Abstracts
OBJECTIVES: (1) Characterize serum (S) and urinary (U) steroid metabolites in complete CYP17 deficiency (cCYP17D); (2) analyze the relative 17α-hydroxylase (17OH) and 17,20-lyase (17,20L) activities in vivo; and (3) comparedata from the two most prevalent mutations in Brazil. SUBJECTS AND METHODS: 20 genotyped cCYP17D patients from a previously reported cohort were homozygous for W406R or R362C; 11 controls were CYP17 wild types (WT). WT and cCYP17D patients had S and U samples drawn to measure: cortisol (F), corticosterone (B), deoxycorticosterone (DOC), 18OH-B, 18OH-DOC, and 17OHP; and tetrahydro (TH)-B, THA, THDOC, THF+5α-THF, TH-cortisone, androsterone, etiocholanolone, 5-pregnenediol, 17OH-pregnenolone and pregnanetriol. RESULTS: Compared to WT, cCYP17D patients had marked elevations of B, DOC, 18OH-B and 18OH-DOC, whereas 17OHP, F and adrenal androgens (AA) were reduced; U steroids parallel S findings. Metabolite ratios revealed that both 17OH and 17,20L activities were impaired in cCYP17D. There were nodifferences between W406R andR362C mutations. CONCLUSIONS: cCYP17D patients show parallel overproduction/overexcretion of 17-deoxysteroids, and marked reduction of F and AA. In addition to 17OH, 17,20-L activity was also impaired in cCYP17D. W406 and R362C mutations disclose similar Sand U patterns.
Congenital adrenal hyperplasia; CYP17; 17-hydroxylase deficiency; 17,20-lyase deficiency; urinary steroid metabolome; corticosterone
OBJETIVOS: (1) Caracterizar os esteroides séricos (S) e urinários (U) na deficiência completa da CYP17 (DcCYP17); (2) analisar as atividades da 17α-hidroxilase (17OH) e 17,20-liase (17, 20 L) in vivo; e (3) comparar as duas mutações mais prevalentes no Brasil. SUJEITOS E MÉTODOS: 20 pacientes genotipados para a DcCYP17, de uma coorte anterior, eram homozigotos para W406R ou R362C (8 cada); 11 controles eram CYP17 wild types (WT). Amostras de S e U foram colhidas dos WT e pacientes para dosagem de: cortisol (F), corticosterona (B), deoxicorticosterona (DOC), 18-OH-B, 18OH-DOC e 17OHP; e tetraidro(TH)-B, THA, TH-DOC, THF+5α-THF, THE, androsterona, etiocolanolona, 5-pregnenediol, 17OH-pregnenolona e pregnanetriol. RESULTADOS: Comparados aos WT, os pacientes com DcCYP17 revelaram elevações acentuadas de B, DOC, 18OHB e 18OHDOC, enquanto 17OHP, F e andrógenos adrenais (AA) estavam reduzidos. Os esteroides na U acompanham os achados no S. As relações de metabólitos mostraram que as atividades de 17OH e 17,20L estavam reduzidas em pacientes com DcCYP17. Não houve diferenças entre pacientes com as mutações W406R e R362C. CONCLUSÕES: Na DcCYP17, a produção e a excreção dos 17-deoxiesteroides estão aumentadas em paralelo, em contraste com a reduzida produção/excreção de F e AA. As atividades da 17OH e 17,20-L estão diminuídas na DcCYP17. As mutações W406 e R362C apresentam achados semelhantes em S e U.
