Abstracts
We report on an adult woman with rare coexistence of acromegaly, pheochromocytoma (PHEO), gastrointestinal stromal tumor (GIST), intestinal polyposis, and thyroid follicular adenoma. At the age of 56, she was diagnosed with acromegaly caused by a pituitary macroadenoma, treated by transsphenoidal surgery, radiotherapy, and octreotide. During routine colonoscopy, multiple polyps were identified as tubular adenomas with high-grade dysplasia on histology. Years later, an abdominal mass of 8.0 x 6.2 cm was detected by routine ultrasound. Surgical exploration revealed an adrenal mass and another tumor adhered to the lesser gastric curvature, which were removed. Pathology confirmed the diagnosis of PHEO and GIST. PHEO immunohistochemistry was negative for GHRH. During follow-up, nodular goiter was found with normal levels of calcitonin and inconclusive cytology. Near-total thyroidectomy was performed, revealing a follicular adenoma. Her family history was negative for all of these tumor types. Genetic analysis for PHEO/paraganglioma genes (SDH A-D, SDHAF2, RET, VHL, TMEM127, and MAX), and pituitary-related genes (AIP, MEN1, and p27) were negative. Though the finding of PHEO and acromegaly with multiple other tumors could be a fortuitous coexistence, we suggest that this case may represent a new variant of MEN syndrome with a de novo germline mutation in a not yet identified gene. Arq Bras Endocrinol Metab. 2012;56(8):507-12
Relatamos o caso de uma mulher com rara coexistência de acromegalia, feocromocitoma (FEO), tumor do estroma gastrointestinal (GIST), polipose intestinal e adenoma folicular de tireoide. Aos 56 anos, ela foi diagnosticada com acromegalia por um macroadenoma hipofisário, tratado com cirurgia transesfenoidal, radioterapia e octreotide. Uma colonoscopia de rotina detectou múltiplos pólipos, que à histologia eram adenomas tubulares com alto grau de displasia. Anos mais tarde, uma ecografia detectou uma massa abdominal de 8.0 x 6.2 cm, que na exploração cirúrgica era uma lesão adrenal e outro tumor aderido à pequena curvatura gástrica. A patologia confirmou os diagnósticos de FEO e GIST. A imuno-histoquímica do FEO foi negativa para GHRH. No seguimento, encontrou-se um bócio nodular com níveis normais de calcitonina e citologia inconclusiva. Após tireoidectomia total o diagnóstico histológico foi de adenoma folicular. A história familiar era negativa para todos esses tumores. As análises genéticas para genes de síndromes de FEO/paragangliomas (SDH A-D, SDHAF2, RET, VHL, TMEM127 e MAX) e para hipofisárias (AIP, MEN1 e p27) foram todas negativas. Embora a presença de FEO e acromegalia com múltiplos outros tumores possa ser uma coexistência fortuita, acreditamos na possibilidade de uma nova variante de NEM com uma mutação germinativa de novo em um gene ainda não identificado Arq Bras Endocrinol Metab. 2012;56(8):507-12
CASE REPORT
Genetic studies in a coexistence of acromegaly, pheochromocytoma, gastrointestinal stromal tumor (GIST) and thyroid follicular adenoma
Estudos genéticos na coexistência de acromegalia, feocromocitoma, tumor do estroma gastrointestinal (GIST) e adenoma folicular de tireoide
César Luiz BoguszewskiI; Tayane Muniz FigheraI; Andressa BornscheinI; Fabricio Machado MarquesII; Judit DénesIII; Eleanor RattenberyIV; Eamonn R. MaherIV; Karen StalsV; Sian EllardV; Marta KorbonitsIII
IEndocrine Division (SEMPR), Department of Internal Medicine, University Hospital at Universidade Federal do Parana (UFPR), Curitiba, Brazil
IIDepartment of Pathology, University Hospital at UFPR, Curitiba, Brazil
IIIDepartment of Endocrinology, Barts and the London School of Medicine, Queen Mary University of London, London, United Kingdom
IVDepartment of Molecular Genetics, Royal Devon and Exeter Foundation Trust, Exeter, EX2 5DW, UK
VCentre for Rare Diseases and Personalised Medicine, University of Birmingham, Birmingham B15 2TT, UK
Correspondence Correspondence to: César Luiz Boguszewski Department of Internal Medicine, SEMPR, Universidade Federal do Parana, Curitiba, PR, Brazil Av. Agostinho Leao Junior, 285 80030-110 Curitiba, PR, Brazil cesarluiz@hc.ufpr.br
