8 small deletion (c.989_992delACCC) associated with extreme phenotype variability. J Clin Endocrinol Metab. 2004;89:5669-74.
- 19. Grasberger H, Ringkananont U, Lefrancois P, Abramowicz M, Vassart G, Refetoff S. Thyroid transcription factor rescues PAX8/p300 synergism impaired by a natural PAX8 paired domain mutation with dominant negative activity. Mol Endocrinol. 2005;19:1779-91.
- 20. Al Taji E, Biebermann H, Límanová Z, Hníková O, Zikmund J, Dame C, et al. Screening for mutations in transcription factors in a Czech cohort of 170 patients with congenital and early-onset hypothyroidism: identification of a novel PAX8 mutation in dominantly inherited early-onset non-autoimmune hypothyroidism. Eur J Endocrinol. 2007;156:521-9.
- 21. Tonacchera M, Banco ME, Montanelli L, Di Cosmo C, Agretti P, De Marco G, et al. Genetic analysis of the PAX8 gene in children with congenital hypothyroidism and dysgenetic or eutopic thyroid glands: identification of a novel sequence variant. Clin Endocrinol (Oxf). 2007;67(1):34-40.
- 22. Esperante SA, Rivolta CM, Miravalle L, Herzovich V, Iorcansky S, Baralle M, et al. Identification and characterization of four PAX8 rare sequence variants (p.T225M, p.L233L, p.G336S and p.A439A) in patients with congenital hypothyroidism and dysgenetic thyroid glands. Clin Endocrinol (Oxf).2008;68(5):828-35.
- 23. Corvilain B, Van Sande J, Dumont JE, Vassart G. Somatic and germline mutations of the TSH receptor and thyroid diseases. Clin Endocrinol (Oxf). 2001;55(2):143-58.
- 24. Refetoff S. Resistance to thyrotropin. J Endocrinol Invest. 2003;26:770-9.
- 25. Xu W, Rould MA, Jun S, Desplan C, Pabo CO. Crystal structure of a paired domain-DNA complex at 2.5A resolution reveals structural basis for PAX developmental mutations. Cell. 1995;80:639-50.
- 26. Mascia A, Nitsch L, Di Lauro R, Zannini M. Hormonal control of the transcription factor PAX8 and its role in the regulation of thyroglobulin gene expression in thyroid cells. J Endocrinol. 2002;172:163-76.
- 27. Esposito C, Miccadei S, Salardi A, Civitareale D. PAX8 activates the enhancer of the human thyroperoxidase gene. Biochem J. 1998;331:37-40.
- 28. Ohno M, Zannini M, Levy O, Carrasco N, Di Lauro R. The paired-domain transcription factor PAX8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol. 1999;19(3):2051-60.
- 29. Poleev A, Fickenscher H, Mundlos S, Winterpacht A, Zabel B, Fidler A, et al. PAX8, a human paired box gene: isolation and expression in developing thyroid, kidney and Wilms' tumors. Development. 1992;116:611-23.
- 30. Stapleton P, Weith A, Urbanek P, Kozmik Z, Busslinger M. Chromosomal localization of seven PAX genes and cloning of a novel family member, PAX-9. Nat Genet. 1993;3:292-8.
- 31. Montanelli L, Tonacchera M. Genetics and phenomics of hypothyroidism and thyroid dys- and agenesis due to PAX8 and TTF1 mutations. Mol Cell Endocrinol. 2010;322:64-71.
Publication Dates
-
Publication in this collection
08 Sept 2010 -
Date of issue
Aug 2010
History
-
Received
10 Apr 2010 -
Accepted
21 July 2010