Hiperplasia adrenal congênita; CYP17; deficiência de 17-hidroxilase; deficiência de 17,20-liase; metaboloma de esteroides urinários; corticosterona
ORIGINAL ARTICLE
Distinctive profile of the 17-hydroxylase and 17,20-lyase activities revealed by urinary steroid metabolomes of patients with CYP17 deficiency
Perfil característico das atividades 17-hidroxilase e 17,20-liase reveladas por meio do metaboloma de esteroides urinários de pacientes com deficiência de CYP17
Marcos S. NeresI; Richard J. AuchusII; Cedric H. L. ShackletonIII; Claudio E. KaterI
IAdrenal and Hypertension Unit, Division of Endocrinology and Metabolism, Department of Medicine, Universidade Federal de São Paulo (Unifesp), São Paulo, SP, Brazil
IIDepartment of Clinical Sciences, University of Texas Southwestern Medical Center, Dallas, Texas, United States
IIIChildren's Hospital of Oakland Research Institute, Oakland, CA, USA, Division of Medical Sciences, University of Birmingham, Institute of Biomedical Research, Birmingham, B152TT, United Kingdom
Correspondence to Correspondence to: Claudio E. Kater Laboratório de Esteroides, Divisão de Endocrinologia Departamento de Medicina, Universidade Federal de São Paulo Rua Pedro de Toledo, 781, 13º andar 04039-032 − São Paulo, SP, Brasil kater@unifesp.br
ABSTRACT
OBJECTIVES: (1) Characterize serum (S) and urinary (U) steroid metabolites in complete CYP17 deficiency (cCYP17D); (2) analyze the relative 17α-hydroxylase (17OH) and 17,20-lyase (17,20L) activities in vivo; and (3) comparedata from the two most prevalent mutations in Brazil.
SUBJECTS AND METHODS: 20 genotyped cCYP17D patients from a previously reported cohort were homozygous for W406R or R362C; 11 controls were CYP17 wild types (WT). WT and cCYP17D patients had S and U samples drawn to measure: cortisol (F), corticosterone (B), deoxycorticosterone (DOC), 18OH-B, 18OH-DOC, and 17OHP; and tetrahydro (TH)-B, THA, THDOC, THF+5α-THF, TH-cortisone, androsterone, etiocholanolone, 5-pregnenediol, 17OH-pregnenolone and pregnanetriol.
RESULTS: Compared to WT, cCYP17D patients had marked elevations of B, DOC, 18OH-B and 18OH-DOC, whereas 17OHP, F and adrenal androgens (AA) were reduced; U steroids parallel S findings. Metabolite ratios revealed that both 17OH and 17,20L activities were impaired in cCYP17D. There were nodifferences between W406R andR362C mutations.
CONCLUSIONS: cCYP17D patients show parallel overproduction/overexcretion of 17-deoxysteroids, and marked reduction of F and AA. In addition to 17OH, 17,20-L activity was also impaired in cCYP17D. W406 and R362C mutations disclose similar Sand U patterns.
Keywords: Congenital adrenal hyperplasia; CYP17; 17-hydroxylase deficiency; 17,20-lyase deficiency; urinary steroid metabolome; corticosterone.
RESUMO
OBJETIVOS: (1) Caracterizar os esteroides séricos (S) e urinários (U) na deficiência completa da CYP17 (DcCYP17); (2) analisar as atividades da 17α-hidroxilase (17OH) e 17,20-liase (17, 20 L) in vivo; e (3) comparar as duas mutações mais prevalentes no Brasil.
SUJEITOS E MÉTODOS: 20 pacientes genotipados para a DcCYP17, de uma coorte anterior, eram homozigotos para W406R ou R362C (8 cada); 11 controles eram CYP17 wild types (WT). Amostras de S e U foram colhidas dos WT e pacientes para dosagem de: cortisol (F), corticosterona (B), deoxicorticosterona (DOC), 18-OH-B, 18OH-DOC e 17OHP; e tetraidro(TH)-B, THA, TH-DOC, THF+5α-THF, THE, androsterona, etiocolanolona, 5-pregnenediol, 17OH-pregnenolona e pregnanetriol.
RESULTADOS: Comparados aos WT, os pacientes com DcCYP17 revelaram elevações acentuadas de B, DOC, 18OHB e 18OHDOC, enquanto 17OHP, F e andrógenos adrenais (AA) estavam reduzidos. Os esteroides na U acompanham os achados no S. As relações de metabólitos mostraram que as atividades de 17OH e 17,20L estavam reduzidas em pacientes com DcCYP17. Não houve diferenças entre pacientes com as mutações W406R e R362C.
CONCLUSÕES: Na DcCYP17, a produção e a excreção dos 17-deoxiesteroides estão aumentadas em paralelo, em contraste com a reduzida produção/excreção de F e AA. As atividades da 17OH e 17,20-L estão diminuídas na DcCYP17. As mutações W406 e R362C apresentam achados semelhantes em S e U.