SUMMARY
We report on an adult woman with rare coexistence of acromegaly, pheochromocytoma (PHEO), gastrointestinal stromal tumor (GIST), intestinal polyposis, and thyroid follicular adenoma. At the age of 56, she was diagnosed with acromegaly caused by a pituitary macroadenoma, treated by transsphenoidal surgery, radiotherapy, and octreotide. During routine colonoscopy, multiple polyps were identified as tubular adenomas with high-grade dysplasia on histology. Years later, an abdominal mass of 8.0 x 6.2 cm was detected by routine ultrasound. Surgical exploration revealed an adrenal mass and another tumor adhered to the lesser gastric curvature, which were removed. Pathology confirmed the diagnosis of PHEO and GIST. PHEO immunohistochemistry was negative for GHRH. During follow-up, nodular goiter was found with normal levels of calcitonin and inconclusive cytology. Near-total thyroidectomy was performed, revealing a follicular adenoma. Her family history was negative for all of these tumor types. Genetic analysis for PHEO/paraganglioma genes (SDH A-D, SDHAF2, RET, VHL, TMEM127, and MAX), and pituitary-related genes (AIP, MEN1, and p27) were negative. Though the finding of PHEO and acromegaly with multiple other tumors could be a fortuitous coexistence, we suggest that this case may represent a new variant of MEN syndrome with a de novo germline mutation in a not yet identified gene. Arq Bras Endocrinol Metab. 2012;56(8):507-12
SUMÁRIO
Relatamos o caso de uma mulher com rara coexistência de acromegalia, feocromocitoma (FEO), tumor do estroma gastrointestinal (GIST), polipose intestinal e adenoma folicular de tireoide. Aos 56 anos, ela foi diagnosticada com acromegalia por um macroadenoma hipofisário, tratado com cirurgia transesfenoidal, radioterapia e octreotide. Uma colonoscopia de rotina detectou múltiplos pólipos, que à histologia eram adenomas tubulares com alto grau de displasia. Anos mais tarde, uma ecografia detectou uma massa abdominal de 8.0 x 6.2 cm, que na exploração cirúrgica era uma lesão adrenal e outro tumor aderido à pequena curvatura gástrica. A patologia confirmou os diagnósticos de FEO e GIST. A imuno-histoquímica do FEO foi negativa para GHRH. No seguimento, encontrou-se um bócio nodular com níveis normais de calcitonina e citologia inconclusiva. Após tireoidectomia total o diagnóstico histológico foi de adenoma folicular. A história familiar era negativa para todos esses tumores. As análises genéticas para genes de síndromes de FEO/paragangliomas (SDH A-D, SDHAF2, RET, VHL, TMEM127 e MAX) e para hipofisárias (AIP, MEN1 e p27) foram todas negativas. Embora a presença de FEO e acromegalia com múltiplos outros tumores possa ser uma coexistência fortuita, acreditamos na possibilidade de uma nova variante de NEM com uma mutação germinativa de novo em um gene ainda não identificado Arq Bras Endocrinol Metab. 2012;56(8):507-12
INTRODUCTION
In the majority of cases, acromegaly is a sporadic disease caused by a GH-secreting pituitary adenoma. More rarely, it is caused by a hypothalamic disorder (hamartoma, choristoma, and ganglioneuroma), or ectopic GHRH secretion by ectopic neoplasias, such as carcinoids, small cell lung cancer, and pancreatic islet cell tumors (1-3). Familial syndromes associated with GH hypersecretion include multiple endocrine neoplasia type 1 (MEN1), in which pituitary, parathyroid, and pancreatic tumors might be associated (4), and the similar MEN4 syndrome caused by germline mutations in the CDKN1B/p27 gene; McCune-Albright syndrome, which involves polyostotic fibrous dysplasia and cutaneous pigmentation (5); and Carney complex, whose clinical picture includes skin pigmentation, mucocutaneous myxomatosis, cardiac myxoma, thyroid and breast lesions (6). Isolated familial acromegaly is described with loss of heterozygosity in chromosome 11q13 (7) and, recently, low-penetrance germline mutations in the aryl hydrocarbon receptor-interacting protein (AIP) gene that were found in individuals who had familial pituitary isolated adenoma (FIPA) predisposition (8-10).
