Resumos
A acromegalia é uma doença associada à elevada morbidade e à redução da expectativa de vida. Em virtude do seu caráter insidioso e do seu não reconhecimento, o diagnóstico é frequentemente realizado com atraso, o que, associado às complicações relacionadas ao excesso do GH/IGF-I, determina elevada morbimortalidade. No entanto, um diagnóstico precoce e um tratamento efetivo minimizam a morbidade e normalizam a taxa de mortalidade. Nesta publicação, o objetivo do Departamento de Neuroendocrinologia da Sociedade Brasileira de Endocrinologia e Metabologia é divulgar quando suspeitar clinicamente da acromegalia e como diagnosticá-la. Além disso, discute-se a maneira mais eficaz e segura de realizar o tratamento da acromegalia, enfatizando que este deve ser realizado em centros de referência. Assim, com base em dados publicados em periódicos de nível científico reconhecido e na experiência dos autores, são apresentadas as recomendações para o diagnóstico e tratamento da doença.
Acromegalia; recomendações; diagnóstico; tratamento
Acromegaly is a disease associated with increased morbidity and reduced life expectancy. Because of its insidious character and its non-recognition, the diagnosis is often made with delay, which, along with the complications related to GH/IGF-I excess, determines high morbidity and mortality. However, an early diagnosis and an effective treatment reduce the morbidity and normalize the mortality rate. In this publication, the goal of Neuroendocrinology Department from Brazilian Society of Endocrinology and Metabolism is to disclose which clinical set should arouse the suspicious of acromegaly and how to diagnose it. Furthermore, we discuss the most effective and safe approach to perform the treatment of acromegaly, emphasizing that it must be carried out in reference centers. Therefore, based on data published in journals with recognized scientific level and authors' experience, recommendations are presented for diagnosis and treatment of the disease.
Acromegaly; recommendations; diagnosis; treatment
REVISÃO
Recomendações do Departamento de Neuroendocrinologia da Sociedade Brasileira de Endocrinologia e Metabologia para o diagnóstico e tratamento da acromegalia no Brasil
Recommendations of Neuroendocrinology Department from Brazilian Society of Endocrinology and Metabolism for diagnosis and treatment of acromegaly in Brazil
Leonardo Vieira NetoI,II; Julio AbuchamIII; Luiz Antônio de AraujoIV; Cesar L. BoguszewskiV; Marcello D. BronsteinVI; Mauro CzepielewskiVII; Raquel S. JalladVI; Nina R. de C. MusolinoVIII; Luciana A. NavesIX; Antônio Ribeiro-Oliveira JúniorX; Lucio VilarXI; Manuel dos S. FariaXII; Mônica R. GadelhaI,XIII
IServiço de Endocrinologia, Hospital Universitário Clementino Fraga Filho (HUCFF), Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brasil
IIServiço de Endocrinologia, Hospital da Lagoa, Rio de Janeiro, RJ, Brasil
IIIUnidade de Neuroendócrino, Escola Paulista de Medicina, Universidade Federal de São Paulo (Unifesp/ EPM), São Paulo, SP, Brasil
IVInstituto de Endocrinologia de Joinville (IEJE/CAD), Joinville, SC, Brasil
VServiço de Endocrinologia e Metabologia, Hospital de Clínicas, Universidade Federal do Paraná (UFPR), Curitiba, PR, Brasil
VIServiço de Endocrinologia, Hospital de Clínicas, Faculdade de Medicina da Universidade de São Paulo (FMUSP), São Paulo, SP, Brasil
VIIServiço de Endocrinologia, Hospital de Clínicas de Porto Alegre, PPG Endocrinologia, Faculdade de Medicina, Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brasil
VIIIDivisão de Neurocirurgia Funcional, Instituto de Psiquiatria, HC-FMUSP, São Paulo, SP, Brasil
IXServiço de Endocrinologia, Hospital Universitário de Brasília, Universidade de Brasília (UnB), Brasília, DF, Brasil
XServiço de Endocrinologia, Hospital de Clínicas, Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, MG, Brasil
XIServiço de Endocrinologia, Hospital de Clínicas, Universidade Federal de Pernambuco (UFPE), Recife, PE, Brasil
XIIServiço de Endocrinologia, Hospital Universitário Presidente Dutra, Universidade Federal do Maranhão (UFMA), São Luís, MA, Brasil
XIIIServiço de Endocrinologia, Instituto Estadual de Diabetes e Endocrinologia Luiz Capriglione (IEDE), Rio de Janeiro, RJ, Brasil
Correspondência para Correspondência para: Mônica R. Gadelha Rua Prof. Rodolpho Paulo Rocco, 255, 9º andar, Ilha do Fundão 21941-913 Rio de Janeiro, RJ, Brasil mgadelha@hucff.ufrj.br
SUMÁRIO
A acromegalia é uma doença associada à elevada morbidade e à redução da expectativa de vida. Em virtude do seu caráter insidioso e do seu não reconhecimento, o diagnóstico é frequentemente realizado com atraso, o que, associado às complicações relacionadas ao excesso do GH/IGF-I, determina elevada morbimortalidade. No entanto, um diagnóstico precoce e um tratamento efetivo minimizam a morbidade e normalizam a taxa de mortalidade. Nesta publicação, o objetivo do Departamento de Neuroendocrinologia da Sociedade Brasileira de Endocrinologia e Metabologia é divulgar quando suspeitar clinicamente da acromegalia e como diagnosticá-la. Além disso, discute-se a maneira mais eficaz e segura de realizar o tratamento da acromegalia, enfatizando que este deve ser realizado em centros de referência. Assim, com base em dados publicados em periódicos de nível científico reconhecido e na experiência dos autores, são apresentadas as recomendações para o diagnóstico e tratamento da doença.
Descritores: Acromegalia; recomendações; diagnóstico; tratamento
SUMMARY
Acromegaly is a disease associated with increased morbidity and reduced life expectancy. Because of its insidious character and its non-recognition, the diagnosis is often made with delay, which, along with the complications related to GH/IGF-I excess, determines high morbidity and mortality. However, an early diagnosis and an effective treatment reduce the morbidity and normalize the mortality rate. In this publication, the goal of Neuroendocrinology Department from Brazilian Society of Endocrinology and Metabolism is to disclose which clinical set should arouse the suspicious of acromegaly and how to diagnose it. Furthermore, we discuss the most effective and safe approach to perform the treatment of acromegaly, emphasizing that it must be carried out in reference centers. Therefore, based on data published in journals with recognized scientific level and authors' experience, recommendations are presented for diagnosis and treatment of the disease.
Keywords: Acromegaly; recommendations; diagnosis; treatment
INTRODUÇÃO
Acromegalia é uma doença sistêmica crônica, decorrente da produção excessiva do hormônio do crescimento (growth hormone GH) e do fator de crescimento semelhante à insulina tipo I (insulin-like growth factor type I IGF-I). Em 98% dos casos, a doença é causada por um adenoma hipofisário secretor de GH (somatotropinoma), em cerca de 2% dos casos pela hipersecreção eutópica ou ectópica do hormônio liberador do GH (growth-hormone releasing hormone GHRH) e muito raramente pela secreção ectópica de GH. A doença é mais comum entre 30 e 50 anos de idade, não havendo predileção por gênero (1,2).
A acromegalia, quando não controlada adequadamente, leva a graves complicações cardiovasculares, respiratórias, metabólicas e neoplásicas (3-7). Reid e cols. (8) analisaram as diferenças clínicas, laboratoriais e radiológicas entre os pacientes acromegálicos com a doença diagnosticada entre os períodos de 1981 a 1994 e 1995 a 2006. Os autores não encontraram diferença entre esses dois períodos com relação ao tempo estimado de início dos sintomas até o diagnóstico, assim como dos níveis de GH e IGF-I e do volume tumoral. Em relação aos sinais, sintomas e comorbidades presentes ao diagnóstico, apenas roncos, apneia, fadiga e pólipos colônicos foram observados com frequência maior no período mais recente. Assim, apesar da melhora nos ensaios de dosagem hormonal, esses achados mostram que o atraso no diagnóstico se manteve inalterado nas últimas três décadas. Isso reflete não apenas o caráter insidioso da doença, mas também o não reconhecimento das características clínicas tanto por parte da população geral quanto pelos profissionais de saúde, de forma que o diagnóstico é frequentemente realizado com importante atraso, ou seja, cerca de oito a dez anos após o aparecimento dos primeiros sinais e sintomas. Esse atraso no diagnóstico, associado aos efeitos do excesso do GH e IGF-I, determina elevada morbimortalidade.
Estudos de metanálises recentes apontam para uma taxa de mortalidade de aproximadamente 1,7 vez maior do que a população geral, quando os níveis de GH e IGF-I se encontram elevados (9,10). Holdaway e cols. mostraram que pacientes com níveis de GH randômico < 2,5 ng/mL avaliados por radioimunoensaio (ou < 1,0 ng/mL por ensaios mais sensíveis) e normalização dos níveis de IGF-I segundo a idade tinham mortalidade próxima da esperada para a população geral. Duração dos sintomas antes do diagnóstico, tempo de doença, idade avançada ao diagnóstico e presença de doença cardiovascular, diabetes melito (DM) e hipertensão arterial sistêmica (HAS) ao diagnóstico são outros fatores que contribuem para o aumento da mortalidade na acromegalia (11). Dessa forma, é prioritária a instituição não apenas de um diagnóstico o mais precocemente possível, mas também de um tratamento efetivo e seguro, capaz de minimizar a morbidade e de normalizar a taxa de mortalidade para a esperada de acordo com a idade e o sexo do paciente.