Descritores: Hiperplasia adrenal congênita; CYP17; deficiência de 17-hidroxilase; deficiência de 17,20-liase; metaboloma de esteroides urinários; corticosterona.
INTRODUCTION
17-Hydroxylase deficiency (17OHD) is generally considered a rare cause of congenital adrenal hyperplasia (CAH). However, it is not so uncommon in specific ethnic groups or geographical areas, as among the Mennonite descendants of Dutch Frieslanders (1) and in Brazil, where 17OHD is considered the second most frequent form of CAH, accounting for 5%-7% of all cases (2,3).
The disease is caused by mutations in the CYP17A1 gene which encodes cytochrome P450c17, an enzyme expressed in the adrenal cortex and the gonads displaying both 17α-hydroxylase and 17,20-lyase activities, among others (4-12). The former activity is carried out by converting pregnenolone and progesterone to 17α-hydroxypregnenolone (17OHPreg) and 17α-hydroxyprogesterone (17OHP), respectively, whereas the latter performs the C17-20 side-chain cleavage of 17OHPreg to yield DHEA, an androgen precursor in the adrenal zona reticulata, ovaries and testes. For maximal activities, the enzyme requires a functional steroid-binding site domain and a functional redox partner-binding site domain for interaction with the electron-transfer protein P450 oxido-reductase and the co-factor cytochrome b5 (7,13-15). Most of the 75 reported mutations in CYP17 (16) occurs in the steroid-binding domain resulting in combined or complete 17α-hydroxylase/17,20-lyase deficiency. Thus, failure to produce sex steroids in these patients results in sexual infantilism and primary amenorrhea in the female (46,XX) and pseudohermaphroditism in the male (46,XY) (17-20). In addition, impaired production of cortisol − typical of complete 17OHD −, leads to increased ACTH secretion that stimulates overproduction of deoxycorticosterone (DOC) and corticosterone (B) − non-17-hydroxylated steroid intermediates with mineralocorticoid activity −, causing severe hypertension and hypokalemia (2,2125). Although the serum steroid profile has been extensively studied in 17OHD, systematic evaluation of the full urinary steroid metabolite (metabolome) in these patients is limited.
In this paper we determined serum levels and the steroid metabolome in a large group of 17OHD patients and examined specific precursor-to-product metabolite ratios in order to: (1) define possible differences concerning both enzymatic activities, (2) compare metabolite ratios in patients with complete 17OHD with those from previously reported patients with isolated 17,20-lyase deficiency (26), and also (3) compare the metabolic pattern between the two most prevalent CYP17 mutations documented in Brazil, the W406R and the R362C.
SUBJECTS AND METHODS
Twenty patients affected with the complete 17OHD form of CAH (twelve 46,XY, and eight 46,XX, all phenotypic female) were selected from a larger cohort of patients previously reported (2). At the time of the study, patients ranged in age from 14 to 42 years (median of 22 yr); in most, clinical diagnosis had been suspected by the absence of secondary sex characteristics and of menstrual periods at the time of puberty, mostly associated with the presence of hypertension and hypokalemia. Complete 17OHD was established by typical laboratory values, as reduced cortisol, aldosterone, and plasma renin activity and elevated LH, FSH, and progesterone, with low to absent sex steroids. All had been genotyped and specific CYP17 mutations identified (R362C, W406R, R362C+W406R, Y329D+splice, Y329X and W406R+M1T) (2).
Eleven normal subjects were also studied as controls (5 male; 6 female, 15 to 57 years of age; median of 26 yr). They were selected among the families of patients with 17OHD, but whose genotyping did not identify mutations in any of the alleles ("wild types").
The study protocol was performed in all subjects and included urinary collections (mostly spot urine samples) to measure steroid metabolites and a morning blood sampling for serum steroids. All patients on ongoing glucocorticoid replacement therapy had their medications discontinued for 72h, in an attempt to reestablish their pre-treatment steroid profile. Five of them had never been treated before.