Pheochromocytomas and paragangliomas are rare neuroendocrine tumors of the adrenal glands and sympathetic and parasympathetic ganglia that occur sporadically or as a part of different hereditary tumor syndromes, including MEN type 2 (MEN2), von Hippel-Lindau disease, neurofibromatosis type 1 (NF1), familial paraganglioma syndrome, and isolated pheochromocytoma/paraganglioma syndromes due to RET, VHL, NF1, SDHA, SDHB, SDHC, SDHD, SDHAF2, MAX, and TMEM127 mutations (11-13). In MEN2a, medullary thyroid cancer (MTC) occurs in nearly 100% of patients, hyperparathyroidism occurs in 20%, while pheochromocytoma is identified in 30%-50%, and it is the primary complaint in only 15% of cases. MEN2b is characterized by the presence of mucosal neuromas, MTC, pheochromocytoma, and marfanoid habitus. Primary hyperparathyroidism is uncommon in this syndrome (13).
The association of pheochromocytoma with pituitary adenomas is very rare. On the other hand, various solid tumors have been described in association with MEN and pheochromocytoma-associated syndromes (4,6,12-14). In this article, we report a female adult patient presenting acromegaly and pheochromocytoma (GHRH-negative) associated with gastrointestinal stromal tumor (GIST), intestinal polyposis, and thyroid follicular adenomas. We discuss whether this case represents a fortuitous association between endocrine and solid tumors, or the possible existence of a novel MEN variant.
SUBJECT AND METHODS
Case report
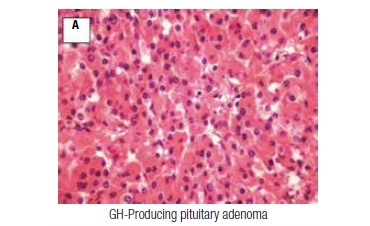
A 56-year-old female patient was initially evaluated in February, 1994, referred from the Neurosurgery Department, where she had a transsphenoidal surgery. She had a typical history of acromegaly, with a progressive enlargement of hands, feet, and lips in the previous years, associated with macroglossia, dizziness, weakness, insomnia, and paresthesias. There was no family history of pituitary or other endocrine or non-endocrine tumors. On physical examination, the patient was hypertensive (160/110 mmHg), obese (body mass index 31 kg/m2), and had acromegalic features (prognathism, prominent forehead, flat nose, and macroglossia). Computerized tomography revealed a pituitary macroadenoma. Pathology of the tumor was consistent with a pure, densely granulated, GH-producing pituitary adenoma (somatotropinoma; Figure 1A). Despite no evidence of residual tumor on a computerized tomography performed one year after pituitary surgery, she had persistent acromegaly (nadir GH level after glucose load of 38 μg/L, and elevated IGF-1 levels). Two years after surgery, radiotherapy (5580 rads in 39 sessions) was carried out and she was started on octreotide, initially 0.1 mg SC three times a day, and soon after, on octreotide LAR 20 mg IM every four weeks, with progressive normalization of serum IGF-1. She developed central hypothyroidism and replacement therapy with levothyroxine was initiated.