Nesta publicação, o objetivo do Departamento de Neuroendocrinologia da Sociedade Brasileira de Endocrinologia e Metabologia é fornecer as recomendações para um diagnóstico adequado e precoce e para um tratamento eficaz e seguro da acromegalia, enfatizando que ambos devem ser realizados em centros de referência. Assim, com base em dados publicados em periódicos de nível científico reconhecido e na experiência dos autores, representantes dos principais centros de referência para tratamento da acromegalia, são apresentadas as recomendações para o diagnóstico e tratamento da doença no Brasil.
DIAGNÓSTICO
Diagnóstico clínico
A acromegalia apresenta curso clínico insidioso, cujas manifestações clínicas decorrem do excesso do GH e/ ou do IGF-I e do efeito de massa do adenoma sobre as estruturas nobres adjacentes à sela túrcica (1).
Como existe um grande atraso no diagnóstico, muitos pacientes já apresentam a doença tipicamente evidente, caracterizada por: fácies acromegálica (protrusão da fronte, acentuação dos malares, aumento do nariz e lábios, acentuação dos sulcos nasolabiais e prognatismo), aumento de extremidades (dedos em "salsicha", aumento do número do calçado), hiperidrose, artralgia, síndrome do túnel do carpo, apneia do sono (roncos noturnos e sonolência diurna), HAS, alterações do metabolismo glicídico (resistência insulínica, intolerância à glicose e DM), visceromegalias (especialmente bócio) e papilomas cutâneos (skin tags), sendo estes últimos importantes marcadores da presença de pólipos colônicos (12). Cumpre mencionar que cerca de 25% dos pacientes com acromegalia apresentam adenoma cossecretor de GH e prolactina (PRL). Nesses casos, além das manifestações relacionadas ao excesso de GH e IGF-I, os pacientes podem também apresentar quadro clínico decorrente da hiperprolactinemia tumoral (1). No entanto, pacientes com doença leve e/ou de curta duração de evolução podem não apresentar um fenótipo tão exuberante quanto o descrito anteriormente, justificando, em parte, a não suspeição e o atraso no diagnóstico nessa subpopulação de pacientes.
Como cerca de 80% dos pacientes apresentam macroadenoma (> 10 mm) ao diagnóstico, não são infrequentes queixas relacionadas ao efeito de compressão do tumor sobre as estruturas vizinhas à região selar, uma vez que pode haver expansão suprasselar (cisterna suprasselar, quiasma óptico, hipotálamo e terceiro ventrículo), infrasselar (seio esfenoidal) e parasselar (seio cavernoso/pares cranianos). Cefaleia é um sintoma comum e pode estar relacionada ao estiramento da dura-máter ou à compressão do quinto par craniano (nervo trigêmeo). Déficit visual, especialmente hemianopsia bitemporal, por compressão do quiasma óptico, também pode ocorrer, sendo, portanto, obrigatória a realização da campimetria visual, de preferência manual (método de Goldmann), em todos os pacientes com macroadenoma com expansão suprasselar. Expansão parasselar (invasão do seio cavernoso) pode comprometer os pares de nervos cranianos oculomotor, troclear, trigêmeo e abducente, determinando quadro clínico variável, dependendo do(s) nervo(s) acometido(s). Expansão infrasselar pode causar fístula liquórica devido à erosão do assoalho selar, levando a um quadro de rinoliquorreia, e pode ser complicada com meningite. Outras consequências do efeito de massa pelo tumor incluem hipopituitarismo (por compressão da adenoipófise normal e/ou da haste hipofisária), hiperprolactinemia (neste caso por compressão da haste) e, mais raramente, hidrocefalia (compressão do terceiro ventrículo).
A tabela 1 reúne os sinais e sintomas associados à acromegalia, os quais, quando presentes, indicam a necessidade de investigação laboratorial da doença.
Diagnóstico laboratorial
A suspeita clínica de acromegalia deve ser confirmada pela avaliação laboratorial com a demonstração de níveis séricos elevados de GH e de IGF-I. Deve-se ressaltar que o GH é secretado de forma pulsátil e, por isso, em indivíduos normais, 70% a 80% dos valores de GH em 24 horas são indetectáveis, mas podem ocorrer picos de secreção com valores que excedem várias vezes o "normal" (13). Por outro lado, pacientes acromegálicos podem apresentar valores de GH dentro da variação dita como "normal" (14). Assim, a interpretação dos valores basais de GH deve ser realizada com cautela, já que não existe uma faixa de normalidade para o GH basal (ou randômico) estabelecida que diferencie indivíduos normais daqueles com acromegalia.
Diferentemente do GH, o IGF-I reflete a secreção integrada do GH, não apresentando flutuação circadiana nem secreção pulsátil, o que o torna uma ferramenta mais robusta para caracterização de atividade de doença. No Brasil, recentemente foram publicados valores de referência para IGF-I para diferentes faixas etárias pelo ensaio Immulite 2000 (15). Causas de resultados falso-positivos na dosagem de IGF-I incluem puberdade, gravidez e hipertireoidismo e de falso-negativos, desnutrição, anorexia nervosa, hipotireoidismo, DM descompensado, doença hepática e renal e estrogenioterapia oral (Tabela 2) (16).
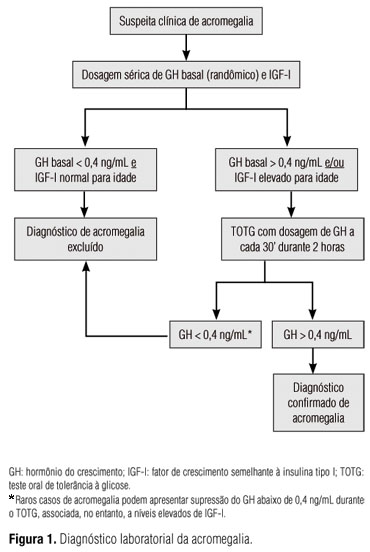
Conforme o consenso realizado em Cortina em 1999 e publicado no ano seguinte (17), o rastreamento é realizado com as dosagens basais de GH e IGF-I: níveis séricos randômicos de GH < 0,4 ng/mL e de IGF-I normais para a idade excluem o diagnóstico de acromegalia (Figura 1), na ausência de fatores que influenciem essas dosagens.
Naqueles pacientes em que o diagnóstico não pode ser afastado com as dosagens basais, está indicado o teste oral de tolerância à glicose (TOTG) com a administração de 75 g de glicose anidra e dosagens subsequentes de GH e glicose a cada 30 minutos por duas horas (17). Entretanto, nos casos em que os níveis de GH basal e IGF-I estão indubitavelmente elevados, o TOTG não é obrigatório (18). De acordo com o novo consenso realizado em Paris e recentemente publicado (18), o GH deve diminuir para valores inferiores a 0,4 ng/mL em qualquer um dos tempos durante o TOTG para que se exclua o diagnóstico de doença ativa (Figura 1). Causas de resultados falso-positivos no TOTG incluem DM descompensado, doença renal ou hepática, hipertireoidismo, anorexia nervosa, desnutrição e adolescência (19). Pacientes com diagnóstico de DM não devem ser submetidos ao TOTG. Raros casos de acromegalia, especialmente pacientes idosos e/ou com microadenoma, podem apresentar supressão do GH abaixo de 0,4 ng/mL durante o TOTG associada, no entanto, com níveis elevados de IGF-I. Nesses casos excepcionais, níveis de IGF-I elevados, na ausência de condições que podem determinar resultados falso-positivos, e juntamente com sinais e sintomas de acromegalia em atividade e presença de um adenoma hipofisário, são suficientes para o diagnóstico de acromegalia (14,20).
Testes dinâmicos com TRH, GHRH ou GnRH não são atualmente empregados, pois não oferecem nenhum benefício adicional ao TOTG ou IGF-I, além de onerarem os custos da investigação e exporem o paciente com macroadenomas ao risco de apoplexia. De forma semelhante, dosagens de IGFBP-3, IGF-I livre, GH urinário e subunidade ácido-lábil também não são utilizadas, por não terem valor cientificamente comprovado na abordagem diagnóstica (17).
A dosagem de GHRH, embora não disponível no Brasil, está indicada nos raros casos em que se suspeita de acromegalia causada por produção ectópica de GHRH. A presença de um segundo tumor, especialmente se na presença de sinais e sintomas relacionados à secreção de outros hormônios (p. ex., catecolaminas, gastrina, serotonina e insulina) e/ou ao efeito de massa tumoral (em outro sítio que não região selar), pode sugerir a possibilidade de acromegalia secundária à secreção de GHRH (para mais detalhes ver referência 2).
