Abstracts
Cowden syndrome is characterized by hamartomatous polyps, trichilemmomas, increased risk of developing neoplasms, and is associated with germline mutations in the PTEN gene. We searched for germline mutations in PTEN in a 49-year-old female patient who presented trichilemmoma with previous history of breast carcinoma, and thyroidectomy for a thyroid nodule. We also searched for somatic mutations in breast and thyroid tumoral tissues. DNA was extracted from peripheral leukocytes, paraffin samples of breast carcinoma, and cytological smears of thyroid nodule fine-needle aspiration biopsy, whose final histopathological diagnosis was adenomatous goiter. PTEN was amplified and sequenced. We identified a novel mutation, due to a T>A inversion at position 159 and A>T inversion at position 160, leading to valine-to-aspartic acid substitution at position 53. The p.Val53Asp was also found in homozygous state in samples obtained from adenocarcinoma breast and thyroid biopsy, denoting loss of heterozygosity. Here, we demonstrated a novel germline mutation in PTEN, as well as somatic loss of the wild-type PTEN allele in breast and thyroid tumors in a patient with Cowden syndrome. Arq Bras Endocrinol Metab. 2012;56(8):592-6
A síndrome de Cowden é caracterizada por pólipos de hamartoma, triquelomomas, risco aumentado em desenvolver neoplasias e está associada a mutações germinativas no gene PTEN. Procuramos por mutação germinativa no PTEN de uma paciente de 49 anos que apresentou triquilemomas com história pregressa de carcinoma de mama e realizou tireoidectomia devido a nódulo de tireoide. Investigamos também uma mutação somática em tecidos tumorais de mama e tireoide. O DNA foi extraído de leucócitos periféricos, de amostras de parafina de carcinoma de mama e exame citológico de nódulo de tireoide obtido de biópsia por agulha fina, cujo diagnóstico histopatológico foi de bócio adenomatoso. O PTEN foi amplificado e sequenciado. Identificou-se uma nova mutação em decorrência de uma inversão de T>A na posição 159 e A>T na posição 160, levando à substituição de valina para ácido aspártico na posição 53. A mutação p.Val53Asp também foi encontrada em estado homozigoto em amostras obtidas do adenocarcinoma de mama e da biópsia de nódulo tireoidiano, denotando perda de heterozigosidade. Portanto, demonstramos uma mutação germinativa no PTEN e também a perda somática do alelo selvagem PTEN no tumor de mama e da tireoide de uma paciente com síndrome de Cowden. Arq Bras Endocrinol Metab. 2012;56(8):592-6
CASE REPORT
New mutation in the PTEN gene in a Brazilian patient with Cowden's syndrome
Nova mutação no gene PTEN em um paciente brasileiro com síndrome de Cowden
Erika U. de LimaI; Iberê C. SoaresII; Debora L. S. DanilovicI; Suemi MaruiI
ILaboratório de Endocrinologia Celular e Molecular, Unidade de Tireoide (LIM/25), Disciplina de Endocrinologia e Metabologia, Faculdade de Medicina da Universidade de São Paulo (FMUSP), São Paulo, SP, Brazil
IIDepartamento de Patologia, FMUSP, São Paulo, SP, Brazil
Correspondence Correspondence to: Suemi Marui Av. Doutor Arnaldo, 455, sala 4345, 4º andar 01246-906 São Paulo, SP, Brazil suemimar@usp.br
SUMMARY
Cowden syndrome is characterized by hamartomatous polyps, trichilemmomas, increased risk of developing neoplasms, and is associated with germline mutations in the PTEN gene. We searched for germline mutations in PTEN in a 49-year-old female patient who presented trichilemmoma with previous history of breast carcinoma, and thyroidectomy for a thyroid nodule. We also searched for somatic mutations in breast and thyroid tumoral tissues. DNA was extracted from peripheral leukocytes, paraffin samples of breast carcinoma, and cytological smears of thyroid nodule fine-needle aspiration biopsy, whose final histopathological diagnosis was adenomatous goiter. PTEN was amplified and sequenced. We identified a novel mutation, due to a T>A inversion at position 159 and A>T inversion at position 160, leading to valine-to-aspartic acid substitution at position 53. The p.Val53Asp was also found in homozygous state in samples obtained from adenocarcinoma breast and thyroid biopsy, denoting loss of heterozygosity. Here, we demonstrated a novel germline mutation in PTEN, as well as somatic loss of the wild-type PTEN allele in breast and thyroid tumors in a patient with Cowden syndrome. Arq Bras Endocrinol Metab. 2012;56(8):592-6