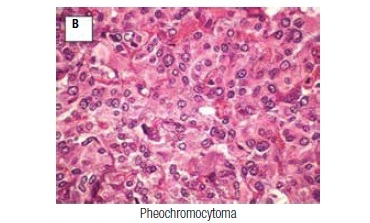
As part of the acromegaly management, colonoscopy was performed six years after surgery, and showed angiodysplasia and multiple polyps with histopathological features of tubular adenomas with high-grade dysplasia. Clinically, she maintained high blood pressure despite the use of multiple anti-hypertensive drugs. Ten years after the pituitary surgery, an abdominal ultrasound found, between the spleen and left kidney, a well-defined solid mass measuring 8.0 x 6.2 cm. Surgical exploration revealed a left-side adrenal mass and another tumor adhered to the lesser gastric curvature, which were removed by means of a left adrenalectomy and subtotal gastrectomy. Pathology confirmed the presence of pheochromocytoma (neuroendocrine tumor with immunohistochemistry positive for chromogranin A and synaptophysin, with absence of necrosis, vascular invasion or capsular invasion; Figure 1B), and revealed a GIST (positive immunohistochemistry for CD 117 and 34; Figure 1C).
Two years later, the patient underwent cervical ultrasound due to clinical suspicion of goiter. The examination showed a poorly defined nodule in the left lobe, with coarse calcifications, measuring 2.3 x 2.3 x 3.4 cm, which was subjected to fine-needle aspiration biopsy in three different occasions, showing inconclusive cytology results. Serum calcitonin levels were normal (< 2 pg/mL). The patient underwent total thyroidectomy and histopathological analysis of the specimen demonstrated a follicular adenoma (Figure 1D).
The patient is now 75 year-old and receives levothyroxine 175 μg/day and octreotide LAR 10 mg monthly with normal GH and IGF-1 levels. She is also on medication for hypercholesterolemia and osteoporosis. Her last magnetic resonance imaging of the pituitary in November, 2010 showed an empty sella.
METHODS AND RESULTS
When the diagnosis of pheochromocytoma was confirmed, we suspected that acromegaly in our patient could be due to ectopic secretion of GHRH by the adrenal tumor. Immunohistochemistry for GHRH was kindly performed by Dr. Michael Thorner (University of Virginia, Charlottesville, VA, USA), according to the technique described elsewhere (15), and the result was negative (Figure 2).
We have looked for possible candidate genes that could explain the coexistence of various tumors in our patient. Genomic deoxyribonucleic acid (DNA) was isolated from peripheral blood. The AIP and MEN1 genes were sequenced with traditional methods, and large deletion was ruled out using multiplex ligation-dependent probe amplification (MLPA; MRC-Holland kit P244-B1, Amsterdam, Netherlands), as described elsewhere (16,17). The current list of pheochromocytoma/paraganglioma genes (SDH A-D, SDHAF2, RET, VHL, TMEM127, and MAX) were studied using exon capture next-generation sequencing, applying the Fluidigm Access Array 48 system. This involves preparing a 48-amplicon library and next-generation sequencing using emulsion PCR and pyrosequencing steps on the Roche GS Junior platform. None of these genes had a mutation in the patient.
DISCUSSION
The coexistence of acromegaly and pheochromocytoma has been first described by Iversen in 1952 (18), and the relationship between these two entities has been explained by fortuitous association, overlapping, or variants of MEN syndromes, or by ectopic production of GHRH by the pheochromocytoma. In a review of pheochromocytomas associated with pituitary adenomas published in the literature between 1964 and 2003, Breckenridge and cols. found a total of 25 cases, and of these, 15 patients had acromegaly (19). The remaining cases included prolactinomas, corticotropinomas, and non-functioning pituitary adenomas.
In some cases, the association was proven to be due to ectopic production of GHRH by the pheochromocytoma (20). Roth and cols. (21) described a patient with pheochromocytoma and high levels of GHRH, and they were able to confirm the adrenal origin of GHRH in the post-mortem examination (20). Similarly, Vieira and cols. (22) described a case of acromegaly secondary to GHRH secretion by a pheochromocytoma that was incidentally discovered in a normotensive patient. The tumor showed positive staining for GHRH. However, the pheochromocytoma of our patient was not a GHRH-secreting tumor, similar to other reports (23), and another reason could probably explain the association. Although we cannot exclude a simple incidental association of two rare diseases in the same person, the probability of such an event is extremely low (23). Therefore, we tried to explore alternative explanations.