Diagnóstico por imagem
Após a confirmação do diagnóstico clínico e laboratorial da acromegalia, o paciente deve ser submetido a um exame de imagem da sela túrcica, ressonância magnética (RM) preferencialmente ou tomografia computadorizada (TC) para identificação e caracterização do adenoma hipofisário quanto ao seu tamanho (micro ou macroadenoma), aspecto (sólido, cístico ou heterogêneo), expansão/invasão extrasselar (supra, infra e paras-selar) e relação com as estruturas adjacentes à sela túrcica (quiasma óptico e seios cavernosos). Esses dados são de extrema valia, pois são preditores de chance de cura ao tratamento cirúrgico e podem orientar a melhor opção terapêutica, como será abordado adiante.
Em pacientes com acromegalia secundária à secreção de GHRH, a RM de sela túrcica pode revelar hipófise normal ou difusamente aumentada, que pode ser erroneamente diagnosticada como macroadenoma. Portanto, um radiologista experiente tem um papel importante na avaliação da RM.
As recomendações dos autores para o diagnóstico da acromegalia encontram-se no quadro 1.
TRATAMENTO
O tratamento da acromegalia, na maioria das vezes, requer abordagem multidisciplinar, envolvendo a participação de endocrinologistas, neurocirurgiões, neurorradiologistas, neuropatologistas e radioterapeutas. O tratamento é considerado efetivo quando preenche os critérios de cura/ controle estabelecidos pelo consenso realizado em Paris em 2009 e recentemente publicado (18), que são: nível randômico (basal) de GH < 1 ng/mL ou nadir de GH < 0,4 ng/mL durante o TOTG e normalização do IGF-I para a idade. É importante ressaltar que os dados sobre a taxa de cura com a cirurgia e sobre a eficácia da terapia medicamentosa ou radioterápica no controle da acromegalia com os novos critérios ainda não estão disponíveis. Dessa forma, os resultados de eficácia das diferentes modalidades terapêuticas aqui apresentados baseiam-se nos critérios de cura do consenso de Cortina (17).
Ainda, para pacientes com valores discrepantes entre GH randômico e IGF-I, pode-se lançar mão do perfil de GH de cinco pontos em vez do seu valor randômico, calculando a média aritmética dos cinco valores do GH coletados a cada 30 minutos (21). Segundo o novo consenso de cura da acromegalia, um valor médio de GH abaixo de 1,0 ng/mL também é utilizado para definir controle bioquímico (18).
Finalmente, outros objetivos almejados com o tratamento incluem ressecção ou controle da massa tumoral com preservação do restante da função adenoipofisária, prevenção de recidivas e controle das complicações, especialmente cardiovasculares, respiratórias e metabólicas.
As opções terapêuticas para a abordagem da acromegalia incluem cirurgia, medicamentos e radioterapia.
Cirurgia
A cirurgia transesfenoidal pode ser o tratamento primário de escolha para acromegalia, sendo a abordagem transcraniana muito raramente empregada. Para um bom resultado do tratamento cirúrgico, é fundamental a experiência do neurocirurgião e, portanto, a cirurgia deve ser feita em centros especializados de referência. Experiência é definida pela realização de pelo menos 50 cirurgias transesfenoidais anualmente (22). Além disso, o resultado cirúrgico também depende do nível pré-operatório de GH e do tamanho e do grau de expansão extrasselar tumoral. Pacientes com níveis muito elevados de GH e com tumores com invasão parasselar apresentam baixa chance de cura (23).
Nomikos e cols. (23) relataram os resultados cirúrgicos em uma grande série de pacientes acompanhados durante 19 anos em um dos melhores centros de referência do mundo. Cura laboratorial foi definida como normalização do GH basal (GH < 2,5 ng/mL), IGF-I normal para idade e supressão do GH < 1,0 ng/mL durante o TOTG. Dos 506 pacientes submetidos à cirurgia transesfenoidal, 57% preencheram os critérios de cura. A taxa de cura se correlacionou com os níveis séricos iniciais de GH, tamanho e invasividade tu-morais. Os melhores resultados foram alcançados com microadenomas e macroadenomas intrasselares, com taxas de cura de 75% e 74%, respectivamente. A taxa de cura foi menor no grupo dos macroadenomas: 45% e 33% para tumores com expansão suprasselar sem e com comprometimento visual, respectivamente, e 42% para aqueles com expansão parasselar. Para os adenomas gigantes (maiores do que 4 cm), a taxa de cura foi de 10%. Durante o acompanhamento por um período de 10,7 anos, a taxa de recorrência foi de 0,4% e a de mortalidade foi de 0,1%.
Apesar da baixa chance de cura cirúrgica em pacientes com macroadenomas com expansão extrasselar, especialmente parasselar, a remoção da maior quantidade possível de tecido tumoral (debulking) pode aumentar a chance de controle dos níveis hormonais com o tratamento medicamentoso adjuvante (24).
A grande vantagem da cirurgia é proporcionar cura imediata, sendo, na maioria das vezes, definitiva. Entretanto, como aproximadamente 60%-70% dos pacientes apresentam macroadenoma com expansão extrasselar ao diagnóstico e as taxas de cura cirúrgica são baixas nessa população, a terapia complementar à cirurgia faz-se necessária em um grande número de pacientes.
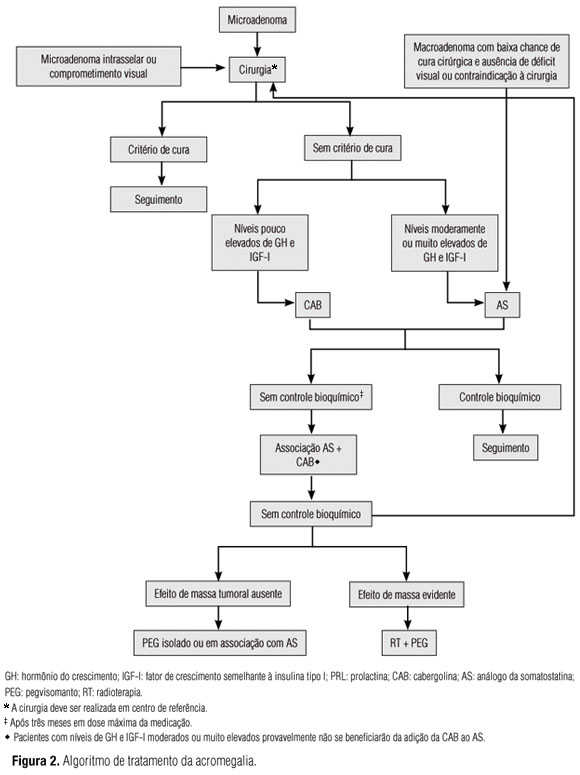
Assim, recomenda-se a cirurgia nos pacientes com micro e macroadenoma intrasselares em que a chance de cura é alta (em torno de 75%) ou ainda naqueles com comprometimento visual (23). Em pacientes com tumores com baixa chance de cura cirúrgica (níveis muito elevados de GH, tumores volumosos e presença de expansão parasselar) ou em pacientes com elevado risco cirúrgico ou que recusem submeter-se ao procedimento, a melhor opção é o tratamento medicamentoso primário com análogo da somatostatina (AS). Em pacientes não controlados com tratamento medicamentoso [AS isoladamente ou em combinação com cabergolina (CAB)], cirurgia de debulking pode ser uma opção. Por outro lado, a cirurgia de debulking também pode ser realizada previamente ao tratamento medicamentoso, levando-se em consideração não apenas o status clínico do paciente, mas também a experiência e a disponibilidade do centro com cada modalidade terapêutica (Quadro 2).
Recomenda-se que, para avaliar a eficácia da cirurgia, dosagens séricas de GH durante o TOTG e IGF-I devem ser realizadas um mês após o procedimento, juntamente com a avaliação de toda a função adenoipofisária. Segundo o novo consenso (18), o paciente é considerado curado quando o nadir do GH durante o TOTG estiver abaixo de 0,4 ng/mL e o valor de IGF-I estiver normal para a idade. Para pacientes com nadir do GH < 0,4 ng/mL, mas com IGF-I elevado, deve-se repetir este último três meses após a cirurgia. Exame de imagem da sela túrcica deve ser solicitado três a seis meses após a cirurgia. Para os pacientes considerados curados, o seguimento é feito apenas com dosagens de GH basal e IGF-I a cada seis meses (Quadro 3). Freda e cols. (25) mostraram que pacientes com nadir de GH > 0,14 ng/mL e níveis normais de IGF-I estão sob maior risco de recidiva do que pacientes com GH < 0,14 ng/mL durante o TOTG pós-operatório e níveis normais de IGF-I. Deve-se estar atento para pacientes que estavam em uso de AS previamente à cirurgia, especialmente se por longo período, pois os níveis hormonais "normais" no pós-operatório podem refletir a terapia supressiva com os AS no período pré-operatório, não caracterizando, portanto, remissão. A avaliação da função adenoipofisária deve ser realizada por meio das dosagens de cortisol, T4 livre, prolactina, LH, FSH e, em homens, da testosterona. Em mulheres, determinação dos ciclos menstruais é suficiente para avaliação da função gonadal.