The study protocol was previously approved by the Committee on Human Research of our institution and informed written consent obtained from all participants.
Analytical methods
All patients and controls had a morning blood sample drawn and were instructed to collect a urinary sample (a few collected 24-hour urine specimens) and keep it under refrigeration until taken to the laboratory. The following serum steroids were measured in the Clinical Research Center at San Francisco General Hospital by radioimmunoassay after HPLC separation: corticosterone (B), deoxycorticosterone (DOC), 18-hydroxyB (18OHB), 18OHDOC, cortisol, aldosterone, and 17OH-progesterone.
From each urinary specimen, a 20 mL aliquot was passed through a C-18 cartridge for steroid metabolite extraction. Cartridges were subsequently mailed to the Children's Hospital of Oakland Research Institute (CHLS) for gas chromatography-mass spectrometry analysis. The compounds of interest were identified and quantified (concentrations expressed as µg/L of urine) and specific steroid ratios were calculated to express specific enzymatic activities (27).
The following urinary metabolites were measured: D5-pregnenediol (5PD) and pregnanediol (PD) (metabolites of the non-17-hydroxylated precursors, pregnenolone and progesterone, respectively); tetrahydro-DOC (THDOC) (metabolite of DOC); tetrahydrocorticosterone (THB) and 5α(allo)-THB plus tetrahydro-11-dehydrocorticosterone or compound A (THA) and 5α(allo)-THA (metabolites of B); 17-hydroxypregnenolone (17HP) and pregnanetriol (PT) (metabolites of 17-hydroxyprogesterone); tetrahydrocortisol (THF) and 5α(allo)-THF plus tetrahydrocortisone (THE) (metabolites of cortisol), as well as the urinary metabolites of sex steroids: androsterone (AN) plus etiocholanolone (ET) (metabolites of androstenedione).
Enzymatic activities were inferred from calculation of the specific precursor-to-product metabolite ratios (Figure 1). For simplicity, the following groups of metabolites: (THB+5αTHB), (THA+5αTHA), (THF+5αTHF), and THE, will be named herein as "Bs", "As", "Fs", and "E", respectively: (1) (5PD+PD)/(AN+ET) and (Bs+As)/(AN+ET) are used as expressions of global CYP17 activity (17-hydroxylase plus 17,20-lyase); (2) (5PD+PD)/(17HP+PT), PD/PT, and (Bs+As)/(Fs+E) stand for the efficiency of 17-hydroxylation solely; and, (3) (17HP+PT)/(AN+ET), PT/(AN+ET), and (Fs+E)/ (AN+ET) indicate specifically 17,20-lyase activity.
The underlined ratios shown above were employed to compare data from our patients with those from a previous publication that studied patients with "isolated 17,20-lyase deficiency" (26).
RESULTS
Patients with 17OHD showed marked elevations (20-to > 100-fold) of serum B, DOC, 18OHB, 18OHDOC and progesterone, as compared to WT (Table 1). Conversely, cortisol and 17OHP were virtually undetectable with normal to reduced aldosterone levels. Adrenal androgens were also typically low (data not shown).
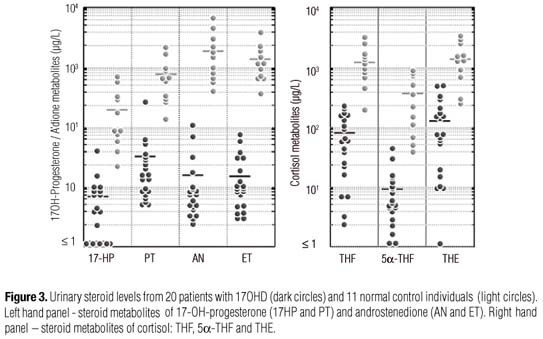
In 17OHD patients, the urinary steroid metabolomes were concordant with their serum findings. Excretion of B metabolites: THA, 5αTHA, THB, and specially 5αTHB were significantly increased, in parallel with THDOC (Figure 2). Accordingly, metabolites of the 17-hydroxylase substrates, pregnenolone and progesterone (5PD and PD) were also significantly increased (Figure 2). In contrast, cortisol metabolites, THF, 5αTHA, and THE, were decreased, as well as 17HP plus PT and AN plus ET (Figure 3).