SUMÁRIO
A síndrome de Cowden é caracterizada por pólipos de hamartoma, triquelomomas, risco aumentado em desenvolver neoplasias e está associada a mutações germinativas no gene PTEN. Procuramos por mutação germinativa no PTEN de uma paciente de 49 anos que apresentou triquilemomas com história pregressa de carcinoma de mama e realizou tireoidectomia devido a nódulo de tireoide. Investigamos também uma mutação somática em tecidos tumorais de mama e tireoide. O DNA foi extraído de leucócitos periféricos, de amostras de parafina de carcinoma de mama e exame citológico de nódulo de tireoide obtido de biópsia por agulha fina, cujo diagnóstico histopatológico foi de bócio adenomatoso. O PTEN foi amplificado e sequenciado. Identificou-se uma nova mutação em decorrência de uma inversão de T>A na posição 159 e A>T na posição 160, levando à substituição de valina para ácido aspártico na posição 53. A mutação p.Val53Asp também foi encontrada em estado homozigoto em amostras obtidas do adenocarcinoma de mama e da biópsia de nódulo tireoidiano, denotando perda de heterozigosidade. Portanto, demonstramos uma mutação germinativa no PTEN e também a perda somática do alelo selvagem PTEN no tumor de mama e da tireoide de uma paciente com síndrome de Cowden. Arq Bras Endocrinol Metab. 2012;56(8):592-6
INTRODUCTION
Cowden syndrome (CS [MIM 158350]), first described by Lloyd and Dennis in 1963 (1), is characterized by hamartomatous polyps of the gastrointestinal tract, mucocutaneous lesions, and increased risk for developing neoplasms, particularly breast, endometrium, thyroid, kidney and colorectal cancers. CS has been estimated to affect around 1 in every 200,000 individuals, although this rate is probably underestimated due to the highly variable phenotype (2). Approximately 80% of CS patients have PTEN mutations. The PTEN gene (10q23.3) [MIM 601728] encodes a ubiquitously expressed tumor suppressor dual-specificity phosphatase that antagonizes the PI3K signalling pathway by means of its lipid phosphatase activity, and negatively regulates the MAPK pathway by means of its protein phosphatase activity (3).
Major organs involved include breast, thyroid, uterus, brain, and mucocutaneous tissues. Breast cancer numbers among the tumor types in which germline PTEN mutation have been documented. Somatic PTEN mutation also occurs, but at a low frequency, and is not associated with CS (4). Thyroid tumors were also found to have somatic deletions of the PTEN gene, predominantly benign forms, suggesting that PTEN might also act as a tumor suppressor for these cancers (5).
Based on the Criteria of the International Cowden Consortium (6), we searched for PTEN mutation in a patient with trichilemmoma, breast carcinoma, and goiter.
CASE REPORT
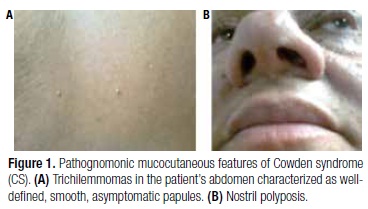
We studied a 49-year-old female patient with pathognomonic criteria of CS: several mucocutaneous lesions measuring 0.2 to 0.4 cm, diagnosed by biopsy as trichilemmoma and nasal mucosal papillomatosis (Figure 1). At 46 years of age, she presented a unilateral breast cancer classified as ductal carcinoma in situ with micropapillary, solid, and cribriform pattern. She was submitted to radio and hormonal (tamoxifen) therapy after surgery. During follow-up, a solid hypoechoic and well-delimited nodule measuring 1.3 x 1.4 x 1.8 cm was noted in the left lobe on thyroid ultrasonography, and the patient was referred to our Endocrinology Department. Fine-needle aspiration biopsy determined the classification of the nodule as Class II of the Bethesda system, and total thyroidectomy was performed, as findings were indicative of CS (7). Histopathological diagnosis was adenomatous goiter. Gastrointestinal mucosa was normal on endoscopy and colonoscopy. The patient's parents and son were both asymptomatic. Nevertheless, DNA analysis was performed for genetic counseling.
DNA analysis
DNA was extracted from peripheral leukocytes as described elsewhere (8). DNA was also extracted from breast cancer tissue obtained from paraffin material and from thyroid cells from FNAB slides, as described elsewhere (9,10).
The PTEN gene was amplified by PCR, including the coding region and exon-intron boundaries, followed by automatic sequencing (ABI Prism 3130 xl, Applied Biosystems, Foster City, CA) and results were compared with the normal sequence. Primers and PCR conditions were amplified as described in table 1.