A mortalidade da cirurgia transesfenoidal, quando realizada por neurocirurgiões com experiência nesse procedimento, é menor que 1%; outras complicações graves, como alteração visual e meningite, ocorrem raramente. Fístula liquórica, diabetes insípido central permanente e sinusite ocorrem em torno de 5% dos casos. Hipopituitarismo e diabetes insípido central transitório manifestam-se na minoria dos casos (23). Deve-se ressaltar, no entanto, que alguns pacientes podem recuperar-se em parte do hipopituitarismo após a cirurgia, por descompressão das células hipofisárias pela remoção do efeito de massa, justificando a avaliação da função adenoipofisária não apenas ao diagnóstico, mas também após o procedimento cirúrgico (23,26).
Tratamento medicamentoso
Três classes de drogas estão disponíveis para o tratamento medicamentoso da acromegalia: agonistas dopaminérgicos, análogos da somatostatina e antagonista do receptor de GH (Tabela 3).
Agonistas dopaminérgicos (AD): Foram a primeira classe de medicamentos usados no tratamento da acromegalia, e as duas drogas comercialmente disponíveis no Brasil são a bromocriptina (BRC) e a CAB. Atuam via receptores dopaminérgicos tipo 2 (DR2), expressos em somatotrofos normais e em células tumorais (27). A BRC foi o primeiro AD usado no tratamento da acromegalia, porém seus resultados foram desapontadores. Jaffe e Barkan (28), em sua revisão da eficácia bioquímica da BRC em mais de 500 acromegálicos em 31 séries, observaram que níveis de GH < 5 ng/mL eram obtidos em 20% dos pacientes, com normalização do IGF-I em apenas 10%. Portanto, não se recomenda o uso da BRC para pacientes com acromegalia.
Em comparação à BRC, a CAB tem maior afinidade de ligação aos DR2 e meia-vida mais prolongada, evitando grandes flutuações nos níveis séricos da droga, o que aumenta sua eficácia terapêutica e reduz a frequência de efeitos colaterais (29). Dessa forma, em virtude de maior eficácia e melhor perfil de tolerabilidade, a CAB pode ser útil no algoritmo de tratamento de alguns pacientes com acromegalia. Por ser uma medicação de custo inferior aos AS, de administração oral e com boa tolerabilidade, recomenda-se que a CAB seja empregada como terapia adjuvante em pacientes com níveis pouco elevados de GH e IGF-I e/ou imunoistoquímica do adenoma mostrando cossecreção GH/ PRL. Além disso, a CAB pode ser associada aos AS em pacientes não controlados com os AS em dose máxima isoladamente (Figura 2; Quadros 4 e 5).
Abs e cols. (30) demonstraram que a cossecreção GH/PRL tem valor preditivo na resposta à monoterapia com a CAB. Além disso, níveis pré-tratamento do GH e IGF-I também se correlacionaram com a resposta a CAB: GH < 20 ng/mL e/ou IGF-I < 750 ng/mL tiveram associação com uma taxa mais elevada de normalização de GH e IGF-I. Embora os dados de eficácia sobre o uso da CAB na acromegalia sejam escassos, a maior casuística (30) que avaliou sua eficácia mostrou uma redução dos níveis de GH para valores abaixo de 2,0 ng/mL e do IGF-I para valores abaixo de 300 ng/ mL em 46% e 39% dos pacientes, respectivamente. Discreta redução do volume tumoral pode ocorrer com a CAB, porém reduções mais significativas podem ser observadas nos adenomas cossecretores GH/PRL (31).
A CAB está disponível em comprimidos de 0,5 mg, e a dose mínima é de 1,5 mg/semana. Com o objetivo de minimizar a frequência e a intensidade dos efeitos colaterais, deve-se iniciar 0,5 mg/semana, elevando-se a dose em um comprimido por semana até atingir a dose de 1,5 mg/semana. A partir de então, a dose deve ser escalonada segundo os níveis de GH e IGF-I mensurados mensalmente durante a titulação da dose. A dose máxima é de 3,5 mg/semana. O volume tumoral deve ser avaliado por meio de exame de imagem a cada 6-12 meses durante o tratamento medicamentoso. Se o volume tumoral não mostrar crescimento nos primeiros anos e o paciente tiver a doença controlada, o exame de imagem pode ser realizado a cada 2-3 anos. Além do controle dos níveis hormonais, a atividade da doença deve ser avaliada clinicamente em toda consulta (cefaleia, fadiga, sudorese excessiva, parestesia, ronco noturno e sonolência diurna, hipertensão arterial e aumento de partes moles). A avaliação da função adenoipofisária deve ser realizada a cada seis meses, conforme descrito anteriormente.
As reações adversas à CAB ocorreram em 13% dos pacientes em uma grande série em que essa droga foi testada (29) e incluem náuseas, tonteiras, cefaleia, constipação intestinal, xerostomia, obstrução nasal e hipotensão postural (28). Schade e cols. (32) mostraram risco elevado de lesão valvar cardíaca em pacientes com doença de Parkinson usando CAB. Esses autores mostraram que esse risco foi dose-dependente e tempo-dependente. Embora lesão valvar clinicamente relevante não tenha sido descrita em pacientes recebendo AD para tratamento de adenomas hipofisários (33), recomenda-se a realização do ecocardiograma transtorácico antes do início da CAB e depois a cada 12 meses durante a terapia (Quadro 4).
Análogos da somatostatina: Os dois análogos sintéticos da somatostatina disponíveis comercialmente são o octreotide (OCT) e o lanreotide (LAN), ambos exercendo seus efeitos mediante a interação com os receptores da somatostatina (SSTR) subtipos 2 (principalmente) e 5, expressos nos somatotropinomas (34,35). As preparações de longa duração disponíveis no Brasil são o OCT-LAR (long acting release) e o LAN Autogel. Constituem, atualmente, a classe de droga de escolha para o tratamento da acromegalia, podendo ser empregados como terapia primária ou adjuvante à cirurgia. A terapia primária com os AS está recomendada para pacientes com baixa chance de cura cirúrgica, com alto risco cirúrgico e naqueles que se recusam à cirurgia (Quadro 5).
Freda e cols. (36), ao avaliarem a eficácia terapêutica dos AS na acromegalia por meio de metanálise, constataram normalização do IGF-I em 67% e diminuição do GH para níveis < 2,5 ng/mL em 57% dos pacientes tratados com OCT-LAR. Além disso, foi observada redução do volume tumoral de pelo menos 10% em 47% dos pacientes em tratamento adjuvante e em 89% daqueles em terapia primária com OCT-LAR. A eficácia do LAN Autogel é semelhante à do OCT-LAR, conforme demonstrado por Murray e Melmed (37). Jallad e cols. (38) avaliaram a eficácia do OCT-LAR em 80 acromegálicos tratados por 6 a 24 meses, dos quais 28 receberam tratamento primário com a droga. Níveis de GH < 2,5 ng/mL e normalização do IGF-I foram alcançados em 59 (74%) e 33 pacientes (41%), respectivamente. Redução do volume tumoral de pelo menos 25% foi considerada significativa, sendo alcançada em 76% dos pacientes que receberam tratamento primário.
Em situações em que a chance de cura cirúrgica é baixa, em pacientes com risco cirúrgico proibitivo e naqueles que se recusam à cirurgia, os AS têm sido cada vez mais empregados como terapia primária (Figura 2). Cozzi e cols. (39) avaliaram o impacto do OCT-LAR como terapia primária em 67 pacientes não selecionados com macroadenoma. Após uma mediana de acompanhamento de 48 meses (6-108 meses), GH < 2,5 ng/mL e IGF-I normal para idade foram alcançados em 69% e 70% dos pacientes, respectivamente. Redução tumoral de 62% ± 31% foi observada em 82% dos pacientes.
Além disso, o impacto da terapia medicamentosa pré-operatória tem sido avaliado por alguns autores. Em teoria, o uso do AS antes da cirurgia combinaria os efeitos sobre a redução do volume tumoral e da atividade da doença, os quais melhorariam os resultados cirúrgicos. No entanto, os dados da literatura ainda não comprovaram o benefício do uso pré-operatório do AS sobre os resultados cirúrgicos e, por isso, não se recomenda de rotina o seu emprego antes da cirurgia com esse objetivo.
Uma vez que os níveis de GH se correlacionam negativamente com a chance de controle bioquímico da doença durante a terapia com AS, a cirurgia de debulking tem sido empregada com o objetivo de aumentar a chance de controle bioquímico com os AS (Figura 2). Jallad e cols. (24) avaliaram o efeito da cirurgia sobre o controle bioquímico da acromegalia em pacientes tratados previamente com OCT-LAR que não atingiram normalização dos níveis hormonais durante a terapia primária. Dos 11 pacientes avaliados, 4 (36%) apresentaram níveis de GH < 2,5 ng/mL e nenhum normalizou o nível de IGF-I durante a terapia primária. No entanto, nessa mesma população, durante a terapia adjuvante, níveis de GH < 2,5 ng/mL e normalização do IGF-I para a idade foram observados em sete (64%) e nove pacientes (82%), respectivamente. Assim, após a cirurgia de debulking, eles observaram que a redução do volume tumoral nesses pacientes melhorou significativamente o resultado da terapia adjuvante com o AS.