Ratios
The (5PD+PD)/(AN+ET) and (Bs+As)/(AN+ET) ratios were significantly elevated (4,500- to 8,500fold) in 17OHD patients as compared to WT, as a result of virtually total CYP17 deficiency (combined 17α-hydroxylase/17,20-lyase activities) (Table 2). The values observed for the latter ratio in patients with "isolated 17,20-lyase deficiency" were intermediate (around 400-fold higher than WT) (26).
When examined in separate, 17α-hydroxylation was predominantly affected over 17,20-lyase activity, as revealed by the disproportionate increase in the (5PD+PD)/(17HP+PT) and (Bs+As)/Fs+E) ratios (> 400-fold), as compared to (17HP+PT)/(AN+ET) and (Fs+E)/(AN+ET) (4- to 10.5-fold) (Table 2). However, 17-hydroxylase activity was slightly (PD/PT) to moderately (Bs+As)/Fs+E) affected in patients with "isolated 17,20-lyase deficiency" (1.7-fold and 37-fold higher than WT, respectively). When the PT/(AN+ET) ratio is considered, 17,20-lyase activity was significantly less impaired in our patients with 17OHD than in those with "isolated 17,20-lyase deficiency" (0.84 [0.64-1.23] vs. 2.25 [1.724.30]), whereas it was similar when the (Fs+E)/(AN+ET) ratio is analyzed (8.37[3.44-13.38] vs. 6.63[3.88-14.94]).
When homozygote 17OHD patients for the W406R and R362C mutations were compared, all individual metabolite values and their respective ratios were similar and not statistically different.
DISCUSSION
In this study we examined the serum steroid levels and the urinary steroid metabolomes from 20 patients with 17OHD, in whom enzymatic activity was proven to be completely abolished by the specific CYP17A1 gene mutations W406R and R362C, as reported elsewhere (2).
The serum steroid profile was comparable to those previously published (8,20,24,25,28) and disclosed marked increases of B, DOC and their 18-hydroxy-derivatives 18OHB and 18OHDOC , the so-called 17-deoxysteroids (from the "mineralocorticoid pathway") of the zona fasciculata.
Also confirmed in this study is the dissociation between the raised levels of 18OHB (aldosterone precursor in the zona glomerulosa) in the presence of reduced aldosterone, a unique finding explained by the production of 18OHB in the zona fasciculata from its abundant precursor corticosterone, stimulated by increased ACTH production (22,23). Since elevated DOC and B play a major role in the pathogenesis of mineralocorticoid hypertension with hypokalemia and chronic renin suppression, zona glomerulosa aldosterone is characteristically reduced in 17OHD (together with a negligible 18OHB contribution from this zone).
Progesterone is an earlier steroid biosynthetic precursor and the major delta-4 substrate for CYP17. It is invariably increased in patients with 17OHD regardless of gender, and also frequently elevated in other adrenal enzymatic deficiencies. The marked progesterone increase found in our patients corroborate data reported by Martin and cols. (28) who suggested that elevated progesterone in the presence of hypergonadotrophic hypogonadism is a simple yet strong evidence for the diagnosis of 17OHD. In isolated 17,20-lyase deficiency progesterone is also elevated, mostly after ACTH stimulation, but suspicion is best raised when the 17OHP to androstenedione ratio is increased, as suggested by Auchus and Miller (6).
The urinary steroid metabolomes evaluated in this study were in agreement with serum findings, implying an appropriate hepatic, renal and peripheral metabolism of the steroid mixture. Urinary metabolites of early biosynthetic steroids, such as pregnenolone and progesterone (5PD and PD), are substantially increased in 17OHD patients. Also, both B metabolite isomers, the reduced (THB, 5αTHB) and the dehydrogenated (THA, 5αTHA) forms, are distinctly elevated, as well as THDOC. The magnitude of metabolite excretion of these steroid is somewhat disproportional and less expressive than that seen with serum levels (25-fold vs. 65-fold higher than WT, respectively), possibly reflecting the broad dispersion into several other metabolites and/or an uneven excretory processing. Nevertheless, the preferential 5α-reduction catabolic pathway remains functionally active, if not increased, in these patients.