Classic MEN syndromes are autosomal dominant diseases consisting of a constellation of endocrine tumors. MEN1 gene is located at chromosome 11q13 and encodes menin, a protein that functions as a tumor suppressor gene. A MEN1-like syndrome (now termed MEN4) has been described with germline mutations in the CDKN1B gene coding for the p27 protein (chromosome 12p13.1-p12) (24). Interestingly, the animal model of this syndrome harbors both pituitary adenomas and pheochromocytomas (24). GH-producing tumors that cause acromegaly occur in 3%-6% of MEN1 patients. The size and invasiveness of MEN1-associated somatotropinomas do not seem to differ significantly from sporadic tumors. However, they may have distinct characteristics, such as earlier manifestation of the tumors, with a MEN1-associated GH/PRL-producing tumor already diagnosed in a 5-year-old boy. More recently, attention has been given to the FIPA (familial isolated pituitary adenomas), and a fifth of these families have a mutation in the AIP gene. There is also evidence that approximately 10% of sporadic macroadenomas occurring before 30 years of age, and 20% of those that are diagnosed in children, harbor an AIP mutation. Typically, AIP-mutation positive somatotroph adenomas respond poorly to somatostatin analogue treatment (25-27). An interplay between RET proto-oncogene and FIPA has been suggested, though the precise mechanism of this interaction has not been characterized yet (28). In our patient, the search for AIP and MEN1 gene mutations showed negative results, excluding the diagnosis of FIPA and MEN1. Accordingly, her acromegaly presented as in most of the sporadic cases, her tumor was not especially aggressive and responded well to medical therapy with a somatostatin analogue.
Different presentations are also observed in pheochromocytomas; only 5% of patients with sporadic tumors are asymptomatic, while half of the patients with MEN2a-associated pheochromocytomas will not have any symptom of the disease (11). Around one third of pheochromocytoma/paraganglioma patients carry a germline mutation (most familial cases, and 10%-20% of the sporadic cases). Predictors for presence of mutation are age < 45 years, multiple tumors, extra-adrenal location, previous head and neck paraganglioma, positive family history, and a malignant tumor (29,30). Apart of persistent hypertension that was resistant to anti-hypertensive therapy, our patient did not have other clinical manifestations of pheochromocytoma. In fact, the tumor was revealed incidentally by an abdominal ultrasound that was performed to investigate gallstones due to the prolonged use of somatostatin analogues. She did not have any predictor of harboring a mutation, and we found her to be negative for SDH A-D, SDHAF2, RET, VHL, TMEM127, and MAX mutations.
Another hypothesis that may explain the coexistence of acromegaly and pheochromocytoma is that it represents a variant of MEN syndromes, with greater heterogeneity of tumor combinations than described in the more well-defined syndromes. Both pituitary adenomas and pheochromocytomas are thought to originate from neural crest-derived cells with common biochemical and ultrastructural features, the so-called APUD cells (31). Accordingly, the occurrence of these tumors in the same person would be related to aberrant neural crest development (32). However, the absence of an autosomal dominant pattern of transmission in families of the reported index cases makes the possibility of a MEN variant unlikely. Moreover, two separate genetic events would be necessary to explain this overlap.