Por fim, os AS podem ser associados com a CAB naqueles pacientes que não obtiveram controle satisfatório apenas com os primeiros (Figura 2). Cozzi e cols. (40) relataram o efeito da terapia combinada de OCT-LAR e CAB em 19 pacientes resistentes à terapia com AS. A terapia combinada proporcionou queda importante dos níveis de GH e IGF-I em 47% dos pacientes, com níveis de GH < 2,5 ng/mL e IGF-I normal em 21% e 42%, respectivamente. Não houve correlação da resposta do GH e IGF-I com nível de PRL ou positividade desse hormônio à imunoistoquímica, diferentemente do que acontece quando a CAB é usada como monoterapia (30). Entretanto, resposta à CAB teve correlação inversa com o nível de GH obtido pelo AS, ou seja, níveis mais baixos de GH durante a terapia com essa droga se correlacionaram positivamente com melhor resposta à terapia combinada. O volume tumoral não foi avaliado no estudo. Jallad e cols. (41) também avaliaram o impacto da associação das duas classes de drogas e observaram que 19 de 34 (56%) pacientes atingiram normalização dos níveis de IGF-I. Não houve correlação da resposta terapêutica com os níveis de PRL, corroborando os achados do estudo de Cozzi e cols. (40). Vilar e cols. (42), assim como Mattar e cols. (43), encontraram resultados similares. Assim, recomenda-se a associação da CAB com os AS em pacientes que não obtiveram controle bioquímico com os AS em dose máxima, particularmente naqueles com níveis de IGF-I leve a moderadamente elevados. Nesses casos, o emprego da CAB deve seguir as mesmas orientações de seu uso como monoterapia.
O OCT-LAR está disponível em ampolas de 10 mg, 20 mg (dose inicial) e 30 mg, de aplicação intramuscular profunda (região glútea) a cada 28 dias. A aplicação da medicação requer treinamento específico e deve ser realizada apenas por profissional de saúde capacitado para tal. O LAN Autogel, de aplicação subcutânea profunda a cada 28 dias, está disponível em ampolas de 90 mg (dose inicial) e 120 mg. O eixo GH/IGF-I deve ser reavaliado a cada três meses, sempre no dia da aplicação e antes dela. Caso sejam preenchidos os critérios laboratoriais de controle da doença (GH basal < 1,0 ng/mL e IGF-I normal para a idade), a dose deve ser mantida. Se, por outro lado, os critérios de controle da doença não forem preenchidos, a dose deve ser aumentada (para 30 mg naqueles recebendo OCT-LAR ou para 120 mg naqueles recebendo LAN Autogel), administrada a cada quatro semanas. Se os níveis de IGF-I estiverem abaixo do limite inferior da normalidade para a idade, deve-se reduzir a dose do OCT-LAR para 10 mg ou aumentar o intervalo entre as aplicações do OCT-LAR 20 mg ou do LAN Autogel 90 mg para cada seis a oito semanas.
Durante o acompanhamento, valores discordantes de GH randômico e IGF-I podem ocorrer em 25%-30% dos pacientes (44). A frequência desse achado diminui quando essas dosagens são realizadas pela mesma metodologia e em laboratórios em que valores normatizados estão disponíveis (15). Caso a discrepância entre GH e IGF-I seja mantida, a conduta deve ser baseada nos níveis de IGF-I, pois reflete a secreção integrada do GH, não apresentando flutuação circadiana, o que o torna uma aferição mais robusta para caracterização de atividade de doença. Uma alternativa é a realização do perfil de GH; um valor médio de GH abaixo de 1,0 ng/mL também é utilizado para definir controle bioquímico. Obviamente, no contexto de discordância, devem-se afastar condições que possam estar falseando os valores encontrados, especialmente para o IGF-I (Tabela 2). A discordância entre os níveis de GH e IGF-I também pode ocorrer em pacientes em tratamento com CAB ou associação CAB e AS e, nesses casos, a conduta é a mesma descrita aqui neste parágrafo.
O volume tumoral deve ser avaliado a cada 6-12 meses durante o tratamento com AS. Se o volume tu-moral não mostrar crescimento nos primeiros anos e o paciente tiver a doença controlada, o exame de imagem pode ser realizado a cada dois a três anos. A atividade da doença também deve ser avaliada clinicamente em toda consulta e a função adenoipofisária, a cada seis meses, como já detalhado anteriormente.
Alterações gastrointestinais (desconforto abdominal, flatulência e aumento do trânsito intestinal) constituem os efeitos colaterais mais comuns dos AS, ocorrendo em metade dos pacientes de forma leve e transitória na maioria das vezes. Colelitíase assintomática ocorre em cerca de 15%. Ultrassonografia de abdome superior deve ser solicitada antes do início do AS e repetida anualmente nos pacientes cujo exame basal foi normal. Na presença de litíase, a abordagem deve ser a mesma empregada para os casos de colelitíase na população geral. Desconforto no local da aplicação, queda transitória de pelos, hipotireoidismo central e bradicardia sinusal assintomática são outros efeitos colaterais (45). O efeito dos AS sobre o metabolismo da glicose é variado e, de certa forma, imprevisível. Assim, é mandatório o acompanhamento com glicemia de jejum e hemoglobina glicosilada (HbA1C) (6).
Antagonistas do receptor de GH: O pegvisomanto (PEG) é o primeiro e único antagonista do receptor de GH (GHR) comercialmente disponível, uma nova classe de drogas, representando importante ferramenta no controle bioquímico da acromegalia. Sua ação é independente das características moleculares e histopatológicas do somatotropinoma. Uma vez que a droga inibe a ligação do GH ao seu receptor, o nível sérico de GH não deve ser usado como marcador bioquímico de sucesso terapêutico, mas sim a normalização dos níveis de IGF-I ajustados para a idade. Dessa forma, os resultados de eficácia do PEG aqui apresentados não sofrem alteração com os novos critérios de controle adotados no último consenso. No Brasil, o uso do PEG está indicado para pacientes em que não foi possível o controle bioquímico da doença com AS em dose máxima isoladamente ou em combinação com a CAB ou para pacientes intolerantes aos AS (Figura 2; Quadro 6).
O primeiro estudo clínico randomizado, duplo-cego, em acromegálicos avaliando a eficácia da medicação, comparado ao placebo, foi realizado por Trainer e cols. (46). Em contraste ao grupo placebo em que os níveis de IGF-I não sofreram modificações, um aumento dose-dependente na frequência de normalização do IGF-I foi observado. Não houve mudança significativa no volume tumoral durante o estudo. Um estudo multicêntrico subsequente (47), que foi uma extensão do estudo acima citado, avaliou a eficácia e a segurança da droga em 152 pacientes tratados por até 18 meses, com uma dose máxima de 40 mg/dia. Oitenta e sete dentre 90 pacientes (97%) tratados por 12 meses ou mais obtiveram normalização do IGF-I, independentemente dos valores de GH e IGF-I pré-tratamento. Dois pacientes com volumosos adenomas apresentaram crescimento tumoral em vigência da terapia. Não está definido, no entanto, se o aumento das dimensões foi secundário à administração de PEG ou relacionado à própria história natural de crescimento do tumor. Um desses pacientes recebeu cotratamento com OCT-LAR, o que permitiu controle bioquímico e do crescimento tumoral (47). Esse exemplo ilustra o efeito sinérgico entre essas duas medicações no controle da doença quando apenas uma droga não é suficiente para tal. Taquifilaxia não foi observada, e baixos níveis de anticorpos antiPEG foram detectados em 17% dos pacientes, porém sem comprometimento da eficácia da medicação (46).
Marazuela e cols. (48) averiguaram a eficácia do PEG como monoterapia em 44 acromegálicos resistentes aos AS. Normalização do IGF-I ocorreu em 84% dos pacientes, porém não foi observada correlação entre a redução ou normalização dos níveis de IGF-I com idade ou níveis de GH e IGF-I pré-tratamento. Entretanto, o percentual médio de redução do IGF-I com o tratamento foi mais pronunciado entre os homens em comparação às mulheres. Além disso, o percentual de controle foi maior no sexo masculino (100% vs. 74%), utilizando-se uma dose média corrigida pelo peso menor (0,19 mg/kg vs. 0,24 mg/kg). Eles observaram também que o percentual médio de redução do IGF-I foi maior no grupo que recebeu radioterapia. Aumento do volume tumoral foi observado em três pacientes entre os meses 15 e 25 após início do PEG. Nenhum desses pacientes tinha apresentado redução de volume tumoral durante a terapia com AS.
Alguns pacientes podem se beneficiar da associação do PEG com os AS, por exemplo, aqueles que apresentam controle do crescimento tumoral com os AS, porém com resposta bioquímica insatisfatória. Além disso, essa associação permite um tratamento com doses menores do PEG. Jorgensen e cols. (49) avaliaram o efeito do tratamento combinado em 11 acromegálicos e observaram que os níveis de IGF-I diminuíram durante a terapia com PEG, porém os menores valores foram alcançados com a terapia combinada. De forma interessante, o tratamento combinado aumentou em 20% as concentrações séricas do PEG. Além disso, níveis plasmáticos de glicose significativamente mais elevados foram observados em pacientes em uso de OCT-LAR em comparação àqueles tratados com PEG.