On the other hand, both cortisol and androgen metabolites are appreciably reduced in patients with complete 17OHD, reflecting their virtually absent production. In addition, the 17-hydroxylated intermediate metabolites, especially 17OH-progesterone (17HP and PT), are also substantially reduced.
The relative enzymatic activities of CYP17 (17-hydroxylase and 17,20-lyase) were estimated by analyzing specific precursor-to-product metabolite ratios. This in vivo assessment has proven useful for the diagnosis of several biosynthetic defects (26,27).
The precursor-to-product metabolite ratios that estimate global enzymatic activity in patients with 17OHD showed very high and unequivocal values in the range of 4,000- to 8,000-fold higher than WT , typical of a complete loss of CYP17 function. A separate evaluation of 17α-hydroxylase and 17,20-lyase activities in the same patients, also demonstrates expressive enzymatic deficiencies (increases of proper metabolite ratios); however, they were at least one and two orders of magnitude less (400- to 900-fold and 3- to 10-fold, respectively) than complete 17OHD, denoting a predominant 17-hydroxylase obliteration. The lower yet significant increased ratios for the latter is not surprising since the amount of substrate for this second reaction (17,20-lyase) would be reduced as a result of deficient 17-hydroxylation.
For comparison with the report by Tiosano and cols. (26) who studied patients initially assumed to have isolated 17,20-lyase deficiency, we examined slightly different metabolite ratios which, nevertheless, express similar enzymatic functions. Using this set of data, global enzymatic activity was much less affected in Tiosano's patients than in ours: although nearly 400fold higher than in WT, the Bs+As/AN+ET ratio in our patients was 12-fold higher than in theirs.
Analysis of the 17-hydroxylase activity alone also confirms the previous findings observed for 17OHD patients as compared to WT (~400-fold higher in the former). However, in the six patients reported by Tio-sano and cols. (26), these ratios reach values 1.7- to 37fold higher than WT, indicating a partial 17-hydroxylase deficiency, an observation already made by the authors.
Concerning 17,20-lyase activity, the PT/(AN+ET) ratio was the only one that discloses higher values in Tiosano's patients than in ours (11-fold vs 4-fold in comparison with WT, respectively), indicating the predominant impairment of 17,20-lyase over 17-hydroxylase, or in other words, a greater availability of 17-hydroxylated products in Tiosano's than in our 17OHD patients. Similar but less evident results could be observed with the Fs+E/AN+ET ratio.
In this regard, it is tempting to speculate that some patients with "complete 17OHD" may in fact have "isolated 17-hydroxylase deficiency", in analogy to those with "isolated 17,20-lyase deficiency" reported by Tiosano in whom the E305G mutation in the CYP17A1 gene was shown in vivo to affect also 17-hydroxylation, although in vitro studies indicate normal activity (29). Indeed, both activities may have an impairment spectrum as a result of different CYP17A1 mutations, since the rarity of "true" isolated 17,20-lyase deficiency (14,15) precludes systematic metabolic studies. Also, in at least one family previously diagnosed as having 17,20-lyase deficiency, reinvestigation showed a homozygous mutation G539R in the gene of P450 oxidoreductase (30).
The urinary metabolome and the metabolite ratios studied in this paper confirm previous in vitro findings of absent 17-hydroxylase and 17,20-lyase activities in 17OHD patients. However, as observed by Tiosano and cols. (26), data derived from these in vivo studies seem more precise than those obtained from enzymatic expression in vitro (26). This observation allows the assumption that many enzymatic processes are under the influence of multiple modulators or modifying factors and characterize the variability of the individual phenotype sometimes associated to the same mutations.
In spite of these observations, data analysis from the two more prevalent mutations in Brazilian patients with 17OHD, both known to abolish CYP17 enzymatic activity, did not evidence any significant differences between groups.
In conclusion, patients with 17OHD disclose parallel overproduction and overexcretion of the adrenocortical 17-deoxysteroids ("the mineralocorticoid pathway of zona fasciculata"), in contrast with a marked decrease in the production and excretion of cortisol and adrenal androgens. In addition to 17-hydroxylation, 17,20-lyase activity is also impaired in complete 17OHD. Homozygote patients for the CYP17 mutations W406 and R362C, both associated with absent 17OH activity, disclosed a similarly abnormal pattern of serum steroids and urinary metabolites.