RESULTS
The DNA sequence from peripheral leukocytes showed a T-to-A inversion at position 159 and an A-to-T inversion at position 160 in exon 2, leading to a valine-to-aspartic acid substitution at position 53 of the protein (p.V53D) (Figure 2A). This p.Val53Asp mutation was also found in the adenocarcinoma breast tissue and in the thyroid nodule, in a homozygous status (Figure 2B). We have not identified this mutation in 200 control alleles. The patient's father also presented this novel mutation in DNA extracted from peripheral leukocytes. Her son and mother showed normal sequences (Figure 2C).
This Valine-53 is a highly conserved amino acid among different species (Figure 3A). Predictive analysis of the protein secondary structure with the p.Val53Asp mutation showed minor structural changes in PTEN: (1) strand elongation at position 124; (2) reduced coil at position 129; (3) coil stretching at positions 123 and 227 of the PTEN protein; (4) coil structure for the strand exchange at position 269; (5) reduced helix elongation at position 290 (Figure 3B).
DISCUSSION
Germline mutations in PTEN have been described in a variety of rare syndromes, collectively known as PTEN hamartoma tumor syndromes (PHTS). The defining clinical feature of PHTS is the presence of hamartomatous tumors, which are the disorganized growth of native cells in native tissues (11). The phenotypic spectrum of PHTS is seen in Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome, and adult Lhermitte-Duclos disease. Lhermitte-Duclos disease is characterized by a dysplastic expansion of ganglion cells within the cerebellum, leading to replacement of the cerebellar internal granule cell layer and, consequently, various degrees of neurological signs, such as macrocephaly, mental retardation, seizures, and tremors (12). Bannayan-Riley-Ruvalcaba syndrome presents macrocephaly, benign hamartomas, pigmented macules of the glans penis, lipomas, hemangiomas, and developmental delay or mental retardation (11). Cowden syndrome is characterized by mucocutaneous features, including trichilemmomas and papillomatous papules. PTEN hamartoma tumor syndromes are inherited in an autosomal dominant manner across all subtypes.
PTEN is a tumor suppressor gene with dual specificity phosphatase activity, and the N-terminal domain contains the phosphatase domain active site, where most PTEN mutations occur (13). PTEN negatively regulates the phosphatidylinositol 3-kinase-AKT and mammalian target of rapamycin (mTOR) signalling pathways, which are critical for cell proliferation, cell cycle progression, and apoptosis. Loss of function in this gene is a risk factor for oncogenesis and PTEN is therefore considered a tumor suppressor gene (3). Loss of function in PTEN activates these pathways and leads to increased cellular growth, migration, proliferation, and survival.
Gastrointestinal polyps, also typically found in CS, are usually asymptomatic and can occur anywhere in the tract, but mostly at the colon (13,14). Cowden syndrome patients are at increased risk of developing breast, thyroid, and endometrium cancer. Lifetime risk of breast cancer in women with CS is estimated to be as high as 50% as compared to 11% within the general population (15,16). As with other hereditary breast cancer syndromes, breast cancer is diagnosed at younger ages compared with the general population. The most frequently observed breast cancer histology is invasive ductal adenocarcinoma. As our patient had no familial history of breast cancer, but presented breast cancer at a young age, the molecular study should clarify the prognosis (16).
Benign thyroid lesions occur in up to 75% of patients with CS and may include adenomas, hamartomas, multinodular goiter, and Hashimoto's thyroiditis (17). If a molecular study is not available, total thyroidectomy is recommended, even after benign cytological diagnosis, given the increased risk of developing subsequent thyroid cancer (6). If molecular study on FNAB material is performed, total thyroidectomy should be indicated, as clinicians are aware of the increased thyroid cancer risk when the PTEN mutation is found. We studied the LOH in thyroid cells obtained from FNAB to clarify whether PTEN had a role in the thyroid cell proliferation observed in our patient. The role of PTEN in her abnormal cellular proliferation was consequently reinforced, despite being a benign lesion (adenomatous goiter).
It is noteworthy that the patient's father also carried the p.V53D mutation and is apparently asymptomatic. To date, he has tested normal on prostate clinical evaluation and serum PSA, as well as on kidney ultrasound and colonoscopy. These exams will be performed annually to exclude prostate, colorectal and kidney cancer, respectively. His thyroid ultrasound was also normal. As phenotype and genotype may be poorly related in CS, he is being followed up closely. As her son does not bear the PTEN mutation, he is not considered at risk. As recommended by the International Cowden Consortium, our patient and her father must undergo cancer screening annually (endometrium, kidney, skin, prostate, colon, and thyroid) (6).