O PEG encontra-se disponível para aplicação subcutânea uma vez ao dia (10 mg, 15 mg e 20 mg). Inicia-se com a dose diária de 10 mg. As concentrações séricas de IGF-I devem ser avaliadas a cada quatro a seis semanas, e então a dose deve ser ajustada com acréscimos ou decréscimos de 5 mg até se alcançarem os níveis séricos de IGF-I normais para a idade, visando manter seus níveis entre zero e +2 desvios-padrão, evitando-se que o IGF-I permaneça no limite inferior do normal, a fim de reduzir o risco da deficiência de GH. A dose máxima diária é de 30 mg. O tratamento não deve ser monitorado ou ajustado com base nas dosagens séricas de GH, por causa da possibilidade de reação cruzada do GH com o PEG. Após estabelecimento da dose do PEG, os níveis de IGF-I devem ser avaliados a cada três a seis meses, juntamente com glicemia de jejum e HbA1C. A dosagem das enzimas hepáticas deve ser solicitada antes e durante o tratamento com PEG. A tabela 4 fornece as orientações ao início do tratamento com PEG, com base nos resultados de testes hepáticos realizados antes do tratamento, e a tabela 5, a conduta com relação ao ajuste da dose do PEG em caso de elevações das enzimas hepáticas observadas durante o tratamento. Nos pacientes com concentrações elevadas de ALT e AST, especialmente em pacientes com história anterior de tratamento com AS, deve-se afastar o diagnóstico de obstrução do trato biliar. Ressonância magnética deve ser solicitada após seis meses do início do PEG e depois anualmente e a função adenoipofisária, avaliada a cada seis meses, como já descrito anteriormente.
Alguns estudos também avaliaram esquemas alternativos de aplicação do PEG, os quais são mais cômodos, garantindo maior adesão ao tratamento. Feenstra e cols. (50) avaliaram a eficácia da combinação do AS (OCT-LAR 30 mg ou LAN Autogel 120 mg, mensalmente) com PEG em dose semanal em 26 acromegálicos resistentes ao OCT-LAR. Concentrações normais de IGF-I foram alcançadas em 18 de 19 pacientes, com uma dose semanal mediana de 60 mg de PEG (variando entre 40 e 80 mg). Esse esquema pode aumentar a adesão e até reduzir o custo do tratamento, sem comprometer sua eficácia.
Alguns estudos, além de mostrarem a eficácia do PEG no controle bioquímico da doença, avaliaram o seu efeito sobre o metabolismo dos carboidratos. Barkan e cols. (51) avaliaram as alterações do controle glicêmico (glicemia de jejum e HbA1C) em pacientes tratados com OCT-LAR que migraram para a terapia com PEG. Ao final do tratamento com PEG (32 semanas), as medianas da glicemia de jejum e da HbA1C reduziram significativamente (-1,4 nmol/L e -0,4%, respectivamente), ocorrendo em acromegálicos com e sem DM. Pacientes diabéticos tiveram a HbA1C reduzida em mais de 1%.
O principal efeito colateral do PEG é a elevação das transaminases hepáticas, ocorrendo de forma dose-independente e frequentemente dentro de três meses do início da medicação. Apesar de aparentemente não se associar a preditores, Neggers e cols. (52) demonstraram que pacientes com DM tinham um risco cinco vezes maior para desenvolver alterações das transaminases hepáticas. Elevações de bilirrubina e fosfatase alcalina, indicadores de grave injúria hepática, não foram observadas (53). Apenas alguns pacientes apresentam aumento das transaminases hepáticas após reinstituição do tratamento. Além disso, entre a maioria dos pacientes que apresenta aumento das transaminases, este ocorre de forma leve, normalizando com o decorrer do tratamento. O mecanismo da injúria hepática não está estabelecido, parecendo estar relacionado com: (a) o retorno das contrações da vesícula biliar com a suspensão do AS, o que poderia determinar refluxo da bile para dentro da via biliar intra-hepática; (b) reação idiossincrática; (c) o processo de associação do polietilenoglicol à molécula do antagonista do receptor de GH. A síndrome de Gilbert é uma condição clínica caracterizada por hiperbilirrubinemia não conjugada leve e intermitente, decorrente da redução da atividade da enzima uridina 5-difosfato glucoroniltransferase (UGT). Bernabeu e cols. (54) avaliaram 36 pacientes tratados com PEG quanto à presença do polimorfismo UGT1A1*28 (genótipo mais frequente na síndrome de Gilbert) e se este influenciaria o desenvolvimento de disfunção hepática durante o tratamento com PEG. Vinte e oito por cento dos pacientes desenvolveram alterações hepáticas durante o uso do PEG, com uma tendência maior entre homens (70% vs. 30%). Interessantemente, carreadores do polimorfismo apresentaram incidência mais elevada de alterações hepáticas (43% vs. 7%). A conduta perante um paciente com elevação das enzimas hepáticas está detalhada nas tabelas 4 e 5.
Uma questão bastante discutida na literatura é se a terapia com PEG está associada com crescimento tu-moral. As possíveis explicações para o aumento do vo-lume tumoral durante a terapia com PEG incluem: (a) ausência de feedback, decorrente da redução dos níveis de IGF-I; (b) efeito direto da droga sobre o tumor; (c) efeito rebote decorrente da suspensão dos AS; (d) padrão de crescimento agressivo do adenoma, independente da modalidade terapêutica empregada. Buhk e cols. (55) avaliaram o curso do volume tumoral durante a terapia com PEG em 61 pacientes, dos quais 45 (74%) completaram 24 meses de acompanhamento. Aumento significativo do volume tumoral (> 25%) foi observado em 3/61 (5%), ocorrendo todos os casos dentro do primeiro ano. Os três pacientes estavam em tratamento com OCT-LAR antes do uso do PEG e nenhum havia recebido radioterapia. Dessa maneira, como em todos os casos o aumento de volume tumoral ocorreu durante o primeiro ano, essas alterações podem correlacionar-se com a mudança de medicação e, assim, ser resultado do recrescimento tumoral induzido pela retirada do AS.
Lipo-hipertrofia também é um efeito colateral do PEG relatado por diferentes autores (56). A ação lipolítica do GH sobre o tecido adiposo é totalmente abolida no local da aplicação, fazendo com que a insulina exerça seu efeito pleno, o que leva à proliferação do tecido adiposo local. Essa alteração é reversível, podendo ser contornada com a intensificação do rodízio entre os locais de aplicação.
Radioterapia
A radioterapia (RT) representa, de forma geral, a última opção no algoritmo de tratamento da acromegalia, reservada principalmente para os casos de tumores não curados com a cirurgia e que não obtiveram controle bioquímico e/ou da massa tumoral com o uso das drogas disponíveis (Figura 2; Quadro 7). Duas modalidades de RT estão disponíveis: convencional e estereotáxica, a qual pode ser fracionada ou em dose única (radiocirurgia).
A RT convencional é administrada em doses fracionadas, quatro a cinco vezes por semana, durante quatro semanas. O controle da hipersecreção hormonal inicia-se em geral após seis meses a dois anos, de forma gradual e, muitas vezes, incompleta (57). Uma vez que a pulsatilidade normal de secreção do GH pode não ser restabelecida após a RT (58), vale a pena ressaltar que valores de GH menores que 2,5 ng/mL não indicam necessariamente controle da doença, pois níveis aumentados de IGF-I são observados em 10% a 40% desses pacientes (59). Controle do crescimento tumoral é observado em mais de 90% dos pacientes, ocorrendo redução do tamanho tumoral em uma parcela dos casos (57).
A RT convencional pode apresentar uma série de efeitos colaterais, sendo os principais hipopituitarismo, dano cerebral (atrofia cerebral, gliose hipotalâmica, alterações de lobo temporal, radionecrose e acidente vascular cerebral) e perda visual. Nos acromegálicos sem deficiência antes da RT, as incidências de hipogonadismo, insuficiência adrenal e hipotireoidismo central são de 50%, 37% e 37%, respectivamente, após a irradiação hipofisária (57). O risco que a literatura relata para desenvolvimento de tumores cerebrais secundários à RT hipofisária é baixo, cerca de 2% após 20 anos do procedimento, isso considerando que o tratamento foi realizado com técnicas antigas; com as novas técnicas, o risco é desconhecido em virtude da falta de estudos de longo prazo (60).
A eficácia da radiocirurgia parece ser semelhante à da RT convencional. Barkan (59) observou, em uma revisão de 12 estudos, que apenas 33% dos pacientes apresentavam níveis de IGF-I normais após radiocirurgia estereotáxica. No entanto, com esta última, parece que os níveis de IGF-I são normalizados mais rapidamente, ocorrendo em média após 1,6 ano na radiocirurgia, contra uma média de 7,1 anos após a RT convencional (61).