Acknowledgements: We thank Dr. Marivânia Costa-Santos for assembling all the urine samples used in this study.
Disclosure: no potential conflict of interest relevant to this article was reported.
Received on Jan/5/2010
Accepted on Aug/2/2010
- 1. Imai T, Yanase T, Waterman MR, Simpson ER, Pratt JJ. Canadian Mennonites and individuals residing in the Friesland region of The Netherlands share the same molecular basis of 17 alpha-hydroxylase deficiency. Hum Genet. 1992;89:95-6.
- 2. Costa-Santos M, Kater CE, Auchus RJ, and The Brazilian CAH Multicenter Study Group. Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab. 2004;89(1):49-60.
- 3. Miller WL. Steroid 17alpha-hydroxylase deficiency not rare everywhere. J Clin Endocrinol Metab. 2004;89:40-2.
- 4. Winter JS, Couch RM, Muller J, Perry YS, Ferreira P, Baydalla L, et al. Combined 17-hydroxylase deficiency and 17,20-desmolase deficiency: evidence for synthesis of a defective cytochrome P450c17. J Clin Endocrinol Metab. 1989;68:309-16.
- 5. Kater CE, Biglieri EG. Disorders of steroid 17alpha-hydroxylase deficiency. Endocrinol Metab Clin North Am 1994;23:341-57.
- 6. Auchus RJ, Miller WL. Molecular modeling of human P450c17 (17alpha-hydroxylase/17,20-lyase): insights into reaction mechanisms and effects of mutations. Mol Endocrinol. 1999;13:1169-82.
- 7. Geller DH, Auchus RJ, Miller WL. P450c17 mutations R347H and R358Q selectively disrupt 17,20-lyase activity by disrupting interactions with P450 oxidoreductase and cytochrome b5. Mol Endocrinol. 1999;13:167-75.
- 8. Auchus RJ. The genetics, pathophysiology and management of human deficiencies of P450c17. Endocrinol Metab Clin North Am. 2001;30:101-19.
- 9. Di Cerbo A, Biason-Lauber A, Savino M, Piemontese MR, Di Giorgio A, Perona M, et al. Combined 17alpha-hydroxylase/17,20-lyase deficiency caused by Phe93Cys mutation in the CYP17 gene. J Clin Endocrinol Metab. 2002;87(2):898-905.
- 10. Van den Akker EL, Koper JW, Boehmer ALM, Themmen APN, Verhoef-Post M, Timmerman MA, et al. Differential inhibition 17alpha-hydroxylase and 17,20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. J Clin Endocrinol Metab. 2002;87(12):5714-21.
- 11. Costa-Santos M, Kater CE, Dias EP, Auchus RJ. Two intronic mutations cause 17-hydroxylase deficiency by disrupting splice acceptor sites: direct demonstration of aberrant splicing and absent enzyme activity by expression of the entire CYP17 gene in HEK-293 cells. J Clin Endocrinol Metab. 2004;89(1):43-8.
- 12. Müsing K, Kaltenbach S, Machicao F, Maser-Gluth C, Hartmann MF, Wudy SA, et al. 17alpha-hydroxylase/17,20-lyase deficiency caused by a novel homozygous mutation (Y27Stop) in the cytochrome CYP17 gene. J Clin Endocrinol Metab. 2005;90(7):4362-5.
- 13. Biason-Lauber A, Leiberman E, Zachmann M. A single amino acid substitution in the putative redox partner-binding site of P450c17 as cause of isolated17,20-lyase deficiency. J Clin Endocrinol Metab. 1997;82:3807-12.
- 14. Geller DH, Auchus RJ, Mendonca BB, Miller WL. The genetic and functional basis of isolated 17,20-lyase deficiency. Nat Genet.1997;17:201-5.
- 15. Miller WL, Auchus RJ, Geller DH. The regulation of 17,20 lyase activity. Steroids. 1997;62:133-42.