Our patient was also diagnosed with intestinal polyposis. The characteristics of her intestinal disease were not indicative of a genetic disorder, but they were likely to be related to the acromegaly (1). The other findings in our case included GIST and thyroid follicular adenoma, tumors that have been observed in various syndromes. Carney complex, often caused by a mutation in the PRKAR1a gene, represents a dominantly inherited MEN syndrome first described in 1985, characterized by spotty skin pigmentation (lentiginosis), cardiac and peripheral myxomas, schwannomas, and endocrine overactivity MEN syndromes. Thyroid follicular neoplasms might be associated with GH-secreting pituitary adenomas in patients with the Carney complex, but in the presence of other endocrine tumors, such as primary pigmented nodular adrenocortical disease (PPNAD), large-cell calcifying Sertoli cell tumors, and Leydig cell tumors (6). In Carney triad (33), a non-inherited rare condition, patients may manifest GIST, lung chondroma, paraganglioma, adrenal cortical adenoma, pheochromocytoma, esophageal leiomyoma, and other abnormalities. More recently, a new association called Carney-Stratakis diad/syndrome was characterized by the association of paragangliomas and GIST, caused by germline mutations of the succinate dehydrogenase (SDH) subunit genes SDHB, SDHC, and SDHD (14,34), which were all negative in our patient. GIST can also occur in NF1, in which pheochromocytoma can be also present in approximately 1% of the patients. Cardinal clinical features of NF1 are multiple dermal neurofibromas, café au lait spots, axillary or inguinal freckling, and hamartomas of the iris (14). NF1 is mostly diagnosed based on the clinical features, but our patient did not have any dermatological/neurological signs that would indicate this disease.
In summary, our patient has an unusual combination of somatotroph pituitary adenoma, pheochromocytoma, follicular thyroid adenoma, multiple colon polyps, and GIST, which makes it likely that she harbors a currently unknown germline mutation predisposing her to multiple tumors in various organs. Based on genetic and clinical tests, we ruled out all the genes known to cause these abnormalities. Next-generation sequencing techniques will possibly become routinely available in the near future to identify the disease-causing abnormalities in cases like this.
Disclosure: no potential conflict of interest relevant to this article was reported.
Received on Aug/1/2012
Accepted on Sept/10/2012
- 1. Ben-Shlomo A, Melmed S. Acromegaly. Endocrinol Metab Clin North Am. 2008;37(1):101-22, viii.
- 2. Taboada GF, van Haute FR, Corrêa LL, Casini AF, Gadelha MR. Etiologic aspects and management of acromegaly. Arq Bras Endocrinol Metabol. 2005;49(5):626-40.
- 3. Garby L, Caron P, Claustrat F, Chanson P, Tabarin A, Rohmer V, et al. Clinical characteristics and outcome of acromegaly induced by ectopic secretion of growth hormone-releasing hormone (GHRH): a French nationwide series of 21 cases. J Clin Endocrinol Metab. 2012;97(6):2093-104.
- 4. Hoff AO, Hauache OM. [Multiple endocrine neoplasia type 1 (MEN 1): clinical, biochemical and molecular diagnosis and treatment of the associated disturbances]. Arq Bras Endocrinol Metabol. 2005;49(5):735-46.
- 5. Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis. 2008;3:12.
- 6. Boikos SA, Stratakis CA. Carney complex: the first 20 years. Curr Opin Oncol. 2007;19(1):24-9.
- 7. Gadelha MR, Prezant TR, Une KN, Glick RP, Moskal SF 2nd, Vaisman M, et al. Loss of heterozygosity on chromosome 11q13 in two families with acromegaly/gigantism is independent of mutations of the multiple endocrine neoplasia type I gene. J Clin Endocrinol Metab. 1999;84(1):249-56.
- 8. Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312(5777):1228-30.
- 9. Beckers A, Daly AF. The clinical, pathological, and genetic features of familial isolated pituitary adenomas. Eur J Endocrinol. 2007;157(4):371-82.
- 10. Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA, et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab. 2007;92(5):1891-6.
- 11. Pereira MA, Souza BF, Freire DS, Lucon AM. [Pheochromocytoma]. Arq Bras Endocrinol Metabol. 2004;48(5):751-75.
- 12. Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18(6):R253-76.
- 13. Maia AL, Gross JL, Punales MK. [Multiple endocrine neoplasia type 2]. Arq Bras Endocrinol Metabol. 2005;49(5):725-34.
- 14. Almeida MQ, Stratakis CA. Solid tumors associated with multiple endocrine neoplasias. Cancer Genet Cytogenet. 2010;203(1):30-6.