Attanasio e cols. (62) avaliaram a eficácia da Gamma Knife em 30 pacientes com um seguimento médio de 46 meses. Os níveis séricos de IGF-I normalizaram em 23% e níveis de GH < 2,5 ng/mL foram alcançados em 37% dos pacientes. Houve redução tumoral (pelo menos 25% do volume inicial) em 58% (11/19 pacientes) após 24 meses de irradiação.
Em conclusão, apesar de a acromegalia ser uma condição com características clínicas muito sugestivas, seu diagnóstico ainda é realizado com significativo atraso. A elevada morbimortalidade associada aos elevados níveis de GH e IGF-I pode ser reduzida para próximo da esperada para a população geral com a instituição de um diagnóstico mais precoce e de uma terapêutica eficaz e segura. No entanto, para um diagnóstico precoce, é fundamental o reconhecimento da doença por parte da população, especialmente dos profissionais de saúde.
A cirurgia transesfenoidal e o tratamento medicamentoso com AS constituem as modalidades terapêuticas de escolha, podendo, ambas, serem utilizadas como terapia primária. Para pacientes com micro ou macroadenomas intrasselares, não existe dúvida de que o tratamento primário deve ser a cirurgia, dada a alta chance de cura. Entretanto, essas altas taxas de cura somente são observadas quando a cirurgia é feita por cirurgiões experientes nesse tipo de procedimento, sendo fundamental o encaminhamento dos pacientes para centros de refe-rência que ofereçam essa possibilidade de sucesso. Já em pacientes com tumores de baixa chance de cura cirúrgica (tumores volumosos ou que invadem seio cavernoso ou elevados níveis de GH), presença de comorbidades que tornam o risco da cirurgia proibitivo e pacientes que recusam a cirurgia, a melhor conduta é o tratamento medicamentoso primário com AS. No entanto, em pacientes não controlados com tratamento medicamentoso, a cirurgia de debulking pode ser uma opção. Por outro lado, o procedimento para debulking também pode ser realizado previamente ao tratamento medicamentoso, levando-se em consideração não apenas o status clínico do paciente, mas também a experiência e a disponibilidade do centro com cada modalidade terapêutica.
Como a maioria dos pacientes tem um macroadenoma à apresentação, a taxa de cura é de aproximadamente 50%, tornando-se necessário o tratamento medicamentoso adjuvante no restante dos pacientes. Os AS, drogas de primeira linha no tratamento medicamentoso da acromegalia, são capazes de controlar os níveis de GH e IGF-I em cerca de 50% dos pacientes. Os AD podem ser usados naqueles com níveis de GH e IGF-I pouco elevados, ou ainda em combinação com os AS quando estes não controlam os níveis hormonais isoladamente. Apesar disso, cerca de 1/4 dos pacientes mantém níveis hormonais elevados, portanto não controlados, expondo-os a complicações cardiovasculares, respiratórias, metabólicas e neoplásicas, as quais são responsáveis pela morbimortalidade elevada. Nessa população, o PEG já foi avaliado como monoterapia ou combinado com AS, com resultados satisfatórios em ambas as situações.
A RT deve ser reservada para pacientes em que o controle da atividade de doença e do volume tumoral não foi possível com a combinação da cirurgia com as drogas atualmente disponíveis.
Assim, em virtude da disponibilidade de diferentes modalidades terapêuticas, aliada ao aprimoramento de técnicas cirúrgicas e de radioterapia e ao surgimento de novas drogas, atualmente o centro que dispõe de todos esses recursos terapêuticos discutidos neste artigo tem o potencial de conseguir o controle da doença em virtualmente 100% dos pacientes.
Declaração: Os autores declaram não haver conflitos de interesse científico neste estudo.
Recebido em 22/Dez/2010
Aceito em 6/Fev/2011
- 1. Donangelo I, Une K, Gadelha M. Diagnóstico e tratamento da acromegalia no Brasil. Arq Bras Endocrinol Metabol. 2003;47(4):331-46.
- 2. Vieira Neto L, Taboada GF, Correa LL, Polo J, Nascimento AF, Chimelli L, et al. Acromegaly secondary to growth hormone-releasing hormone secreted by an incidentally discovered pheochromocytoma. Endocr Pathol. 2007;18(1):46-52.
- 3. Colao A, Ferone D, Marzullo P, Lombardi G. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev. 2004;25(1):102-52.
- 4. Lima GA, Gomes EM, Nunes RC, Vieira Neto L, Sieiro AP, Brabo EP, et al. Osteosarcoma and acromegaly: a case report and review of the literature. J Endocrinol Invest. 2006;29(11):1006-11.
- 5. Casini AF, Araujo PB, Fontes R, Xavier SS, Gadelha MR. [Cardiac morphology and performance alterations and analysis of determinant factors of left ventricular hypertrophy in 40 patients with acromegaly]. Arq Bras Endocrinol Metabol. 2006;50(1):82-90.
- 6. Correa LL, Taboada GF, Van Haute FR, Casini AF, Balarini GA, Vieira Neto L, et al. [Evaluation of glucose metabolism in acromegalic patients before and after treatment with octreotide LAR]. Arq Bras Endocrinol Metabol. 2008;52(1):55-64.
- 7. Van Haute FR, Taboada GF, Correa LL, Lima GA, Fontes R, Riello AP, et al. Prevalence of sleep apnea and metabolic abnormalities in patients with acromegaly and analysis of cephalometric parameters by magnetic resonance imaging. Eur J Endocrinol. 2008;158(4):459-65.
- 8. Reid TJ, Post KD, Bruce JN, Nabi Kanibir M, Reyes-Vidal CM, Freda PU. Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol (Oxf). 2010;72(2):203-8.
- 9. Dekkers OM, Biermasz NR, Pereira AM, Romijn JA, Vandenbroucke JP. Mortality in acromegaly: a metaanalysis. J Clin Endocrinol Metab. 2008;93(1):61-7.
- 10. Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol. 2008;159(2):89-95.
- 11. Holdaway IM, Rajasoorya RC, Gamble GD. Factors influencing mortality in acromegaly. J Clin Endocrinol Metab. 2004;89(2):667-74.
- 12. Leavitt J, Klein I, Kendricks F, Gavaler J, VanThiel DH. Skin tags: a cutaneous marker for colonic polyps. Ann Intern Med. 1983;98(6):928-30.
- 13. Boguszewski CL. O laboratório no diagnóstico e seguimento da acromegalia. Arq Bras Endocrinol Metab. 2002;46(1):34-44.
- 14. Dimaraki EV, Jaffe CA, DeMott-Friberg R, Chandler WF, Barkan AL. Acromegaly with apparently normal GH secretion: implications for diagnosis and follow-up. J Clin Endocrinol Metab. 2002;87(8):3537-42.
- 15. Rosario PW. Normal values of serum IGF-1 in adults: results from a Brazilian population. Arq Bras Endocrinol Metabol. 2010;54(5):477-81.
- 16. Clemmons DR, Van Wyk JJ. Factors controlling blood concentration of somatomedin C. Clin Endocrinol Metab. 1984;13(1):113-43.
- 17. Giustina A, Barkan A, Casanueva FF, Cavagnini F, Frohman L, Ho K, et al. Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab. 2000;85(2):526-9.
- 18. Giustina A, Chanson P, Bronstein MD, Klibanski A, Lamberts S, Casanueva FF, et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010;95(7):3141-8.
- 19. Freda PU. Current concepts in the biochemical assessment of the patient with acromegaly. Growth Horm IGF Res. 2003;13(4):171-84.
- 20. Ribeiro-Oliveira Jr A, Faje AT, Barkan AL. Limited utility of oral glucose tolerance test in biochemically-active acromegaly. Eur J Endocrinol. 2011;164(1):17-22. Epub 2010 Oct 6.
- 21. Taboada GF, Correa LL, Oliveira Machado E, Van Haute FR, Casini AF, Balarini GA, et al. Two hour mean GH is not superior to basal GH for the follow-up of acromegalic patients treated with Octreotide LAR. Growth Horm IGF Res. 2007;17(1):77-81.
- 22. Melmed S, Colao A, Barkan A, Molitch M, Grossman AB, Kleinberg D, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. 2009;94(5):1509-17.
- 23. Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical 'cure'. Eur J Endocrinol. 2005;152(3):379-87.
- 24. Jallad RS, Musolino NR, Kodaira S, Cescato VA, Bronstein MD. Does partial surgical tumour removal influence the response to octreotide-LAR in acromegalic patients previously resistant to the somatostatin analogue? Clin Endocrinol (Oxf). 2007;67(2):310-5.
- 25. Freda PU, Nuruzzaman AT, Reyes CM, Sundeen RE, Post KD. Significance of "abnormal" nadir growth hormone levels after oral glucose in postoperative patients with acromegaly in remission with normal insulin-like growth factor-I levels. J Clin Endocrinol Metab. 2004;89(2):495-500.
- 26. Fatemi N, Dusick JR, Mattozo C, McArthur DL, Cohan P, Boscardin J, et al. Pituitary hormonal loss and recovery after transsphenoidal adenoma removal. Neurosurgery. 2008;63(4):709-18; discussion 18-9.