-
16The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff. Available in: www.hgmd.cf.ac.uk Accessed on: 10 June 2009.
- 17. Biglieri EG, Herron MA, Brust N. 17-hydroxylation deficiency in man. J Clin Invest. 1966;45:1946-54.
- 18. Zachmann M, Vollmin JA, Hamilton W, Prader A. Steroid 17,20-desmolase deficiency: a new cause of male pseudohermaphroditism. Clin Endocrinol (Oxf). 1972;1:369-85.
- 19. Yanase T, Simpson ER, Waterman MR. 17alpha-hydroxylase/17,20-lyase deficiency: from clinical investigation to molecular definition. Endocr Rev. 1991;12:91-108.
- 20. Taniyama M, Tanabe M, Saito H, Ban Y, Nawata H, Yanase T. Subtle 17alpha-hydroxylase/17,20-lyase deficiency with homozygous Y201N mutation in a infertile women. J Clin Endocrinol Metab. 2005;90(5):2508-11.
- 21. Wei J-Q, Wei J-L, Li W-C, Bi Y-S, Wei F-C. Genotype of five Chinese patients with 17alpha-hydroxylase deficiency diagnosed through high-performance liquid chromatography serum adrenal profile: identification of two novel CYP17 mutations. J Clin Endocrinol Metab. 2006;91(9):3647-53.
- 22. Kater CE, Biglieri EG, Brust N, Chang B, Hirai J. The unique patterns of plasma aldosterone and 18-hydroxycorticosterone concentrations in the 17 alpha-hydroxylase deficiency syndrome. J Clin Endocrinol Metab. 1982;55(2):295-302.
- 23. Kater CE, Biglieri EG. Zona fasciculata origin of 18-hydroxycorticosterone in the chronically suppressed zona glomerulosa. J Clin Endocrinol Metab. 1982;55(4):628-33.
- 24. Yang J, Cui B, Sun S, Shi T, Zheng S, Bi Y, et al. Genotype-phenotype correlation in eight Chinese 17alpha-hydroxylase/17,20-lyase deficiency patients with five novel Mutations of CYP17A1 gene. J Clin Endocrinol Metab. 2006;91(9):3619-25.
- 25. Rosa S, Duff C, Meyer M, Lang-Muritano M, Balercia G, Boscaro M, et al. P450c17 deficiency: clinical and molecular characterization of six patients. J Clin Endocrinol Metab. 2007;92(3):1000-7.
- 26. Tiosano D, Knopf C, Koren I, Levanon N, Hartmann MF, Hochberg Z, et al. Metabolic evidence for impaired 17alpha-hydroxylase activity in a kindred bearing the E305G mutation for isolate 17,20-lyase activity. Eur J Endocrinol. 2008;158:385-92.
- 27. Caulfield MP, Lynn T, Gottschalk ME, Jones KL, Taylor NF, Malunowicz EM, et al. The diagnosis of congenital adrenal hyperplasia in the newborn by gas chromatography/mass spectrometry analysis of random urine specimens J Clin Endocrinol Metab. 2002;87(8):3682-90.
- 28. Martin RM, Lin CJ, Costa EMF, De Oliveira ML, Carrilho A, Villar H, et al. P450c17 deficiency in Brazilian patients: biochemical diagnosis through progesterone levels confirmed by CYP17 genotyping. J Clin Endocrinol Metab. 2003;88(12):5739-46.
- 29. Sherbet DP, Tiosano D, Kwist KM, Hochberg Z, Auchus RJ. CYP17 mutation E305G causes isolated 17,20-lyase deficiency by selectively altering substrate binding. J Biol Chem. 2003;278:48563-9.
- 30. Hershkovitz E, Parvari R, Wudy SA, Hartmann MF, Gomes LG, Loewental N, et al. Homozygous mutation G539R in the gene for P450 oxidoreductase in a family previously diagnosed as having 17,20-lyase deficiency. J Clin Endocrinol Metab. 2008;93(9):3584-8.
Publication Dates
-
Publication in this collection
08 Feb 2011 -
Date of issue
Dec 2010
History
-
Received
05 Jan 2010 -
Accepted
02 Aug 2010