Most of the described mutations are frameshift or stop codon mutations throughout the whole gene, except exons 1 and 9 (17). A mutational hotspot was found in exon 5, which encodes the phosphatase catalytic core motif (18). The p.Val53Asp mutation described here is located at exon 2 of the PTEN gene, and is located in a highly conserved amino acid (Figure 3A). Twelve different germline mutations have been identified in exon 2 and are distributed as shown in Figure 4, all related to patients with CS (15). Given the absence of a described mutation in 200 normal alleles, p.V53 has shown to be a highly conserved amino acid among species, and changes in secondary mutated protein structure corroborate to link phenotype and genotype (Figure 3). Furthermore, deletion of one amino acid (D52) was described previously, and was associated with CS (19).
In conclusions, we identified a novel germline p.Val53Asp mutation in the PTEN gene in a patient with Cowden syndrome, and demonstrated its involvement in breast cancer and thyroid lesions. Clinical management of patients with CS should include early and frequent screening for associated malignancies.
Disclosure: no potential conflict of interest relevant to this article was reported.
Received on July/25/2012
Accepted on Nov/5/2012
- 1. Lloyd KM, Dennis M. Cowden's disease. A possible new symptom complex with multiple system involvement. Ann Intern Med. 1963(1);58:136-42.
- 2. Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. 2004;41(5):323-6.
- 3. Pezzolesi MG, Zbuk KM, Waite KA, Eng C. Comparative genomic and functional analyses reveal a novel cis-acting PTEN regulatory element as a highly conserved functional E-box motif deleted in Cowden syndrome. Hum Mol Genet. 2007;16(9):1058-71.
- 4. Rhei E, Kang L, Bogomolniy F, Federici MG, Borgen PI, Boyd J. Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in primary breast carcinomas. Cancer Res. 1997;57(17):3657-9.
- 5. Dahia PL, Marsh DJ, Zheng Z, Zedenius J, Komminoth P, Frisk T, et al. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 1997;57(21):4710-3.
- 6. Blumenthal GM, Dennis PA. PTEN hamartoma tumor syndromes. Eur J Hum Genet. 2008;16(11):1289-300.
- 7. Cibas ES, Ali SZ. The Bethesda System for Reporting Thyroid Cytopathology. Thyroid. 2009;19(11):1159-65.
- 8. Abrao MG, Billerbeck AE, Nishi MY, Marui S, Mendonca BB. [Standardization of DNA extraction with NaCl from oral mucosa cells: application in PROP1 gene study]. Arq Bras Endocrinol Metabol. 2005;49(6):978-82.
- 9. Bielawski K, Zaczek A, Lisowska U, Dybikowska A, Kowalska A, Falkiewicz B. The suitability of DNA extracted from formalin-fixed, paraffin-embedded tissues for double differential polymerase chain reaction analysis. Int J Mol Med. 2001;8(5):573-8.
- 10. Troncone G, Cozzolino I, Fedele M, Malapelle U, Palombini L. Preparation of thyroid FNA material for routine cytology and BRAF testing: a validation study. Diagn Cytopathol. 2010;38(3):172-6.
- 11. Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ, Ahmed SF, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8(8):1461-72.
- 12. Zhou XP, Marsh DJ, Morrison CD, Chaudhury AR, Maxwell M, Reifenberger G, et al. Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. Am J Hum Genet. 2003;73(5):1191-8.
- 13. Pilarski R, Stephens JA, Noss R, Fisher JL, Prior TW. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet. 2011;48(8):505-12.
- 14. Nelen MR, Padberg GW, Peeters EA, Lin AY, van den Helm B, Frants RR, et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet. 1996;13(1):114-6.
- 15. Agrawal S, Eng C. Differential expression of novel naturally occurring splice variants of PTEN and their functional consequences in Cowden syndrome and sporadic breast cancer. Hum Mol Genet. 2006;15(5):777-87.
- 16. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16(1):64-7.
- 17. Harach HR, Soubeyran I, Brown A, Bonneau D, Longy M. Thyroid pathologic findings in patients with Cowden disease. Ann Diagn Pathol. 1999;3(6):331-40.
- 18. Bonneau D, Longy M. Mutations of the human PTEN gene. Hum Mutat. 2000;16(2):109-22.
- 19. Tan MH, Mester J, Peterson C, Yang Y, Chen JL, Rybicki LA, et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011;88(1):42-56.
Correspondence to:
Publication Dates
-
Publication in this collection
02 Jan 2013 -
Date of issue
Nov 2012
History
-
Received
25 July 2012 -
Accepted
05 Nov 2012