- 27. Neto LV, Machado Ede O, Luque RM, Taboada GF, Marcondes JB, Chimelli LM, et al. Expression analysis of dopamine receptor subtypes in normal human pituitaries, nonfunctioning pituitary adenomas and somatotropinomas, and the association between dopamine and somatostatin receptors with clinical response to octreotide-LAR in acromegaly. J Clin Endocrinol Metab. 2009;94(6):1931-7.
- 28. Jaffe CA, Barkan AL. Treatment of acromegaly with dopamine agonists. Endocrinol Metab Clin North Am. 1992;21(3):713-35.
- 29. Verhelst J, Abs R, Maiter D, Van den Bruel A, Vandeweghe M, Velkeniers B, et al. Cabergoline in the treatment of hyperprolactinemia: a study in 455 patients. J Clin Endocrinol Metab. 1999;84(7):2518-22.
- 30. Abs R, Verhelst J, Maiter D, Van Acker K, Nobels F, Coolens JL, et al. Cabergoline in the treatment of acromegaly: a study in 64 patients. J Clin Endocrinol Metab. 1998;83(2):374-8.
- 31. Vilar L, Czepielewsk MA, Naves LA, Rollin GA, Casulari LA, Coelho CE. Substantial shrinkage of adenomas cosecreting growth hormone and prolactin with use of cabergoline therapy. Endocr Pract. 2007;13(4):396-402.
- 32. Schade R, Andersohn F, Suissa S, Haverkamp W, Garbe E. Dopamine agonists and the risk of cardiac-valve regurgitation. N Engl J Med. 2007;356(1):29-38.
- 33. Lancellotti P, Livadariu E, Markov M, Daly AF, Burlacu MC, Betea D, et al. Cabergoline and the risk of valvular lesions in endocrine disease. Eur J Endocrinol. 2008;159(1):1-5.
- 34. Taboada GF, Luque RM, Neto LV, Machado Ede O, Sbaffi BC, Domingues RC, et al. Quantitative analysis of somatostatin receptor subtypes (1-5) gene expression levels in somatotropinomas and correlation to in vivo hormonal and tumor volume responses to treatment with octreotide LAR. Eur J Endocrinol. 2008;158(3):295-303.
- 35. Vieira Neto L, Taboada GF, Gadelha MR. Somatostatin receptors subtypes 2 and 5, dopamine receptor type 2 expression and gsp status as predictors of octreotide LAR responsiveness in acromegaly. Arq Bras Endocrinol Metabol. 2008;52(8):1288-95.
- 36. Freda PU, Katznelson L, Van der Lely AJ, Reyes CM, Zhao S, Rabinowitz D. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2005;90(8):4465-73.
- 37. Murray RD, Melmed S. A critical analysis of clinically available somatostatin analog formulations for therapy of acromegaly. J Clin Endocrinol Metab. 2008;93(8):2957-68.
- 38. Jallad RS, Musolino NR, Salgado LR, Bronstein MD. Treatment of acromegaly with octreotide-LAR: extensive experience in a Brazilian institution. Clin Endocrinol (Oxf). 2005;63(2):168-75.
- 39. Cozzi R, Montini M, Attanasio R, Albizzi M, Lasio G, Lodrini S, et al. Primary treatment of acromegaly with octreotide LAR: a long-term (up to nine years) prospective study of its efficacy in the control of disease activity and tumor shrinkage. J Clin Endocrinol Metab. 2006;91(4):1397-403.
- 40. Cozzi R, Attanasio R, Lodrini S, Lasio G. Cabergoline addition to depot somatostatin analogues in resistant acromegalic patients: efficacy and lack of predictive value of prolactin status. Clin Endocrinol (Oxf). 2004;61(2):209-15.
- 41. Jallad RS, Bronstein MD. Optimizing medical therapy of acromegaly: beneficial effects of cabergoline in patients uncontrolled with long-acting release octreotide. Neuroendocrinology. 2009;90(1):82-92.
- 42. Vilar L, Azevedo MF, Naves LA, Casulari LA, Albuquerque JL, Montenegro RM, et al. Role of the addition of cabergoline to the management of acromegalic patients resistant to longterm treatment with octreotide LAR. Pituitary. 2010 Nov 21.
- 43. Mattar P, Alves Martins MR, Abucham J. Short- and long-term efficacy of combined cabergoline and octreotide treatment in controlling igf-I levels in acromegaly. Neuroendocrinology. 2010;92(2):120-7.
- 44. Machado EO, Taboada GF, Neto LV, Van Haute FR, Correa LL, Balarini GA, et al. Prevalence of discordant GH and IGF-I levels in acromegalics at diagnosis, after surgical treatment and during treatment with octreotide LAR. Growth Horm IGF Res. 2008;18(5):389-93.
- 45. Freda PU. Somatostatin analogs in acromegaly. J Clin Endocrinol Metab. 2002;87(7):3013-8.
- 46. Trainer PJ, Drake WM, Katznelson L, Freda PU, Herman-Bonert V, Van der Lely AJ, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med. 2000;342(16):1171-7.
- 47. Van der Lely AJ, Hutson RK, Trainer PJ, Besser GM, Barkan AL, Katznelson L, et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet. 2001;358(9295):1754-9.
- 48. Marazuela M, Lucas T, Alvarez-Escola C, Puig-Domingo M, Garcia de la Torre N, Miguel-Novoa P, et al. Long-term treatment of acromegalic patients resistant to somatostatin analogs with the GH receptor antagonist pegvisomant: its efficacy in relation to gender and previous radiotherapy. Eur J Endocrinol. 2009;160(4):535-42. Epub 2009 Jan 15.
- 49. Jorgensen JO, Feldt-Rasmussen U, Frystyk J, Chen JW, Kristensen LO, Hagen C, et al. Cotreatment of acromegaly with a somatostatin analog and a growth hormone receptor antagonist. J Clin Endocrinol Metab. 2005;90(10):5627-31.
- 50. Feenstra J, De Herder WW, Ten Have SM, Van den Beld AW, Feelders RA, Janssen JA, et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet. 2005;365(9471):1644-6.
- 51. Barkan AL, Burman P, Clemmons DR, Drake WM, Gagel RF, Harris PE, et al. Glucose homeostasis and safety in patients with acromegaly converted from long-acting octreotide to pegvisomant. J Clin Endocrinol Metab. 2005;90(10):5684-91.
- 52. Neggers SJ, Van Aken MO, Janssen JA, Feelders RA, De Herder WW, Van der Lely AJ. Long-term efficacy and safety of combined treatment of somatostatin analogs and pegvisomant in acromegaly. J Clin Endocrinol Metab. 2007;92(12):4598-601.
- 53. Roelfsema F, Biermasz NR, Pereira AM, Romijn JA. The role of pegvisomant in the treatment of acromegaly. Expert Opin Biol Ther. 2008;8(5):691-704.
- 54. Bernabeu I, Marazuela M, Lucas T, Loidi L, Alvarez-Escola C, Luque-Ramirez M, et al. Pegvisomant-induced liver injury is related to the UGT1A1*28 polymorphism of Gilbert's syndrome. J Clin Endocrinol Metab. 2010;95(5):2147-54.
- 55. Buhk JH, Jung S, Psychogios MN, Goricke S, Hartz S, Schulz-Heise S, et al. Tumor volume of growth hormone-secreting pituitary adenomas during treatment with pegvisomant: a prospective multicenter study. J Clin Endocrinol Metab. 2010;95(2):552-8.
- 56. Bonert VS, Kennedy L, Petersenn S, Barkan A, Carmichael J, Melmed S. Lipodystrophy in patients with acromegaly receiving pegvisomant. J Clin Endocrinol Metab. 2008;93(9):3515-8.
- 57. Jaffe CA. Reevaluation of conventional pituitary irradiation in the therapy of acromegaly. Pituitary. 1999;2(1):55-62.
- 58. Peacey SR, Shalet SM. Growth hormone pulsatility in acromegaly following radiotherapy. Pituitary. 1999;2(1):63-9.
- 59. Barkan AL. Radiotherapy in acromegaly: the argument against. Clin Endocrinol (Oxf). 2003;58(2):132-5.
- 60. Brada M, Ford D, Ashley S, Bliss JM, Crowley S, Mason M, et al. Risk of second brain tumour after conservative surgery and radiotherapy for pituitary adenoma. BMJ. 1992;304(6838):1343-6.
- 61. Jackson IM, Noren G. Role of gamma knife therapy in the management of pituitary tumors. Endocrinol Metab Clin North Am. 1999;28(1):133-42.
- 62. Attanasio R, Epaminonda P, Motti E, Giugni E, Ventrella L, Cozzi R, et al. Gamma-knife radiosurgery in acromegaly: a 4-year follow-up study. J Clin Endocrinol Metab. 2003;88(7):3105-12.
Correspondência para:
Datas de Publicação
-
Publicação nesta coleção
04 Maio 2011 -
Data do Fascículo
Mar 2011
Histórico
-
Recebido
22 Dez 2010 -
Aceito
06 Fev 2011