- 15. Asa SL, Kovacs K, Thorner MO, Leong DA, Rivier J, Vale W. Immunohistological localization of growth hormone-releasing hormone in human tumors. J Clin Endocrinol Metab. 1985;60(3):423-7.
- 16. Igreja S, Chahal HS, King P, Bolger GB, Srirangalingam U, Guasti L, et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum Mutat. 2010;31(8):950-60.
- 17. Ellard S, Hattersley AT, Brewer CM, Vaidya B. Detection of an MEN1 gene mutation depends on clinical features and supports current referral criteria for diagnostic molecular genetic testing. Clin Endocrinol (Oxf). 2005;62(2):169-75.
- 18. Iversen K. Acromegaly associated with phaeochromocytoma. Acta Med Scand. 1952;142(1):1-5.
- 19. Breckenridge SM, Hamrahian AH, Faiman C, Suh J, Prayson R, Mayberg M. Coexistence of a pituitary macroadenoma and pheochromocytoma--a case report and review of the literature. Pituitary. 2003;6(4):221-5.
- 20. Sano T, Saito H, Yamasaki R, Hosoi E, Kameyama K, Saito S, et al. Production and secretion of immunoreactive growth hormone-releasing factor by pheochromocytomas. Cancer. 1986;57(9):1788-93.
- 21. Roth KA, Wilson DM, Eberwine J, Dorin RI, Kovacs K, Bensch KG, et al. Acromegaly and pheochromocytoma: a multiple endocrine syndrome caused by a plurihormonal adrenal medullary tumor. J Clin Endocrinol Metab. 1986;63(6):1421-6.
- 22. Vieira Neto L, Taboada GF, Corrêa LL, Polo J, Nascimento AF, Chimelli L, et al. Acromegaly secondary to growth hormone-releasing hormone secreted by an incidentally discovered pheochromocytoma. Endocr Pathol. 2007;18(1):46-52.
- 23. Sleilati GG, Kovacs KT, Honasoge M. Acromegaly and pheochromocytoma: report of a rare coexistence. Endocr Pract. 2002;8(1):54-60.
- 24. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Höfler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103(42):15558-63.
- 25. Chahal HS, Trivellin G, Leontiou CA, Alband N, Fowkes RC, Tahir A, et al. Somatostatin analogs modulate AIP in somatotroph adenomas: the role of the ZAC1 pathway. J Clin Endocrinol Metab. 2012;97(8):E1411-20.
- 26. Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab. 2008;93(6):2390-401.
- 27. Daly AF, Tichomirowa MA, Petrossians P, Heliövaara E, Jaffrain-Rea ML, Barlier A, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab. 2010;95(11):E373-83.
- 28. Vargiolu M, Fusco D, Kurelac I, Dirnberger D, Baumeister R, Morra I, et al. The tyrosine kinase receptor RET interacts in vivo with aryl hydrocarbon receptor-interacting protein to alter survivin availability. J Clin Endocrinol Metab. 2009;94(7):2571-8.
- 29. Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44(5):328-33.
- 30. Erlic Z, Rybicki L, Peczkowska M, Golcher H, Kann PH, Brauckhoff M, et al. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res. 2009;15(20):6378-85.
- 31. Pearse AG. The cytochemistry and ultrastructure of polypeptide hormone-producing cells of the APUD series and the embryologic, physiologic and pathologic implications of the concept. J Histochem Cytochem. 1969;17(5):303-13.
- 32. Bolande RP. The neurocristopathies: a unifying concept of disease arising in neural crest maldevelopment. Human Pathology. 1974;5(4):409-29.
- 33. Carney JA. Carney triad: a syndrome featuring paraganglionic, adrenocortical, and possibly other endocrine tumors. J Clin Endocrinol Metab. 2009;94(10):3656-62.
- 34. McWhinney SR, Pasini B, Stratakis CA. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357(10):1054-6.
Correspondence to:
Publication Dates
-
Publication in this collection
02 Jan 2013 -
Date of issue
Nov 2012
History
-
Received
01 Aug 2012 -
Accepted
10 Sept 2012